
Peroxisome Proliferator-Activated Receptor α Has a Protective Effect on Fatty Liver Caused by Excessive Sucrose Intake

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Diet
2.3. Serum Chemistry
2.4. Glucose and Insulin Tests
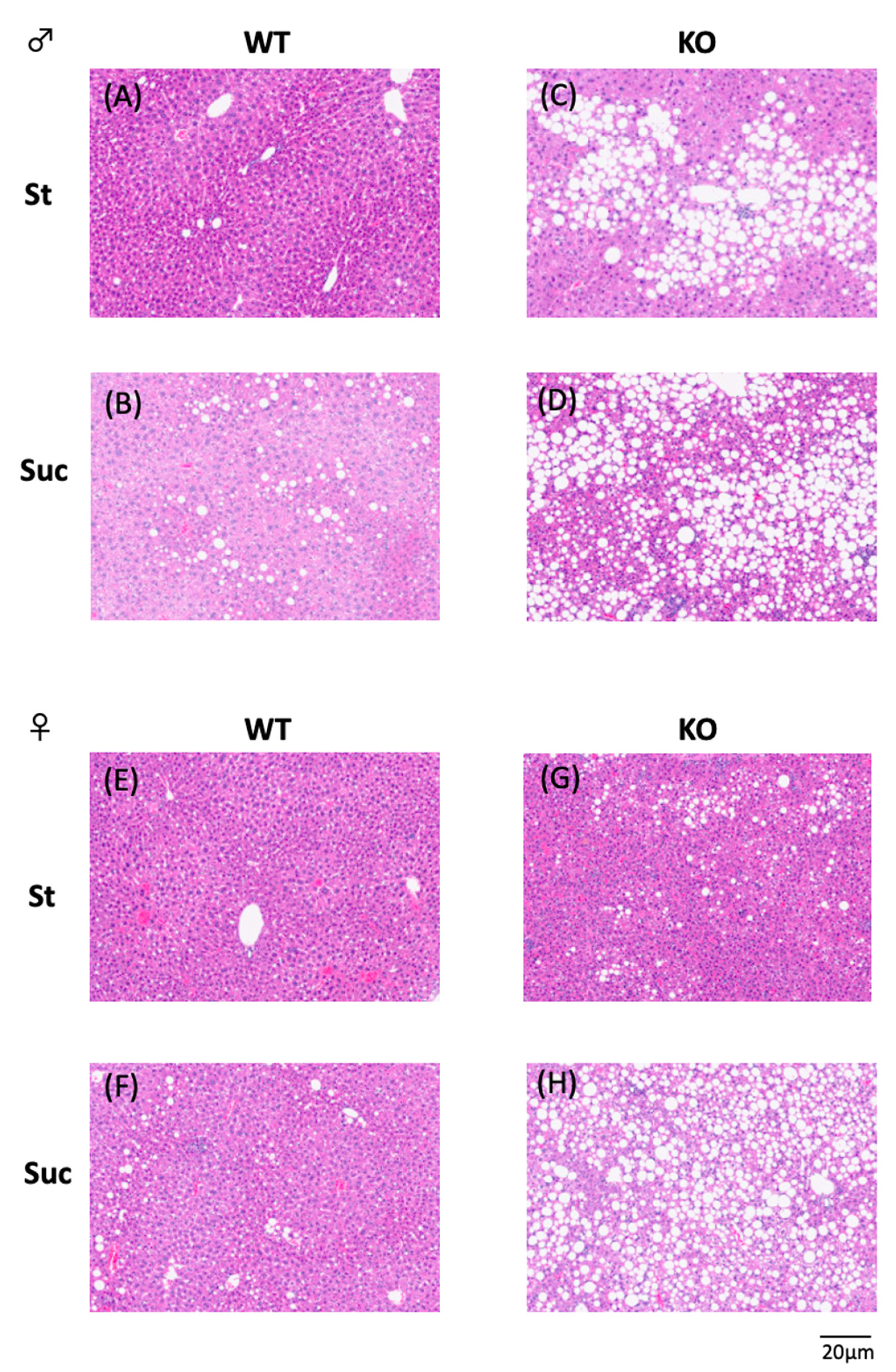
2.5. Hepatic Histology
2.6. Quantitative Real-Time PCR
2.7. Statistical Analysis
3. Results
3.1. Body Weight and Tissue Weights
3.2. Serum Analysis
3.3. Hepatic Lipid Analysis
3.4. Hepatic mRNA Expression Analysis
3.5. Glucose Tolerance Test and Insulin Tolerance Test
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Francque, S.M.; Marchesini, G.; Kautz, A.; Walmsley, M.; Dorner, R.; Lazarus, J.V.; Zelber-Sagi, S.; Hallsworth, K.; Busetto, L.; Frühbeck, G.; et al. Non-alcoholic fatty liver disease: A patient guideline. JHEP Rep. 2021, 3, 100322. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Farahvash, M.J.; Janney, C.G.; Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107, 1103–1109. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H. Sterol regulatory element-binding protein family as global regulators of lipid synthetic genes in energy metabolism. Vitam. Horm. 2002, 65, 167–194. [Google Scholar] [PubMed]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Investig. 1998, 101, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, J.; Jackson, S.M.; Kim, J.B.; Spiegelman, B.M.; Edwards, P.A. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J. Biol. Chem. 1997, 272, 7298–7305. [Google Scholar] [CrossRef] [PubMed]
- Kast, H.R.; Nguyen, C.M.; Anisfeld, A.M.; Ericsson, J.; Edwards, P.A. CTP:phosphocholine cytidylyltransferase, a new sterol- and SREBP-responsive gene. J. Lipid Res. 2001, 42, 1266–1272. [Google Scholar] [CrossRef]
- Shimano, H.; Horton, J.D.; Hammer, R.E.; Shimomura, I.; Brown, M.S.; Goldstein, J.L. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Investig. 1996, 98, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Salehi, S.; Cheung, P.; Homko, C.; Song, W.; Loveland-Jones, C.; Jayarajan, S. Comparison of in vivo effects of insulin on SREBP-1c activation and INSIG-1/2 in rat liver and human and rat adipose tissue. Obesity 2013, 21, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef]
- Ye, J.; DeBose-Boyd, R.A. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004754. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Nakamori, A.; Sasaki, E.; Wada, S.; Ezaki, O. Fish oil prevents sucrose-induced fatty liver but exacerbates high-safflower oil-induced fatty liver in ddy mice. Hepatology 2007, 46, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Kishimoto, K.; Miura, S.; Ezaki, O. Dietary beta-conglycinin prevents fatty liver induced by a high-fat diet by a decrease in peroxisome proliferator-activated receptor gamma2 protein. J. Nutr. Biochem. 2012, 23, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Hillgartner, F.B.; Salati, L.M.; Goodridge, A.G. Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiol. Rev. 1995, 75, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Dutcher, A.K.; Towle, H.C. Glucose and insulin function through two distinct transcription factors to stimulate expression of lipogenic enzyme genes in liver. J. Biol. Chem. 2001, 276, 9437–9445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.M.; Liu, Z.; Towle, H.C. Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J. Biol. Chem. 1995, 270, 21991–21997. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Mannaerts, G.P. Peroxisomal lipid metabolism. Annu. Rev. Nutr. 1994, 14, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx, J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta 1996, 1302, 93–109. [Google Scholar] [CrossRef]
- Inoue, M.; Ohtake, T.; Motomura, W.; Takahashi, N.; Hosoki, Y.; Miyoshi, S.; Suzuki, Y.; Saito, H.; Kohgo, Y.; Okumura, T. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun. 2005, 336, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [Green Version]
- Patsouris, D.; Reddy, J.K.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Regnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouche, E.; Lasserre, F.; Naylies, C.; Betoulieres, C.; et al. Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ikaga, R.; Ogawa, H.; Yamazaki, T. Different expressions of clock genes in fatty liver induced by high-sucrose and high-fat diets. Chronobiol. Int. 2021, 38, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yasui, K.; Tani, Y.; Yamada, H. Acyl-CoA oxidase from Candida tropicalis. Biochem. Biophys. Res. Commun. 1979, 91, 108–113. [Google Scholar] [CrossRef]
- Allain, C.C.; Poon, L.S.; Chan, C.S.; Richmond, W.; Fu, P.C. Enzymatic determination of total serum cholesterol. Clin. Chem. 1974, 20, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Spayd, R.W.; Bruschi, B.; Burdick, B.A.; Dappen, G.M.; Eikenberry, J.N.; Esders, T.W.; Figueras, J.; Goodhue, C.T.; LaRossa, D.D.; Nelson, R.W.; et al. Multilayer film elements for clinical analysis: Applications to representative chemical determinations. Clin. Chem. 1978, 24, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Okawa, S.; Takahashi, M. The effects on weight loss and gene expression in adipose and hepatic tissues of very-low carbohydrate and low-fat isoenergetic diets in diet-induced obese mice. Nutr. Metab. 2016, 13, 78. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Gulick, T.; Cresci, S.; Caira, T.; Moore, D.D.; Kelly, D.P. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc. Natl. Acad. Sci. USA 1994, 91, 11012–11016. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Li, C.Y.; Bina, H.A.; Lynes, S.E.; Michael, M.D.; Shanafelt, A.B.; Kharitonenkov, A.; Wasserman, D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 2009, 150, 4084–4093. [Google Scholar] [CrossRef] [Green Version]
- Motomura, W.; Inoue, M.; Ohtake, T.; Takahashi, N.; Nagamine, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem. Biophys. Res. Commun. 2006, 340, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Dobrzyn, A.; Man, W.C.; Chu, K.; Sampath, H.; Kim, H.J.; Ntambi, J.M. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J. Biol. Chem. 2004, 279, 25164–25171. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Knight, B.L.; Wiggins, D.; Humphreys, S.M.; Gibbons, G.F. Disturbances in the normal regulation of SREBP-sensitive genes in PPAR alpha-deficient mice. J. Lipid Res. 2001, 42, 328–337. [Google Scholar] [CrossRef]
- Kang, J.H.; Yun, S.I.; Park, M.H.; Park, J.H.; Jeong, S.Y.; Park, H.O. Anti-obesity effect of Lactobacillus gasseri BNR17 in high-sucrose diet-induced obese mice. PLoS ONE 2013, 8, e54617. [Google Scholar] [CrossRef]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D., Jr. Loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; Shapiro, J.; Abraham, N.G.; Kappas, A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity 2014, 22, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lei, T.; Huang, J.F.; Wang, S.B.; Zhou, L.L.; Yang, Z.Q.; Chen, X.D. The link between fibroblast growth factor 21 and sterol regulatory element binding protein 1c during lipogenesis in hepatocytes. Mol. Cell. Endocrinol. 2011, 342, 41–47. [Google Scholar] [CrossRef]
- Cha, D.R.; Han, J.Y.; Su, D.M.; Zhang, Y.; Fan, X.; Breyer, M.D.; Guan, Y. Peroxisome proliferator-activated receptor-alpha deficiency protects aged mice from insulin resistance induced by high-fat diet. Am. J. Nephrol. 2007, 27, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARgamma2 is an initial event to induce fatty liver in response to a diet high in butter: PPARgamma2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Costet, P.; Legendre, C.; More, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef] [PubMed]
- Edvardsson, U.; Ljungberg, A.; Linden, D.; William-Olsson, L.; Peilot-Sjogren, H.; Ahnmark, A.; Oscarsson, J. PPARalpha activation increases triglyceride mass and adipose differentiation-related protein in hepatocytes. J. Lipid Res. 2006, 47, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Jalouli, M.; Carlsson, L.; Ameen, C.; Linden, D.; Ljungberg, A.; Michalik, L.; Eden, S.; Wahli, W.; Oscarsson, J. Sex difference in hepatic peroxisome proliferator-activated receptor alpha expression: Influence of pituitary and gonadal hormones. Endocrinology 2003, 144, 101–109. [Google Scholar] [CrossRef]
- Trottier, J.; Caron, P.; Straka, R.J.; Barbier, O. Profile of serum bile acids in noncholestatic volunteers: Gender-related differences in response to fenofibrate. Clin. Pharmacol. Ther. 2011, 90, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bryzgalova, G.; Hedman, E.; Khan, A.; Efendic, S.; Gustafsson, J.A.; Dahlman-Wright, K. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: A possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol. Endocrinol. 2006, 20, 1287–1299. [Google Scholar] [CrossRef]
- Bryzgalova, G.; Lundholm, L.; Portwood, N.; Gustafsson, J.A.; Khan, A.; Efendic, S.; Dahlman-Wright, K. Mechanisms of antidiabetogenic and body weight-lowering effects of estrogen in high-fat diet-fed mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E904–E912. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.; Jornayvaz, F.R.; Lee, H.Y.; Kanda, S.; Guigni, B.A.; Kahn, M.; Samuel, V.T.; Carvalho, C.R.; Petersen, K.F.; Jurczak, M.J.; et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology 2013, 154, 1021–1028. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dietary Constituents | St | Suc |
|---|---|---|
| g/100 g | ||
| Safflower oil | 1.0 | 1.0 |
| Butter | 3.6 | 3.6 |
| Casein | 19.7 | 19.7 |
| α-Starch | 66.3 | 16.5 |
| Sucrose | - | 49.8 |
| Vitamin mix (AIN-93) | 1.0 | 1.0 |
| Mineral mix (AIN-93) | 3.5 | 3.5 |
| Cellulose powder | 5.0 | 5.0 |
| L-Cystine | 0.3 | 0.3 |
| en% | ||
| Fat | 10 | 10 |
| Carbohydrate | 70 | 70 |
| Protein | 20 | 20 |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| 36b4 | GGCCCTGCACTCTCGCTTTC | TGCCAGGACGCGCTTGT |
| Acc1 | GGACAGACTGATCGCAGAGAAAG | TGGAGAGCCCCACACACA |
| Adrp | AAGAGGCCAAACAAAAGAGCCAGGAGACCA | ACCCTGAATTTTCTGGTTGGCACTGTGCAT |
| Cd36 | AATGGCACAGACGCAGCCT | GGTTGTCTGGATTCTGGA |
| Cpt1 | GCACTGCAGCTCGCACATTACAA | CTCAGACAGTACCTCCTTCAGGAAA |
| Fas | GCTGCGGAAACTTCAGGAAAT | AGAGACGTGTCACTCCTGGACTT |
| Fgf21 | ATGGAATGGATGAGATCTAGAGTTGG | TCTTGGTCGTCATCTGTGTAGAGG |
| Mcad | GATCGCAATGGGTGCTTTTGATAGAA | AGCTGATTGGCAATGTCTCCAGCAAA |
| Pparα | CCTCAGGGTACCACTACGGAGT | GCCGAATAGTTCGCCGAA |
| Pparγ1 | GAGTGTGACGACAAGATTTG | GGTGGGCCAGAATGGCATCT |
| Pparγ2 | TCTGGGAGATTCTCCTGTTGA | GGTGGGCCAGAATGGCATCT |
| Scd1 | CCCCTGCGGATCTTCCTTAT | AGGGTCGGCGTGTGTTTCT |
| Srebp-1c | GGAGCCATGGATTGCACATT | CCTGTCTCACCCCCAGCATA |
| WT | KO | Two-Way ANOVA p Value | |||||
|---|---|---|---|---|---|---|---|
| St | Suc | St | Suc | Strain | Diet | Strain × Diet | |
| Male | |||||||
| n | 6 | 6 | 6 | 6 | |||
| Total Energy Intake (kcal/day/mouse) | 16.6 ± 0.4 | 14.6 ± 0.4 | 15.7 ± 0.4 | 14.7 ± 1.3 | 0.271 | 0.001 | 0.225 |
| Weight (g) | |||||||
| BW at start | 34.8 ± 2.0 | 34.9 ± 1.3 | 33.3 ± 1.3 | 34.9 ± 2.6 | 0.433 | 0.390 | 0.413 |
| BW | 36.0 ± 2.8 | 38.0 ± 1.6 | 35.7 ± 2.2 | 40.6 ± 5.4 | 0.503 | 0.055 | 0.397 |
| Liver | 1.300 ± 0.144 | 1.704 ± 0.137 | 1.746 ± 0.183 | 2.209 ± 0.345 | <0.001 | 0.001 | 0.793 |
| Epididymal WAT | 1.388 ± 0.261 | 1.716 ± 0.107 | 1.267 ± 0.253 | 1.566 ± 0.468 | 0.381 | 0.054 | 0.924 |
| Retroperitoneal WAT | 0.311 ± 0.074 | 0.404 ± 0.057 | 0.207 ± 0.030 | 0.282 ± 0.098 | 0.005 | 0.028 | 0.795 |
| Mesenteric WAT | 0.493 ± 0.099 | 0.712 ± 0.071 | 0.461 ± 0.089 | 0.682 ± 0.271 | 0.698 | 0.012 | 0.998 |
| Subcutaneous WAT | 0.858 ± 0.216 | 0.996 ± 0.159 | 0.487 ± 0.054 | 0.711 ± 0.375 | 0.012 | 0.139 | 0.716 |
| BAT | 0.208 ± 0.039 | 0.209 ± 0.024 | 0.162 ± 0.014 | 0.191 ± 0.035 | 0.046 | 0.323 | 0.382 |
| Quadriceps | 0.294 ± 0.025 | 0.312 ± 0.016 | 0.289 ± 0.021 | 0.307 ± 0.051 | 0.755 | 0.247 | 0.998 |
| Gastrocnemius | 0.247 ± 0.023 | 0.241 ± 0.024 | 0.240 ± 0.018 | 0.267 ± 0.017 | 0.380 | 0.326 | 0.134 |
| Female | |||||||
| n | 6 | 6 | 6 | 6 | |||
| Total Energy Intake (kcal/day/mouse) | 15.7 ± 0.8 | 13.4 ± 1.1 | 14.3 ± 0.3 | 12.9 ± 0.4 | 0.009 | <0.001 | 0.228 |
| Weight (g) | |||||||
| BW at start | 26.4 ± 1.1 | 26.2 ± 1.0 | 26.0 ± 1.1 | 26.1 ± 1.0 | 0.621 | 0.935 | 0.750 |
| BW | 27.1 ± 0.8 | 26.5 ± 0.9 | 26.2 ± 1.6 | 26.6 ± 1.6 | 0.495 | 0.893 | 0.387 |
| Liver | 1.090 ± 0.128 | 1.251 ± 0.050 | 1.308 ± 0.110 | 1.469 ± 0.222 | 0.004 | 0.026 | 0.998 |
| Periuterine WAT | 0.715 ± 0.202 | 0.529 ± 0.261 | 1.008 ± 0.239 | 1.262 ± 0.273 | <0.001 | 0.765 | 0.065 |
| Retroperitoneal WAT | 0.098 ± 0.017 | 0.085 ± 0.026 | 0.069 ± 0.020 | 0.071 ± 0.028 | 0.057 | 0.634 | 0.463 |
| Mesenteric WAT | 0.243 ± 0.043 | 0.255 ± 0.047 | 0.306 ± 0.065 | 0.363 ± 0.074 | 0.005 | 0.217 | 0.420 |
| Subcutaneous WAT | 0.381 ± 0.094 | 0.295 ± 0.051 | 0.317 ± 0.065 | 0.283 ± 0.036 | 0.230 | 0.063 | 0.409 |
| BAT | 0.109 ± 0.019 | 0.096 ± 0.009 | 0.107 ± 0.015 | 0.097 ± 0.011 | 0.905 | 0.097 | 0.874 |
| Quadriceps | 0.263 ± 0.022 | 0.283 ± 0.031 | 0.255 ± 0.023 | 0.258 ± 0.025 | 0.177 | 0.343 | 0.475 |
| Gastrocnemius | 0.212 ± 0.023 | 0.198 ± 0.012 | 0.192 ± 0.009 | 0.183 ± 0.010 | 0.016 | 0.106 | 0.729 |
| WT | KO | Two-Way ANOVA p Value | |||||
|---|---|---|---|---|---|---|---|
| St | Suc | St | Suc | Strain | Diet | Strain × Diet | |
| Male | |||||||
| Glucose (mg/dL) | 110.2 ± 6.9 | 114.2 ± 6.6 | 88.4 ± 6.1 | 94.8 ± 4.5 | <0.001 | 0.107 | 0.698 |
| TG (mg/dL) | 123.5 ± 25.1 | 137.2 ± 20.6 | 120.8 ± 15.4 | 173.0 ± 32.3 | 0.191 | 0.015 | 0.132 |
| TC (mg/dL) | 97.0 ± 21.7 | 119.8 ± 46.7 | 96.5 ± 5.7 | 141.0 ± 26.2 | 0.487 | 0.034 | 0.465 |
| NEFA (mEq/L) | 0.481 ± 0.052 | 0.662 ± 0.134 | 0.533 ± 0.076 | 0.851 ± 0.140 | 0.039 | <0.001 | 0.219 |
| Female | |||||||
| Glucose (mg/dL) | 104.2 ± 5.0 | 107.3 ± 10.5 | 94.2 ± 7.8 | 94.5 ± 4.1 | 0.003 | 0.607 | 0.679 |
| TG (mg/dL) | 76.3 ± 10.6 | 76.0 ± 20.0 | 106.7 ± 19.2 | 80.3 ± 17.9 | 0.041 | 0.107 | 0.117 |
| TC (mg/dL) | 86.0 ± 10.2 | 97.9 ± 15.3 | 98.2 ± 18.9 | 102.3 ± 10.9 | 0.220 | 0.232 | 0.558 |
| NEFA (mEq/L) | 0.539 ± 0.143 | 0.500 ± 0.216 | 0.982 ± 0.231 | 1.112 ± 0.144 | <0.001 | 0.601 | 0.339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazaki, T.; Ihato, M. Peroxisome Proliferator-Activated Receptor α Has a Protective Effect on Fatty Liver Caused by Excessive Sucrose Intake. Biomedicines 2022, 10, 2199. https://doi.org/10.3390/biomedicines10092199
Yamazaki T, Ihato M. Peroxisome Proliferator-Activated Receptor α Has a Protective Effect on Fatty Liver Caused by Excessive Sucrose Intake. Biomedicines. 2022; 10(9):2199. https://doi.org/10.3390/biomedicines10092199
Chicago/Turabian StyleYamazaki, Tomomi, and Megumi Ihato. 2022. "Peroxisome Proliferator-Activated Receptor α Has a Protective Effect on Fatty Liver Caused by Excessive Sucrose Intake" Biomedicines 10, no. 9: 2199. https://doi.org/10.3390/biomedicines10092199
APA StyleYamazaki, T., & Ihato, M. (2022). Peroxisome Proliferator-Activated Receptor α Has a Protective Effect on Fatty Liver Caused by Excessive Sucrose Intake. Biomedicines, 10(9), 2199. https://doi.org/10.3390/biomedicines10092199