

Modifiable Innate Biology within the Gut–Brain Axis for Alzheimer’s Disease
 ,
,  and
and 
{kind=link}
Abstract
1. Introduction
2. Targeting Innate Immunity in Alzheimer’s Disease
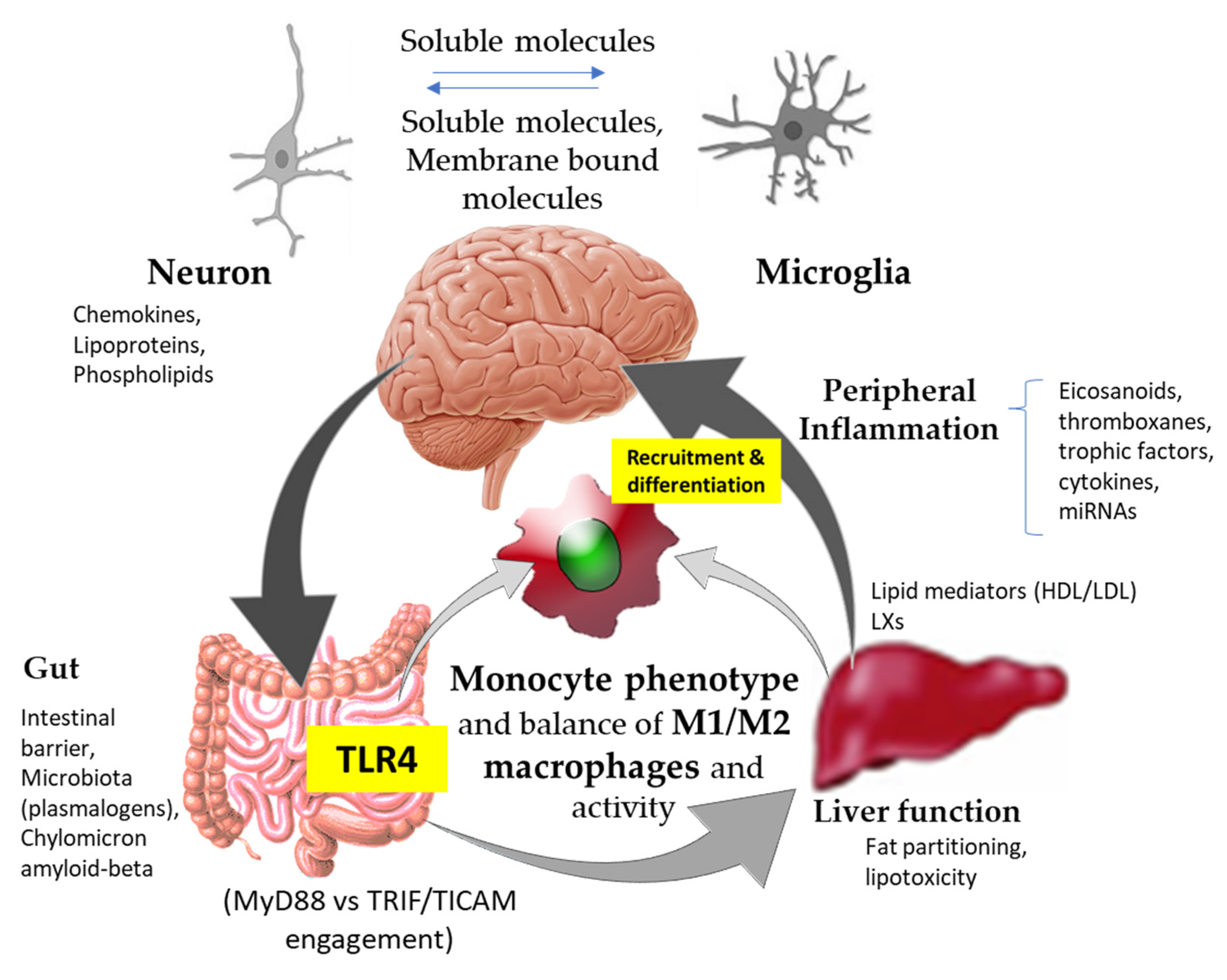
2.1. Gut–Liver–Brain Axis
2.2. Innate Immune Receptors
3. A Journey between Modifiable Factors within the Gut–Liver–Brain Axis for AD
3.1. Innate Immunity and Lipid Metabolism
3.2. Innate Immunity within the Gut–Liver Axis
3.3. Microbial Influence
4. Modifiable Proinflammatory Lipid Mediators in AD
4.1. Fatty Acids: Eicosanoids, Thromboxanes and Prostaglandins
4.2. Inflammatory Regulation
5. The Innate Immune Reflex of Metabolic Interventions in Innate Immunity in AD
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the Global Burden of Alzheimer’s Disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The Global Prevalence of Dementia: A Systematic Review and Metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Novikova, G.; Kapoor, M.; TCW, J.; Abud, E.M.; Efthymiou, A.G.; Chen, S.X.; Cheng, H.; Fullard, J.F.; Bendl, J.; Liu, Y.; et al. Integration of Alzheimer’s Disease Genetics and Myeloid Genomics Identifies Disease Risk Regulatory Elements and Genes. Nat. Commun. 2021, 12, 1610. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Risk Factors for Alzheimer’s Disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.K.; Lin, S.L.; Schooling, C.M. Re-Thinking Alzheimer’s Disease Therapeutic Targets Using Gene-Based Tests. EBioMedicine 2018, 37, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Pons, V.; Rivest, S. Targeting Systemic Innate Immune Cells as a Therapeutic Avenue for Alzheimer Disease. Pharmacol. Rev. 2022, 74, 1–17. [Google Scholar] [CrossRef]
- Rabiee, N.; Bagherzadeh, M.; Rabiee, M. A Perspective to the Correlation Between Brain Insulin Resistance and Alzheimer: Medicinal Chemistry Approach. Curr. Diabetes Rev. 2019, 15, 255–258. [Google Scholar] [CrossRef]
- Liu, Y.; Thalamuthu, A.; Mather, K.A.; Crawford, J.; Ulanova, M.; Wong, M.W.K.; Pickford, R.; Sachdev, P.S.; Braidy, N. Plasma Lipidome Is Dysregulated in Alzheimer’s Disease and Is Associated with Disease Risk Genes. Transl. Psychiatry 2021, 11, 344. [Google Scholar] [CrossRef]
- García-Domínguez, I.; Veselá, K.; García-Revilla, J.; Carrillo-Jiménez, A.; Roca-Ceballos, M.A.; Santiago, M.; de Pablos, R.M.; Venero, J.L. Peripheral Inflammation Enhances Microglia Response and Nigral Dopaminergic Cell Death in an in Vivo MPTP Model of Parkinson’s Disease. Front. Cell. Neurosci. 2018, 12, 398. [Google Scholar] [CrossRef]
- Muñoz Fernández, S.S.; Lima Ribeiro, S.M. Nutrition and Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 677–697. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Selma-Gracia, R.; Megušar, P.; Haros, C.M.; Laparra, J.M. Immunonutritional Bioactives from Chenopodium quinoa and Salvia Hispanica L. Flour Positively Modulate Insulin Resistance and Preserve Alterations in Peripheral Myeloid Population. Nutrients 2021, 13, 1537. [Google Scholar] [CrossRef] [PubMed]
- Srdić, M.; Ovčina, I.; Fotschki, B.; Haros, C.M.; Llopis, J.M.L. C. Quinoa and S. Hispanica L. Seeds Provide Immunonutritional Agonists to Selectively Polarize Macrophages. Cells 2020, 9, 593. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Jin, J.; Lim, J.-E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.-D.; Tahara, K.; Lalonde, R.; Fukuchi, K. TLR4 Mutation Reduces Microglial Activation, Increases Aβ Deposits and Exacerbates Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Brown, D.; Saiz, B. Chenopodium Quinoa and Salvia hispanica Provide Immunonutritional Agonists to Ameliorate Hepatocarcinoma Severity under a High-Fat Diet. Nutrients 2020, 12, 1946. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty Food, Fatty Acids, and Microglial Priming in the Adult and Aged Hippocampus and Amygdala. Brain Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef]
- Pallebage-Gamarallage, M.M.S.; Takechi, R.; Lam, V.; Galloway, S.; Dhaliwal, S.; Mamo, J.C.L. Post-Prandial Lipid Metabolism, Lipid-Modulating Agents and Cerebrovascular Integrity: Implications for Dementia Risk. Atheroscler. Suppl. 2010, 11, 49–54. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, W.; Guo, Y.; Xu, J.; Yin, M. CX3CL1/CX3CR1 in Alzheimer’s Disease: A Target for Neuroprotection. Biomed Res. Int. 2016, 2016, 8090918. [Google Scholar] [CrossRef]
- Bouzas, A.; Giménez-Bastida, J.A.; García-Tejedor, A.; Haros, C.M.; de Cedrón, M.G.; de Molina, A.R.; Laparra, J.M. Intestinal Intervention Strategy Targeting Myeloid Cells to Improve Hepatic Immunity during Hepatocarcinoma Development. Biomedicines 2021, 9, 1633. [Google Scholar] [CrossRef]
- Matsuda, S.; Matsuda, Y.; D’Adamio, L. CD74 Interacts with APP and Suppresses the Production of Aβ. Mol. Neurodegener. 2009, 4, 41. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.-V.; Doty, K.R.; Town, T. Innate Immunity Fights Alzheimer’s Disease. Trends Neurosci. 2015, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 Pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Doran, S.J.; Barrett, J.P.; Henry, R.J.; Ma, E.L.; Faden, A.I.; Loane, D.J. Chronic Alterations in Systemic Immune Function after Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1419–1436. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Landreth, G.E. The Role of Microglia in Amyloid Clearance from the AD Brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- D’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia Contribute to the Propagation of Aβ into Unaffected Brain Tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia Convert Aggregated Amyloid-β into Neurotoxic Forms through the Shedding of Microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Toyoda, T.; Abe, K.; Hamaguchi, T.; Ono, K.; et al. Rosmarinic Acid Suppresses Alzheimer’s Disease Development by Reducing Amyloid β Aggregation by Increasing Monoamine Secretion. Sci. Rep. 2019, 9, 8711. [Google Scholar] [CrossRef]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular Signal-Regulated Kinase Regulates Microglial Immune Responses in Alzheimer’s Disease. J. Neurosci. Res. 2021, 99, 1704–1721. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A Key Player in Microglial Biology and Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Glebov, K.; Wunderlich, P.; Karaca, I.; Walter, J. Functional Involvement of γ-Secretase in Signaling of the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2). J. Neuroinflamm. 2016, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Rosciszewski, G.; Cadena, V.; Murta, V.; Lukin, J.; Villarreal, A.; Roger, T.; Ramos, A.J. Toll-Like Receptor 4 (TLR4) and Triggering Receptor Expressed on Myeloid Cells-2 (TREM-2) Activation Balance Astrocyte Polarization into a Proinflammatory Phenotype. Mol. Neurobiol. 2018, 55, 3875–3888. [Google Scholar] [CrossRef] [PubMed]
- Munawara, U.; Catanzaro, M.; Xu, W.; Tan, C.; Hirokawa, K.; Bosco, N.; Dumoulin, D.; Khalil, A.; Larbi, A.; Lévesque, S.; et al. Hyperactivation of Monocytes and Macrophages in MCI Patients Contributes to the Progression of Alzheimer’s Disease. Immun. Ageing 2021, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liang, Y.; Zhang, X.; Xu, J.; Lin, J.; Zhang, R.; Kang, K.; Liu, C.; Zhao, C.; Zhao, M. Low-Density Lipoprotein Cholesterol and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2020, 12, 5. [Google Scholar] [CrossRef]
- Van der Tuin, S.J.L.; Li, Z.; Berbée, J.F.P.; Verkouter, I.; Ringnalda, L.E.; Neele, A.E.; van Klinken, J.B.; Rensen, S.S.; Fu, J.; de Winther, M.P.J.; et al. Lipopolysaccharide Lowers Cholesteryl Ester Transfer Protein by Activating F4/80+Clec4f+Vsig4+Ly6C− Kupffer Cell Subsets. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Martinez, A.E.; Weissberger, G.; Kuklenyik, Z.; He, X.; Meuret, C.; Parekh, T.; Rees, J.C.; Parks, B.A.; Gardner, M.S.; King, S.M.; et al. The Small HDL Particle Hypothesis of Alzheimer’s Disease. Alzheimer’s Dement. 2022. [Google Scholar] [CrossRef]
- Singh, R.K.; Haka, A.S.; Asmal, A.; Barbosa-Lorenzi, V.C.; Grosheva, I.; Chin, H.F.; Xiong, Y.; Hla, T.; Maxfield, F.R. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 86–102. [Google Scholar] [CrossRef]
- Rönnemaa, E.; Zethelius, B.; Vessby, B.; Lannfelt, L.; Byberg, L.; Kilander, L. Serum Fatty-Acid Composition and the Risk of Alzheimer’s Disease: A Longitudinal Population-Based Study. Eur. J. Clin. Nutr. 2012, 66, 885–890. [Google Scholar] [CrossRef]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between Fatty Acid Metabolism in the Brain and Alzheimer Disease Neuropathology and Cognitive Performance: A Nontargeted Metabolomic Study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated Fatty Acid Activates but Polyunsaturated Fatty Acid Inhibits Toll-like Receptor 2 Dimerized with Toll-like Receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef]
- Junker, Y.; Zeissig, S.; Kim, S.-J.; Barisani, D.; Wieser, H.; Leffler, D.A.; Zevallos, V.; Libermann, T.A.; Dillon, S.; Freitag, T.L.; et al. Wheat Amylase Trypsin Inhibitors Drive Intestinal Inflammation via Activation of Toll-like Receptor 4. J. Exp. Med. 2012, 209, 2395–2408. [Google Scholar] [CrossRef] [PubMed]
- Fotschki, B.; Garcia-Tejedor, A.; Nieto Fuentes, J.A.; Laparra, J.M. Immunonutritional Protease Inhibitors from T. Durum and A. Sativa Display Metabolic Similarities When Assayed on Human Macrophage-like Cells. Int. J. Mol. Sci. 2021, 22, 8307. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhou, G.; Chen, P.; Wang, Y.; Han, J.; Chen, M.; He, Y.; Zhang, S. Which Long Noncoding RNAs and Circular RNAs Contribute to Inflammatory Bowel Disease? Cell Death Dis. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Lorca, C.; Laparra, J.M.; Céspedes, M.V.; Casaní, L.; Florit, S.; Jové, M.; Mota-Martorell, N.; Vilella, E.; Gallart-Palau, X.; Serra, A. Industrial By-Products As a Novel Circular Source of Biocompatible Extracellular Vesicles. Adv. Funct. Mater. 2022, 32, 2202700. [Google Scholar] [CrossRef]
- Lu, P.; Sodhi, C.P.; Yamaguchi, Y.; Jia, H.; Prindle, T.; Fulton, W.B.; Vikram, A.; Bibby, K.J.; Morowitz, M.J.; Hackam, D.J. Intestinal Epithelial Toll-like Receptor 4 Prevents Metabolic Syndrome by Regulating Interactions between Microbes and Intestinal Epithelial Cells in Mice. Mucosal Immunol. 2018, 11, 727–740. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Scholtzova, H.; Kascsak, R.J.; Bates, K.A.; Boutajangout, A.; Kerr, D.J.; Meeker, H.C.; Mehta, P.D.; Spinner, D.S.; Wisniewski, T. Induction of Toll-Like Receptor 9 Signaling as a Method for Ameliorating Alzheimer’s Disease-Related Pathology. J. Neurosci. 2009, 29, 1846–1854. [Google Scholar] [CrossRef]
- Budikhina, A.S.; Murugina, N.E.; Maximchik, P.V.; Dagil, Y.A.; Nikolaeva, A.M.; Balyasova, L.S.; Murugin, V.V.; Selezneva, E.M.; Pashchenkova, Y.G.; Chkadua, G.Z.; et al. Interplay between NOD1 and TLR4 Receptors in Macrophages: Nonsynergistic Activation of Signaling Pathways Results in Synergistic Induction of Proinflammatory Gene Expression. J. Immunol. 2021, 206, 2206–2220. [Google Scholar] [CrossRef]
- Takahashi, Y.; Isuzugawa, K.; Murase, Y.; Imai, M.; Yamamoto, S.; Iizuka, M.; Akira, S.; Bahr, G.M.; Momotani, E.-I.; Hori, M.; et al. Up-Regulation of NOD1 and NOD2 through TLR4 and TNF-Alpha in LPS-Treated Murine Macrophages. J. Vet. Med. Sci. 2006, 68, 471–478. [Google Scholar] [CrossRef]
- Ishii, M.; Iadecola, C. Adipocyte-Derived Factors in Age-Related Dementia and Their Contribution to Vascular and Alzheimer Pathology. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 966–974. [Google Scholar] [CrossRef]
- Pichiah, P.B.T.; Sankarganesh, D.; Arunachalam, S.; Achiraman, S. Adipose-Derived Molecules–Untouched Horizons in Alzheimer’s Disease Biology. Front. Aging Neurosci. 2020, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Maurya, C.K.; Arha, D.; Rai, A.K.; Singh, S.; Varshney, S.; Schertzer, J.D.; Tamrakar, A.K. Nod1-Mediated Lipolysis Promotes Diacylglycerol Accumulation and Successive Inflammation via PKCδ-IRAK Axis in Adipocytes. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Cisbani, G.; Rivest, S. Targeting Innate Immunity to Protect and Cure Alzheimer’s Disease: Opportunities and Pitfalls. Mol. Psychiatry 2021, 26, 5504–5515. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Medicherla, S.; Sheikh, N.; Terry, B.; Phillipps, A.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Targeted Lipidomics of Fontal Cortex and Plasma Diacylglycerols (DAG) in Mild Cognitive Impairment and Alzheimer’s Disease: Validation of DAG Accumulation Early in the Pathophysiology of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Patrick, R.P. Role of Phosphatidylcholine-DHA in Preventing APOE4-Associated Alzheimer’s Disease. FASEB J. 2019, 33, 1554–1564. [Google Scholar] [CrossRef]
- Purohit, J.S.; Hu, P.; Burke, S.J.; Collier, J.J.; Chen, J.; Zhao, L. The Effects of NOD Activation on Adipocyte Differentiation. Obesity 2013, 21, 737–747. [Google Scholar] [CrossRef]
- Heneka, M.T.; Reyes-Irisarri, E.; Hüll, M.; Kummer, M.P. Impact and Therapeutic Potential of PPARs in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 643–650. [Google Scholar] [CrossRef]
- Marwarha, G.; Ghribi, O. Leptin Signaling and Alzheimer’s Disease. Am. J. Neurodegener. Dis. 2012, 1, 245–265. [Google Scholar]
- Gonzalez-Cotto, M.; Palmieri, E.M.; Baseler, W.A.; Rice, C.M.; McVicar, D.W. Trem2 Supports the Metabolic Program of Alternative Activated Macrophages. J. Immunol. 2020, 204, 73. [Google Scholar]
- Baitsch, D.; Bock, H.H.; Engel, T.; Telgmann, R.; Müller-Tidow, C.; Varga, G.; Bot, M.; Herz, J.; Robenek, H.; von Eckardstein, A.; et al. Apolipoprotein E Induces Antiinflammatory Phenotype in Macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1160–1168. [Google Scholar] [CrossRef]
- Onodera, T.; Futai, E.; Kan, E.; Abe, N.; Uchida, T.; Kamio, Y.; Kaneko, J. Phosphatidylethanolamine Plasmalogen Enhances the Inhibiting Effect of Phosphatidylethanolamine on γ-Secretase Activity. J. Biochem. 2015, 157, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Hossain, M.S.; Sejimo, S.; Akashi, K. Plasmalogens Inhibit Endocytosis of Toll-like Receptor 4 to Attenuate the Inflammatory Signal in Microglial Cells. Mol. Neurobiol. 2019, 56, 3404–3419. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Krüppel-Like Factors in Metabolic Homeostasis and Cardiometabolic Disease. Front. Cardiovasc. Med. 2018, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Rivers, S.L.; Klip, A.; Giacca, A. NOD1: An Interface Between Innate Immunity and Insulin Resistance. Endocrinology 2019, 160, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, H.E.; Bai, Y.-M.; Tsai, S.-J.; Su, T.-P.; Chen, T.-J.; Wang, Y.-P.; Chen, M.-H. Inflammatory Bowel Disease Is Associated with Higher Dementia Risk: A Nationwide Longitudinal Study. Gut 2021, 70, 85–91. [Google Scholar] [CrossRef]
- Van Welden, S.; Selfridge, A.C.; Hindryckx, P. Intestinal Hypoxia and Hypoxia-Induced Signalling as Therapeutic Targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 596–611. [Google Scholar] [CrossRef]
- Shen, G.-M.; Zhao, Y.-Z.; Chen, M.-T.; Zhang, F.-L.; Liu, X.-L.; Wang, Y.; Liu, C.-Z.; Yu, J.; Zhang, J.-W. Hypoxia-Inducible Factor-1 (HIF-1) Promotes LDL and VLDL Uptake through Inducing VLDLR under Hypoxia. Biochem. J. 2012, 441, 675–683. [Google Scholar] [CrossRef]
- Kheirandish, L.; Row, B.W.; Li, R.C.; Brittian, K.R.; Gozal, D. Apolipoprotein E-Deficient Mice Exhibit Increased Vulnerability to Intermittent Hypoxia-Induced Spatial Learning Deficits. Sleep 2005, 28, 1412–1417. [Google Scholar] [CrossRef]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The Gut Microbiota Suppresses Insulin-Mediated Fat Accumulation via the Short-Chain Fatty Acid Receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef]
- Cox, N.; Crozet, L.; Holtman, I.R.; Loyher, P.-L.; Lazarov, T.; White, J.B.; Mass, E.; Stanley, E.R.; Elemento, O.; Glass, C.K.; et al. Diet-Regulated Production of PDGFcc by Macrophages Controls Energy Storage. Science 2021, 373. [Google Scholar] [CrossRef]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [PubMed]
- Krishna, K.; Behnisch, T.; Sajikumar, S. Inhibition of Histone Deacetylase 3 Restores Amyloid-β Oligomer-Induced Plasticity Deficit in Hippocampal CA1 Pyramidal Neurons. J. Alzheimer’s Dis. 2016, 51, 783–791. [Google Scholar] [CrossRef]
- Nakajima, A.; Nakatani, A.; Hasegawa, S.; Irie, J.; Ozawa, K.; Tsujimoto, G.; Suganami, T.; Itoh, H.; Kimura, I. The Short Chain Fatty Acid Receptor GPR43 Regulates Inflammatory Signals in Adipose Tissue M2-Type Macrophages. PLoS ONE 2017, 12, e0179696. [Google Scholar] [CrossRef] [PubMed]
- Abuelezz, S.A.; Hendawy, N. HMGB1/RAGE/TLR4 Axis and Glutamate as Novel Targets for PCSK9 Inhibitor in High Fat Cholesterol Diet Induced Cognitive Impairment and Amyloidosis. Life Sci. 2021, 273, 119310. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.U.; Woodling, N.S.; Shi, J.; Andreasson, K.I. Inflammatory Cyclooxygenase Activity and PGE2 Signaling in Models of Alzheimer’s Disease. Curr. Immunol. Rev. 2015, 11, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Prostaglandin E2 Signalling Is Implicated in Inflammation Early in the Alzheimer Disease Course. Nat. Rev. Neurol. 2012, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Robinson, K.J.; Liu, L.; James, M.; Yao, Y.; Xie, S.X.; Brunden, K.R. Inflammatory Eicosanoids Increase Amyloid Precursor Protein Expression via Activation of Multiple Neuronal Receptors. Sci. Rep. 2015, 5, 18286. [Google Scholar] [CrossRef]
- Womack, T.R.; Vollert, C.T.; Ohia-Nwoko, O.; Schmitt, M.; Montazari, S.; Beckett, T.L.; Mayerich, D.; Murphy, M.P.; Eriksen, J.L. Prostacyclin Promotes Degenerative Pathology in a Model of Alzheimer’s Disease. Front. Cell. Neurosci. 2022, 16, 769347. [Google Scholar] [CrossRef]
- Weng, J.; Zhao, G.; Weng, L.; Guan, J.; Alzheimer’s Disease Neuroimaging Initiative. Aspirin Using Was Associated with Slower Cognitive Decline in Patients with Alzheimer’s Disease. PLoS ONE 2021, 16, e0252969. [Google Scholar] [CrossRef] [PubMed]
- Thoonsen, H.; Richard, E.; Bentham, P.; Gray, R.; van Geloven, N.; De Haan, R.J.; Van Gool, W.A.; Nederkoorn, P.J. Aspirin in Alzheimer’s Disease. Stroke 2010, 41, 2690–2692. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Bezugla, Y.; Kolada, A.; Kamionka, S.; Bernard, B.; Scheibe, R.; Dieter, P. COX-1 and COX-2 Contribute Differentially to the LPS-Induced Release of PGE2 and TxA2 in Liver Macrophages. Prostaglandins Other Lipid Mediat. 2006, 79, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.-M.; Zong, Y.; Sun, L.; Guo, J.-Z.; Zhang, W.; He, Y.; Song, R.; Wang, W.-M.; Xiao, C.-J.; Lu, D. Resveratrol Inhibits Inflammatory Responses via the Mammalian Target of Rapamycin Signaling Pathway in Cultured LPS-Stimulated Microglial Cells. PLoS ONE 2012, 7, e32195. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, G.-Y.; Choi, Y.H. Naringenin Attenuates the Release of Pro-Inflammatory Mediators from Lipopolysaccharide-Stimulated BV2 Microglia by Inactivating Nuclear Factor-ΚB and Inhibiting Mitogen-Activated Protein Kinases. Int. J. Mol. Med. 2012, 30, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, A.N.; Giannopoulos, P.F.; Praticò, D. The Direct Role of 5-Lipoxygenase on Tau Pathology, Synaptic Integrity and Cognition in a Mouse Model of Tauopathy. Transl. Psychiatry 2017, 7, 1288. [Google Scholar] [CrossRef] [PubMed]
- Joshi, Y.B.; Di Meco, A.; Praticó, D. Modulation of Amyloid-β Production by Leukotriene B 4 via the γ-Secretase Pathway. J. Alzheimer’s Dis. 2014, 38, 503–506. [Google Scholar] [CrossRef]
- Rahman, S.O.; Singh, R.K.; Hussain, S.; Akhtar, M.; Najmi, A.K. A Novel Therapeutic Potential of Cysteinyl Leukotrienes and Their Receptors Modulation in the Neurological Complications Associated with Alzheimer’s Disease. Eur. J. Pharmacol. 2019, 842, 208–220. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; González-Sarrías, A.; Laparra, J.M.; Schneider, C.; Espín, J.C. Targeting Mammalian 5-Lipoxygenase by Dietary Phenolics as an Anti-Inflammatory Mechanism: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 7937. [Google Scholar] [CrossRef]
- Nakashima, F.; Suzuki, T.; Gordon, O.N.; Golding, D.; Okuno, T.; Giménez-Bastida, J.A.; Yokomizo, T.; Schneider, C. Biosynthetic Crossover of 5-Lipoxygenase and Cyclooxygenase-2 Yields 5-Hydroxy-PGE2 and 5-Hydroxy-PGD2. JACS Au 2021, 1, 1380–1388. [Google Scholar] [CrossRef]
- Griesser, M.; Suzuki, T.; Tejera, N.; Mont, S.; Boeglin, W.E.; Pozzi, A.; Schneider, C. Biosynthesis of Hemiketal Eicosanoids by Cross-over of the 5-Lipoxygenase and Cyclooxygenase-2 Pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 6945–6950. [Google Scholar] [CrossRef]
- Schneider, C.; Boeglin, W.E.; Yin, H.; Stec, D.F.; Voehler, M. Convergent Oxygenation of Arachidonic Acid by 5-Lipoxygenase and Cyclooxygenase-2. J. Am. Chem. Soc. 2006, 128, 720–721. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Griesser, M.; Boeglin, W.E.; Suzuki, T.; Schneider, C. Convergence of the 5-LOX and COX-2 Pathways: Heme-Catalyzed Cleavage of the 5S-HETE-Derived Di-Endoperoxide into Aldehyde Fragments. J. Lipid Res. 2009, 50, 2455–2462. [Google Scholar] [CrossRef]
- Boer, R.E.; Giménez-Bastida, J.A.; Boutaud, O.; Jana, S.; Schneider, C.; Sulikowski, G.A. Total Synthesis and Biological Activity of the Arachidonic Acid Metabolite Hemiketal E2. Org. Lett. 2018, 20, 4020–4022. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Bastida, J.A.; Shibata, T.; Uchida, K.; Schneider, C. Roles of 5-Lipoxygenase and Cyclooxygenase-2 in the Biosynthesis of Hemiketals E2 and D2 by Activated Human Leukocytes. FASEB J. 2017, 31, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Bastida, J.A.; González-Sarrías, A.; Espín, J.C.; Schneider, C. Inhibition of 5-Lipoxygenase-Derived Leukotrienes and Hemiketals as a Novel Anti-Inflammatory Mechanism of Urolithins. Mol. Nutr. Food Res. 2020, 64, 2000129. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, X.; Sun, L.; Schultzberg, M.; Hjorth, E. Can Inflammation Be Resolved in Alzheimer’s Disease? Ther. Adv. Neurol. Disord. 2018, 11, 1756286418791107. [Google Scholar] [CrossRef]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.-M.; Lein, P.J.; et al. Polyunsaturated Fatty Acids and Fatty Acid-Derived Lipid Mediators: Recent Advances in the Understanding of Their Biosynthesis, Structures, and Functions. Prog. Lipid Res. 2022, 86, 101165. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, X.; Hjorth, E.; Colas, R.A.; Schroeder, L.; Granholm, A.-C.; Serhan, C.N.; Schultzberg, M. Pro-Resolving Lipid Mediators Improve Neuronal Survival and Increase Aβ42 Phagocytosis. Mol. Neurobiol. 2016, 53, 2733–2749. [Google Scholar] [CrossRef]
- Mas, E.; Croft, K.D.; Zahra, P.; Barden, A.; Mori, T.A. Resolvins D1, D2, and Other Mediators of Self-Limited Resolution of Inflammation in Human Blood Following n-3 Fatty Acid Supplementation. Clin. Chem. 2012, 58, 1476–1484. [Google Scholar] [CrossRef]
- Fiala, M.; Halder, R.C.; Sagong, B.; Ross, O.; Sayre, J.; Porter, V.; Bredesen, D.E. ω-3 Supplementation Increases Amyloid-β Phagocytosis and Resolvin D1 in Patients with Minor Cognitive Impairment. FASEB J. 2015, 29, 2681–2689. [Google Scholar] [CrossRef]
- Phillips, M.A.; Childs, C.E.; Calder, P.C.; Rogers, P.J. No Effect of Omega-3 Fatty Acid Supplementation on Cognition and Mood in Individuals with Cognitive Impairment and Probable Alzheimer’s Disease: A Randomised Controlled Trial. Int. J. Mol. Sci. 2015, 16, 24600–24613. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-Y.; Cheng, C.; Satyanarayanan, S.K.; Chiu, L.-T.; Chien, Y.-C.; Chuu, C.-P.; Lan, T.-H.; Su, K.-P. Omega-3 Fatty Acids and Blood-Based Biomarkers in Alzheimer’s Disease and Mild Cognitive Impairment: A Randomized Placebo-Controlled Trial. Brain Behav. Immun. 2022, 99, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Bastida, J.A.; Ávila-Gálvez, M.Á.; Espín, J.C.; González-Sarrías, A. Evidence for Health Properties of Pomegranate Juices and Extracts beyond Nutrition: A Critical Systematic Review of Human Studies. Trends Food Sci. Technol. 2021, 114, 410–423. [Google Scholar] [CrossRef]
- Moré, M.I.; Freitas, U.; Rutenberg, D. Positive Effects of Soy Lecithin-Derived Phosphatidylserine plus Phosphatidic Acid on Memory, Cognition, Daily Functioning, and Mood in Elderly Patients with Alzheimer’s Disease and Dementia. Adv. Ther. 2014, 31, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, M.; Nakao, H.; Nakamura, M.; Tazaki, F.; Maebuchi, M.; Ibuki, M.; Takeda, M. Effect of Multicomponent Exercise and Nutrition Support on the Cognitive Function of Older Adults: A Randomized Controlled Trial. Clin. Interv. Aging 2019, 14, 2145–2153. [Google Scholar] [CrossRef]
- Jang, C.H.; Oh, J.; Lim, J.S.; Kim, H.J.; Kim, J.-S. Fermented Soy Products: Beneficial Potential in Neurodegenerative Diseases. Foods 2021, 10, 636. [Google Scholar] [CrossRef]
- Cortés-Martín, A.; Selma, M.V.; Tomás-Barberán, F.A.; González-Sarrías, A.; Espín, J.C. Where to Look into the Puzzle of Polyphenols and Health? The Postbiotics and Gut Microbiota Associated with Human Metabotypes. Mol. Nutr. Food Res. 2020, 64, 1900952. [Google Scholar] [CrossRef]
- García-Villalba, R.; Giménez-Bastida, J.A.; Cortés-Martín, A.; Ávila-Gálvez, M.Á.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C.; González-Sarrías, A. Urolithins: A Comprehensive Update on Their Metabolism, Bioactivity, and Associated Gut Microbiota. Mol. Nutr. Food Res. 2022, 2101019. [Google Scholar] [CrossRef]
- Tsai, C.-F.; Chen, G.-W.; Chen, Y.-C.; Shen, C.-K.; Lu, D.-Y.; Yang, L.-Y.; Chen, J.-H.; Yeh, W.-L. Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients 2021, 14, 67. [Google Scholar] [CrossRef]
- Wang, F.; Xia, J.-J.; Shen, L.-J.; Jiang, T.-T.; Li, W.-L.; You, D.-L.; Chang, Q.; Hu, S.-Y.; Wang, L.; Wu, X. Curcumin Attenuates Intracerebral Hemorrhage-Induced Neuronal Apoptosis and Neuroinflammation by Suppressing JAK1/STAT1 Pathway. Biochem. Cell Biol. 2022, 100, 236–245. [Google Scholar] [CrossRef]
- Jiang, T.; Xu, S.; Shen, Y.; Xu, Y.; Li, Y. Genistein Attenuates Isoflurane-Induced Neuroinflammation by Inhibiting TLR4-Mediated Microglial-Polarization in Vivo and in Vitro. J. Inflamm. Res. 2021, 14, 2587–2600. [Google Scholar] [CrossRef] [PubMed]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol Mitigates Lipopolysaccharide- and Aβ-Mediated Microglial Inflammation by Inhibiting the TLR4/NF-ΚB/STAT Signaling Cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Le, K.; Song, Z.; Deng, J.; Peng, X.; Zhang, J.; Wang, L.; Zhou, L.; Bi, H.; Liao, Z.; Feng, Z. Quercetin Alleviates Neonatal Hypoxic-Ischemic Brain Injury by Inhibiting Microglia-Derived Oxidative Stress and TLR4-Mediated Inflammation. Inflamm. Res. 2020, 69, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yang, G.; Liu, J. Phloretin Attenuates Behavior Deficits and Neuroinflammatory Response in MPTP Induced Parkinson’s Disease in Mice. Life Sci. 2019, 232, 116600. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yuan, C.; Wang, G.; Luo, J.; Ma, H.; Xu, L.; Mu, Y.; Li, Y.; Seeram, N.P.; Huang, X.; et al. Urolithins Attenuate LPS-Induced Neuroinflammation in BV2Microglia via MAPK, Akt, and NF-ΚB Signaling Pathways. J. Agric. Food Chem. 2018, 66, 571–580. [Google Scholar] [CrossRef]
- Lee, G.; Park, J.-S.; Lee, E.-J.; Ahn, J.-H.; Kim, H.-S. Anti-Inflammatory and Antioxidant Mechanisms of Urolithin B in Activated Microglia. Phytomedicine 2019, 55, 50–57. [Google Scholar] [CrossRef]
- Shi, Q.; Zheng, Y.; Wang, L.; Xue, Y.; Yang, Y. Curcumin Suppresses Neuroinflammation to Protect Neurons by Preventing NLRP3 Inflammasome Activation. Eur. J. Inflamm. 2021, 19, 20587392211058616. [Google Scholar] [CrossRef]
- Toney, A.M.; Albusharif, M.; Works, D.; Polenz, L.; Schlange, S.; Chaidez, V.; Ramer-Tait, A.E.; Chung, S. Differential Effects of Whole Red Raspberry Polyphenols and Their Gut Metabolite Urolithin A on Neuroinflammation in BV-2 Microglia. Int. J. Environ. Res. Public Health 2021, 18, 68. [Google Scholar] [CrossRef]
- Kang, G.; Kong, P.-J.; Yuh, Y.-J.; Lim, S.-Y.; Yim, S.-V.; Chun, W.; Kim, S.-S. Curcumin Suppresses Lipopolysaccharide-Induced Cyclooxygenase-2 Expression by Inhibiting Activator Protein 1 and Nuclear Factor κB Bindings in BV2 Microglial Cells. J. Pharmacol. Sci. 2004, 94, 325–328. [Google Scholar] [CrossRef]
- Yu, Y.; Shen, Q.; Lai, Y.; Park, S.Y.; Ou, X.; Lin, D.; Jin, M.; Zhang, W. Anti-Inflammatory Effects of Curcumin in Microglial Cells. Front. Pharmacol. 2018, 9, 386. [Google Scholar] [CrossRef]
- Ávila-Gálvez, M.Á.; González-Sarrías, A.; Espín, J.C. In Vitro Research on Dietary Polyphenols and Health: A Call of Caution and a Guide on How To Proceed. J. Agric. Food Chem. 2018, 66, 7857–7858. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; Tomás-Barberán, F.A.; Espín, J.C. Neuroprotective Effects of Bioavailable Polyphenol-Derived Metabolites against Oxidative Stress-Induced Cytotoxicity in Human Neuroblastoma SH-SY5Y Cells. J. Agric. Food Chem. 2017, 65, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Huang, H.; Mo, X.; Zhu, Y.; Chen, X.; Li, X.; Peng, X.; Xu, Z.; Chen, L.; Rong, S.; et al. Quercetin-3-O-Glucuronide Alleviates Cognitive Deficit and Toxicity in Aβ1-42-Induced AD-Like Mice and SH-SY5Y Cells. Mol. Nutr. Food Res. 2021, 65, 2000660. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A Promotes Mitophagy and Suppresses NLRP3 Inflammasome Activation in Lipopolysaccharide-Induced BV2 Microglial Cells and MPTP-Induced Parkinson’s Disease Model. Neuropharmacology 2022, 207, 108963. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Mitophagy Enhancers against Phosphorylated Tau-Induced Mitochondrial and Synaptic Toxicities in Alzheimer Disease. Pharmacol. Res. 2021, 174, 105973. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, J.; Qiu, J.; Wang, L.; Zhuo, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A Protects Dopaminergic Neurons in Experimental Models of Parkinson’s Disease by Promoting Mitochondrial Biogenesis through the SIRT1/PGC-1α Signaling Pathway. Food Funct. 2022, 13, 375–385. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Mahnken, J.D.; Welch, P.; Bothwell, R.; Koppel, S.; Jackson, R.L.; Burns, J.M.; Swerdlow, R.H. A Mitochondrial Biomarker-Based Study of S-Equol in Alzheimer’s Disease Subjects: Results of a Single-Arm, Pilot Trial. J. Alzheimer’s Dis. 2017, 59, 291–300. [Google Scholar] [CrossRef]
- Sekikawa, A.; Higashiyama, A.; Lopresti, B.J.; Ihara, M.; Aizenstein, H.; Watanabe, M.; Chang, Y.; Kakuta, C.; Yu, Z.; Mathis, C.; et al. Associations of Equol-Producing Status with White Matter Lesion and Amyloid-β Deposition in Cognitively Normal Elderly Japanese. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12089. [Google Scholar] [CrossRef]
- Yao, J.; Zhao, L.; Mao, Z.; Chen, S.; Wong, K.C.; To, J.; Brinton, R.D. Potentiation of Brain Mitochondrial Function by S-Equol and R/S-Equol Estrogen Receptor β-Selective PhytoSERM Treatments. Brain Res. 2013, 1514, 128–141. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Lin, S.-H.; Hidayah, K.; Lin, C.-I. Equol Pretreatment Protection of SH-SY5Y Cells against Aβ (25–35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression. Nutrients 2019, 11, 2356. [Google Scholar] [CrossRef]
- Li, K.; Teng, C.; Min, Q. Advanced Nanovehicles-Enabled Delivery Systems of Epigallocatechin Gallate for Cancer Therapy. Front. Chem. 2020, 8, 573297. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Sun, R.; Wang, Q.; Chen, D.; Yu, B.; He, J.; Yu, J.; Luo, J.; Luo, Y.; Yan, H.; et al. L-Isoleucine Administration Alleviates DSS-Induced Colitis by Regulating TLR4/MyD88/NF-ΚB Pathway in Rats. Front. Immunol. 2021, 12, 817583. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; van Lent, D.M.; Wolfsgruber, S.; Weinhold, L.; Kleineidam, L.; Bickel, H.; Scherer, M.; Eisele, M.; van den Bussche, H.; Wiese, B.; et al. Prospective Associations between Single Foods, Alzheimer’s Dementia and Memory Decline in the Elderly. Nutrients 2018, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Mietelska-Porowska, A.; Domańska, J.; Want, A.; Więckowska-Gacek, A.; Chutorański, D.; Koperski, M.; Wojda, U. Induction of Brain Insulin Resistance and Alzheimer’s Molecular Changes by Western Diet. Int. J. Mol. Sci. 2022, 23, 4744. [Google Scholar] [CrossRef] [PubMed]
- Gannon, O.J.; Robison, L.S.; Salinero, A.E.; Abi-Ghanem, C.; Mansour, F.M.; Kelly, R.D.; Tyagi, A.; Brawley, R.R.; Ogg, J.D.; Zuloaga, K.L. High-Fat Diet Exacerbates Cognitive Decline in Mouse Models of Alzheimer’s Disease and Mixed Dementia in a Sex-Dependent Manner. J. Neuroinflamm. 2022, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, D.; Kasperek, K.; Rękawek, P.; Piątkowska-Chmiel, I. The Therapeutic Role of Ketogenic Diet in Neurological Disorders. Nutrients 2022, 14, 1952. [Google Scholar] [CrossRef]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in Regional Cerebral Blood Flow Associated with a 45 day Course of the Ketogenic Agent, Caprylidene, in Patients with Mild to Moderate Alzheimer’s Disease: Results of a Randomized, Double-Blinded, Pilot Study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a Medium-Chain Triglyceride-Based Ketogenic Formula on Cognitive Function in Patients with Mild-to-Moderate Alzheimer’s Disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.-A.; St-Pierre, V.; Myette-Côté, É.; Langlois, F.; Roy, M.; Morin, M.-C.; Bocti, C.; Fulop, T.; Godin, J.-P.; et al. A Ketogenic Drink Improves Cognition in Mild Cognitive Impairment: Results of a 6-Month RCT. Alzheimer’s Dement. 2021, 17, 543–552. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized Crossover Trial of a Modified Ketogenic Diet in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef]
- Myette-Côté, É.; St-Pierre, V.; Beaulieu, S.; Castellano, C.-A.; Fortier, M.; Plourde, M.; Bocti, C.; Fulop, T.; Cunnane, S.C. The Effect of a 6-Month Ketogenic Medium-Chain Triglyceride Supplement on Plasma Cardiometabolic and Inflammatory Markers in Mild Cognitive Impairment. Prostaglandins, Leukot. Essent. Fat. Acids 2021, 169, 102236. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcos Pasero, H.; García Tejedor, A.; Giménez-Bastida, J.A.; Laparra Llopis, J.M. Modifiable Innate Biology within the Gut–Brain Axis for Alzheimer’s Disease. Biomedicines 2022, 10, 2098. https://doi.org/10.3390/biomedicines10092098
Marcos Pasero H, García Tejedor A, Giménez-Bastida JA, Laparra Llopis JM. Modifiable Innate Biology within the Gut–Brain Axis for Alzheimer’s Disease. Biomedicines. 2022; 10(9):2098. https://doi.org/10.3390/biomedicines10092098
Chicago/Turabian StyleMarcos Pasero, Helena, Aurora García Tejedor, Juan Antonio Giménez-Bastida, and José Moisés Laparra Llopis. 2022. "Modifiable Innate Biology within the Gut–Brain Axis for Alzheimer’s Disease" Biomedicines 10, no. 9: 2098. https://doi.org/10.3390/biomedicines10092098
APA StyleMarcos Pasero, H., García Tejedor, A., Giménez-Bastida, J. A., & Laparra Llopis, J. M. (2022). Modifiable Innate Biology within the Gut–Brain Axis for Alzheimer’s Disease. Biomedicines, 10(9), 2098. https://doi.org/10.3390/biomedicines10092098