
Machine Learning Data Analysis Highlights the Role of Parasutterella and Alloprevotella in Autism Spectrum Disorders

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Gathering and Bioinformatic Data Analysis
2.2. Statistical Software and Packages
2.3. Data Filtering, Normalization, Multivariate Data Analysis and Batch-Effect Correction
2.4. ML Algorithms: Optimization, Training and Evaluation
- True Positive (TP): an ASD sample correctly predicted as an ASD sample;
- True Negative (TN): an HC sample correctly predicted as an HC sample;
- False Positive (FP): an ASD sample erroneously predicted as an HC sample;
- False Negative (FN): an HC sample erroneously predicted as an ASD sample.
2.5. Evaluation of Feature Importance, Feature Selection and Feature Contribution
2.6. Dataset Analysis Based on Control Selection
3. Results
3.1. Number of Samples Analyzed and Preliminary Filters
3.2. Analysis of Beta-Diversity and Evaluation of the Batch Effect
3.3. Preliminary Results of the Random Forest on Three Datasets
3.4. Evaluation of Algorithm Metrics and Evaluation of the Control Selection Role
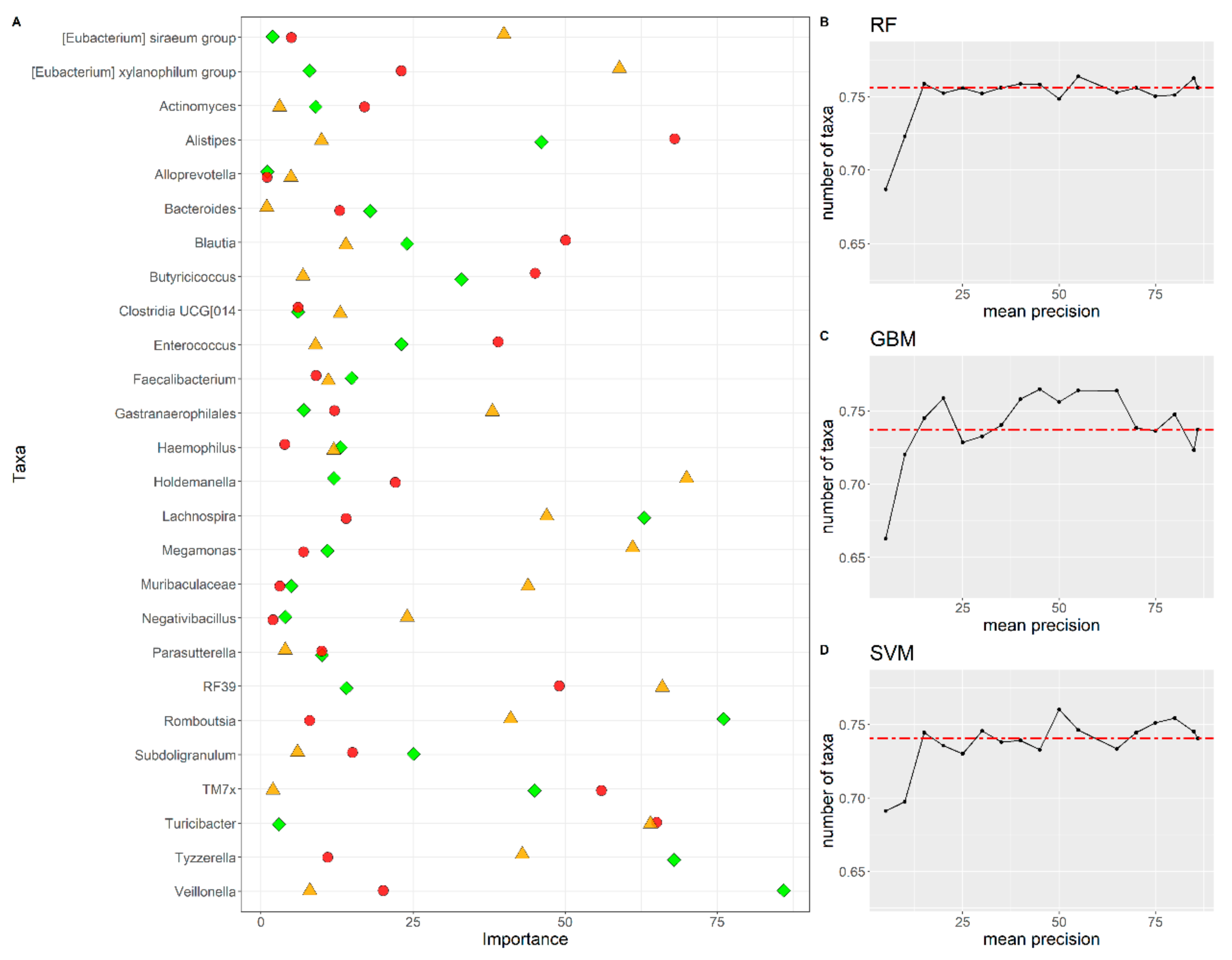
3.5. Feature Importance and Comparison of Features among Algorithms
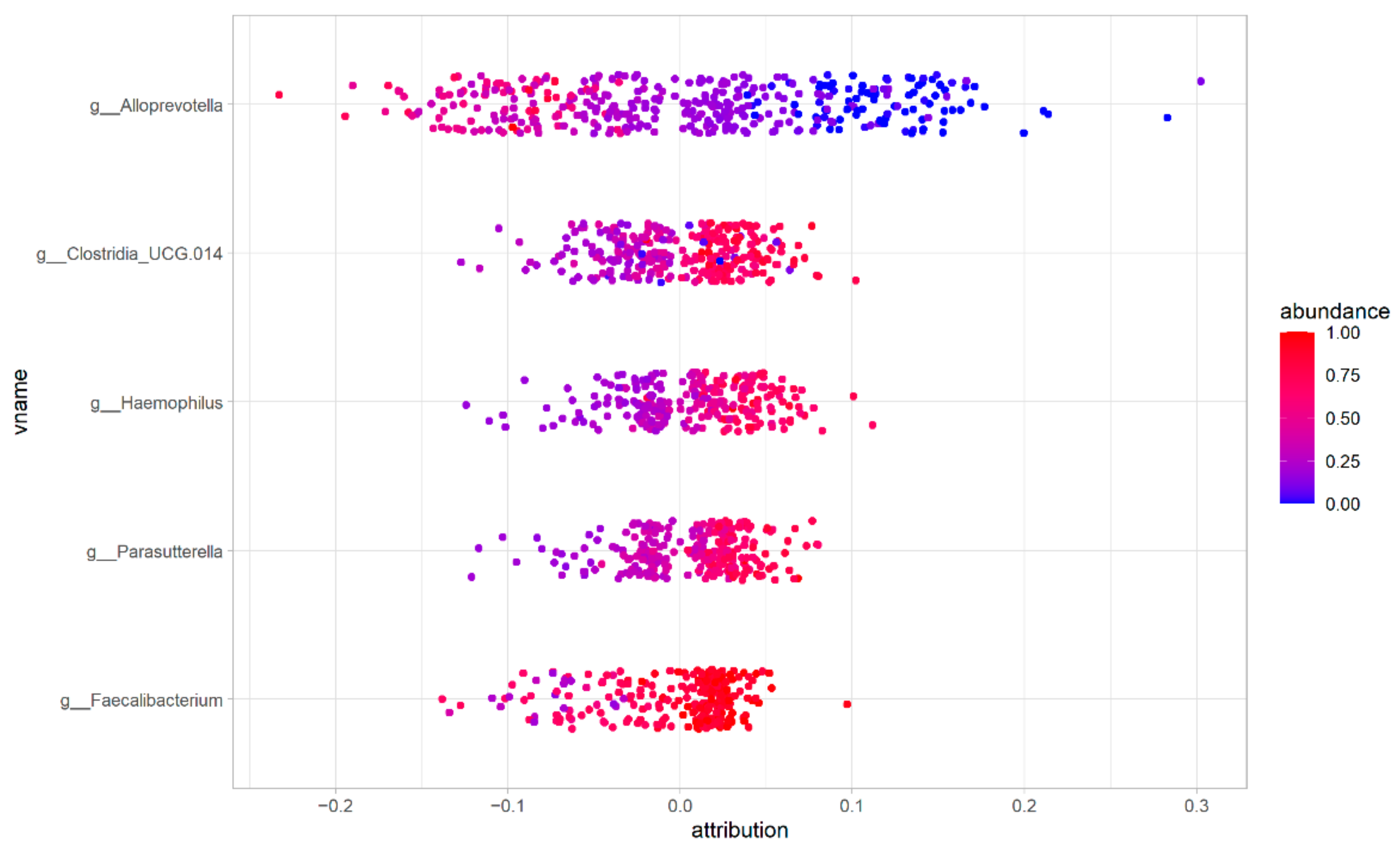
3.6. Feature Contribution to the ASD/HC Phenotype Classification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGP | American Gut Project |
| ASD | Autism Spectrum Disorder |
| ASVs | Amplicon Sequence Variants |
| FN | False Negative |
| FP | False Positive |
| GABA | Gamma-aminobutyric acid |
| GBM | Gradient Boosting Machine |
| HC | Healthy Controls |
| ML | Machine Learning |
| PCA | Principal Coordinate Analysis |
| PCoA | Principal Component Analysis |
| RF | Random Forest |
| SCFAs | Short Chain Fatty Acids |
| SHAP | SHapley Additive exPlanations algorithm |
| SVA | Surrogate Variable Analysis |
| SVM | Support Vector Machine |
| TN | True Negative |
| TNR | True Negative Rate |
| TP | True Positive |
| TPR | True Positive Rate |
References
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of Small Intestinal Microbiota in Liver Cirrhosis and Its Association with Etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, Y.M.; Borcherding, D.; Kanthasamy, A.; Kim, H.J.; Willette, A.A.; Jergens, A.; Allenspach, K.; Mochel, J.P. The Gut-Brain Axis in Neurodegenerative Diseases and Relevance of the Canine Model: A Review. Front. Aging Neurosci. 2019, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Pulikkan, J.; Mazumder, A.; Grace, T. Role of the Gut Microbiome in Autism Spectrum Disorders. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Valicenti-McDermott, M.; McVicar, K.; Rapin, I.; Wershil, B.K.; Cohen, H.; Shinnar, S. Frequency of Gastrointestinal Symptoms in Children with Autistic Spectrum Disorders and Association with Family History of Autoimmune Disease. J. Dev. Behav. Pediatr. 2006, 27, S128–S136. [Google Scholar] [CrossRef]
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271. [Google Scholar] [CrossRef]
- de Theije, C.G.M.; Wopereis, H.; Ramadan, M.; van Eijndthoven, T.; Lambert, J.; Knol, J.; Garssen, J.; Kraneveld, A.D.; Oozeer, R. Altered Gut Microbiota and Activity in a Murine Model of Autism Spectrum Disorders. Brain Behav. Immun. 2014, 37, 197–206. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618.e17. [Google Scholar] [CrossRef]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Pérez, A.; Peterson, V.; et al. Microbiota-Related Changes in Bile Acid & Tryptophan Metabolism Are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [CrossRef]
- Pulikkan, J.; Maji, A.; Dhakan, D.B.; Saxena, R.; Mohan, B.; Anto, M.M.; Agarwal, N.; Grace, T.; Sharma, V.K. Gut Microbial Dysbiosis in Indian Children with Autism Spectrum Disorders. Microb. Ecol. 2018, 76, 1102–1114. [Google Scholar] [CrossRef]
- Averina, O.V.; Kovtun, A.S.; Polyakova, S.I.; Savilova, A.M.; Rebrikov, D.V.; Danilenko, V.N. The Bacterial Neurometabolic Signature of the Gut Microbiota of Young Children with Autism Spectrum Disorders. J. Med. Microbiol. 2020, 69, 558–571. [Google Scholar] [CrossRef]
- Dan, Z.; Mao, X.; Liu, Q.; Guo, M.; Zhuang, Y.; Liu, Z.; Chen, K.; Chen, J.; Xu, R.; Tang, J.; et al. Altered Gut Microbial Profile Is Associated with Abnormal Metabolism Activity of Autism Spectrum Disorder. Gut Microbes 2020, 11, 1246–1267. [Google Scholar] [CrossRef]
- Zurita, M.F.; Cárdenas, P.A.; Sandoval, M.E.; Peña, M.C.; Fornasini, M.; Flores, N.; Monaco, M.H.; Berding, K.; Donovan, S.M.; Kuntz, T.; et al. Analysis of Gut Microbiome, Nutrition and Immune Status in Autism Spectrum Disorder: A Case-Control Study in Ecuador. Gut Microbes 2020, 11, 453–464. [Google Scholar] [CrossRef]
- Coretti, L.; Paparo, L.; Riccio, M.P.; Amato, F.; Cuomo, M.; Natale, A.; Borrelli, L.; Corrado, G.; Comegna, M.; Buommino, E.; et al. Gut Microbiota Features in Young Children with Autism Spectrum Disorders. Front. Microbiol. 2018, 9, 3146. [Google Scholar] [CrossRef]
- Son, J.S.; Zheng, L.J.; Rowehl, L.M.; Tian, X.; Zhang, Y.; Zhu, W.; Litcher-Kelly, L.; Gadow, K.D.; Gathungu, G.; Robertson, C.E.; et al. Comparison of Fecal Microbiota in Children with Autism Spectrum Disorders and Neurotypical Siblings in the Simons Simplex Collection. PLoS ONE 2015, 10, e0137725. [Google Scholar] [CrossRef]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal Flora and Gastrointestinal Status in Children with Autism-Comparisons to Typical Children and Correlation with Autism Severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef]
- Kang, D.W.; Ilhan, Z.E.; Isern, N.G.; Hoyt, D.W.; Howsmon, D.P.; Shaffer, M.; Lozupone, C.A.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Differences in Fecal Microbial Metabolites and Microbiota of Children with Autism Spectrum Disorders. Anaerobe 2018, 49, 121–131. [Google Scholar] [CrossRef]
- Vernocchi, P.; Ristori, M.V.; Guerrera, S.; Guarrasi, V.; Conte, F.; Russo, A.; Lupi, E.; Albitar-Nehme, S.; Gardini, S.; Paci, P.; et al. Gut Microbiota Ecology and Inferred Functions in Children With ASD Compared to Neurotypical Subjects. Front. Microbiol. 2022, 13, 871086. [Google Scholar] [CrossRef]
- Ding, X.; Xu, Y.; Zhang, X.; Zhang, L.; Duan, G.; Song, C.; Li, Z.; Yang, Y.; Wang, Y.; Wang, X.; et al. Gut Microbiota Changes in Patients with Autism Spectrum Disorders. J. Psychiatr. Res. 2020, 129, 149–159. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Petitpierre, G.; Luisier, A.C.; Bensafi, M. Eating Behavior in Autism: Senses as a Window towards Food Acceptance. Curr. Opin. Food Sci. 2021, 41, 210–216. [Google Scholar] [CrossRef]
- Li, R.; Li, L.; Xu, Y.; Yang, J. Machine Learning Meets Omics: Applications and Perspectives. Brief. Bioinform. 2022, 23, bbab460. [Google Scholar] [CrossRef]
- Maurya, N.S.; Kushwaha, S.; Chawade, A.; Mani, A. Transcriptome Profiling by Combined Machine Learning and Statistical R Analysis Identifies TMEM236 as a Potential Novel Diagnostic Biomarker for Colorectal Cancer. Sci. Rep. 2021, 11, 14304. [Google Scholar] [CrossRef]
- Dias-Audibert, F.L.; Navarro, L.C.; de Oliveira, D.N.; Delafiori, J.; Melo, C.F.O.R.; Guerreiro, T.M.; Rosa, F.T.; Petenuci, D.L.; Watanabe, M.A.E.; Velloso, L.A.; et al. Combining Machine Learning and Metabolomics to Identify Weight Gain Biomarkers. Front. Bioeng. Biotechnol. 2020, 8, 6. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Ghannam, R.B.; Techtmann, S.M. Machine Learning Applications in Microbial Ecology, Human Microbiome Studies, and Environmental Monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef]
- West, P.R.; Amaral, D.G.; Bais, P.; Smith, A.M.; Egnash, L.A.; Ross, M.E.; Palmer, J.A.; Fontaine, B.R.; Conard, K.R.; Corbett, B.A.; et al. Metabolomics as a Tool for Discovery of Biomarkers of Autism Spectrum Disorder in the Blood Plasma of Children. PLoS ONE 2014, 9, e112445. [Google Scholar] [CrossRef]
- Oh, D.H.; Kim, I.B.; Kim, S.H.; Ahn, D.H. Predicting Autism Spectrum Disorder Using Blood-Based Gene Expression Signatures and Machine Learning. Clin. Psychopharmacol. Neurosci. 2017, 15, 47–52. [Google Scholar] [CrossRef]
- Wu, T.; Wang, H.; Lu, W.; Zhai, Q.; Zhang, Q.; Yuan, W.; Gu, Z.; Zhao, J.; Zhang, H.; Chen, W. Potential of Gut Microbiome for Detection of Autism Spectrum Disorder. Microb. Pathog. 2020, 149, 104568. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2017; Volume 30, pp. 4766–4775. [Google Scholar]
- Pietrucci, D.; Teofani, A.; Unida, V.; Cerroni, R.; Biocca, S.; Stefani, A.; Desideri, A. Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach. Brain Sci. 2020, 10, 242. [Google Scholar] [CrossRef]
- Que, Y.; Cao, M.; He, J.; Zhang, Q.; Chen, Q.; Yan, C.; Lin, A.; Yang, L.; Wu, Z.; Zhu, D.; et al. Gut Bacterial Characteristics of Patients With Type 2 Diabetes Mellitus and the Application Potential. Front. Immunol. 2021, 12, 3218. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Gallins, P. A Review and Tutorial of Machine Learning Methods for Microbiome Host Trait Prediction. Front. Genet. 2019, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- de Angelis, M.; Francavilla, R.; Piccolo, M.; De Giacomo, A.; Gobbetti, M. Autism Spectrum Disorders and Intestinal Microbiota. Gut Microbes 2015, 6, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef]
- Castrignanò, T.; Gioiosa, S.; Flati, T.; Cestari, M.; Picardi, E.; Chiara, M.; Fratelli, M.; Amente, S.; Cirilli, M.; Tangaro, M.A.; et al. ELIXIR-IT HPC@CINECA: High Performance Computing Resources for the Bioinformatics Community. BMC Bioinform. 2020, 21, 352. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. Babraham Bioinformatics. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 November 2021).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-In-One FASTQ Preprocessor. In Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2018; Volume 34, pp. i884–i890. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2′s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The SVA Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Mcglinn, D.; Minchin, P.R.; Hara, R.B.O.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2016. Available online: https://github.com/vegandevs/vegan (accessed on 1 November 2021).
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-Filtering Vastly Improves Diversity Estimates from Illumina Amplicon Sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.A.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome Differential Abundance Methods Produce Different Results across 38 Datasets. Nat. Commun. 2022, 13, 342. [Google Scholar] [CrossRef]
- Dwiyanto, J.; Hussain, M.H.; Reidpath, D.; Ong, K.S.; Qasim, A.; Lee, S.W.H.; Lee, S.M.; Foo, S.C.; Chong, C.W.; Rahman, S. Ethnicity Influences the Gut Microbiota of Individuals Sharing a Geographical Location: A Cross-Sectional Study from a Middle-Income Country. Sci. Rep. 2021, 11, 2618. [Google Scholar] [CrossRef]
- Leeming, E.R.; Louca, P.; Gibson, R.; Menni, C.; Spector, T.D.; Le Roy, C.I. The Complexities of the Diet-Microbiome Relationship: Advances and Perspectives. Genome Med. 2021, 13, 10. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate Analyses in Microbial Ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Roguet, A.; Eren, A.M.; Newton, R.J.; McLellan, S.L. Fecal Source Identification Using Random Forest. Microbiome 2018, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Feres, M.; Louzoun, Y.; Haber, S.; Faveri, M.; Figueiredo, L.C.; Levin, L. Support Vector Machine-Based Differentiation between Aggressive and Chronic Periodontitis Using Microbial Profiles. Int. Dent. J. 2018, 68, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Liu, Y.Y. Comparative Study of Classifiers for Human Microbiome Data. Med. Microecol. 2020, 4, 100013. [Google Scholar] [CrossRef]
- James, G.; Witten, D.; Hastie, T.; Tibishirani, R. An Introduction to Statistical Learning with Applications in R; Spinger: Berlin/Heidelberg, Germany, 2013; ISBN 978-1-4614-7137-0. [Google Scholar]
- Chicco, D. Ten Quick Tips for Machine Learning in Computational Biology. BioData Min. 2017, 10, 35. [Google Scholar] [CrossRef]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-Analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef]
- Mladenić, D.; Brank, J.; Grobelnik, M.; Milic-Frayling, N. Feature Selection Using Linear Classifier Weights: Interaction with Classification Models. In Proceedings of the Sheffield SIGIR-Twenty-Seventh Annual International ACM SIGIR Conference on Research and Development in Information Retrieval; Association for Computing Machinery: New York, NY, USA, 2004; pp. 234–241. [Google Scholar]
- Carrieri, A.P.; Haiminen, N.; Maudsley-Barton, S.; Gardiner, L.J.; Murphy, B.; Mayes, A.E.; Paterson, S.; Grimshaw, S.; Winn, M.; Shand, C.; et al. Explainable AI Reveals Changes in Skin Microbiome Composition Linked to Phenotypic Differences. Sci. Rep. 2021, 11, 4565. [Google Scholar] [CrossRef]
- Levi Mortera, S.; Vernocchi, P.; Basadonne, I.; Zandonà, A.; Chierici, M.; Durighello, M.; Marzano, V.; Gardini, S.; Gasbarrini, A.; Urbani, A.; et al. A Metaproteomic-Based Gut Microbiota Profiling in Children Affected by Autism Spectrum Disorders. J. Proteom. 2022, 251, 104407. [Google Scholar] [CrossRef]
- Finnicum, C.T.; Beck, J.J.; Dolan, C.V.; Davis, C.; Willemsen, G.; Ehli, E.A.; Boomsma, D.I.; Davies, G.E.; De Geus, E.J.C. Cohabitation Is Associated with a Greater Resemblance in Gut Microbiota Which Can Impact Cardiometabolic and Inflammatory Risk. BMC Microbiol. 2019, 19, 230. [Google Scholar] [CrossRef]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced Incidence of Prevotella and Other Fermenters in Intestinal Microflora of Autistic Children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef]
- Tomova, A.; Husarova, V.; Lakatosova, S.; Bakos, J.; Vlkova, B.; Babinska, K.; Ostatnikova, D. Gastrointestinal Microbiota in Children with Autism in Slovakia. Physiol. Behav. 2015, 138, 179–187. [Google Scholar] [CrossRef]
- Qiao, Y.; Wu, M.; Feng, Y.; Zhou, Z.; Chen, L.; Chen, F. Alterations of Oral Microbiota Distinguish Children with Autism Spectrum Disorders from Healthy Controls. Sci. Rep. 2018, 8, 1597. [Google Scholar] [CrossRef]
- Liu, S.; Li, E.; Sun, Z.; Fu, D.; Duan, G.; Jiang, M.; Yu, Y.; Mei, L.; Yang, P.; Tang, Y.; et al. Altered Gut Microbiota and Short Chain Fatty Acids in Chinese Children with Autism Spectrum Disorder. Sci. Rep. 2019, 9, 287. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal Microbiota and Metabolome of Children with Autism and Pervasive Developmental Disorder Not Otherwise Specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabrò, A.; et al. New Evidences on the Altered Gut Microbiota in Autism Spectrum Disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef]
- Maigoro, A.Y.; Lee, S. Gut Microbiome-Based Analysis of Lipid a Biosynthesis in Individuals with Autism Spectrum Disorder: An in Silico Evaluation. Nutrients 2021, 13, 688. [Google Scholar] [CrossRef]
- Ma, Q.; Li, Y.; Wang, J.; Li, P.; Duan, Y.; Dai, H.; An, Y.; Cheng, L.; Wang, T.; Wang, C.; et al. Investigation of Gut Microbiome Changes in Type 1 Diabetic Mellitus Rats Based on High-Throughput Sequencing. Biomed. Pharmacother. 2020, 124, 109873. [Google Scholar] [CrossRef]
- Cheng, M.; Sun, Y.; Wang, L.; Tan, L.; Jin, H.; Yan, S.; Li, S.; Xiao, X. Integrative Analysis of Microbiome and Metabolome in Rats with Gest-Aid plus Oral Liquid Supplementation Reveals Mechanism of Its Healthcare Function. Food Qual. Saf. 2021, 5, fyab010. [Google Scholar] [CrossRef]
- Rosenberg, E. The Family Prevotellaceae. In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea; Springer: Berlin/Heidelberg, Germany, 2014; pp. 825–827. ISBN 9783642301230. [Google Scholar]
- Ho, L.K.H.; Tong, V.J.W.; Syn, N.; Nagarajan, N.; Tham, E.H.; Tay, S.K.; Shorey, S.; Tambyah, P.A.; Law, E.C.N. Gut Microbiota Changes in Children with Autism Spectrum Disorder: A Systematic Review. Gut Pathog. 2020, 12, 6. [Google Scholar] [CrossRef]
- Srikantha, P.; Hasan Mohajeri, M. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 2115. [Google Scholar] [CrossRef]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Gérard, C.; van Nhieu, J.T.; Furet, J.P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced Diversity of Faecal Microbiota in Crohn’s Disease Revealed by a Metagenomic Approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Muehlbauer, A.L.; Alazizi, A.; Burns, M.B.; Findley, A.; Messina, F.; Gould, T.J.; Cascardo, C.; Pique-Regi, R.; Blekhman, R.; et al. Gut Microbiota Has a Widespread and Modifiable Effect on Host Gene Regulation. mSystems 2019, 4, e00323-18. [Google Scholar] [CrossRef] [PubMed]
- Morotomi, M. The Family Sutterellaceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9783642301, pp. 1005–1012. ISBN 9783642301971. [Google Scholar]
- Cheung, S.G.; Goldenthal, A.R.; Uhlemann, A.C.; Mann, J.J.; Miller, J.M.; Sublette, M.E. Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 2019, 10, 34. [Google Scholar] [CrossRef]
- Amirkhanzadeh Barandouzi, Z.; Starkweather, A.R.; Henderson, W.A.; Gyamfi, A.; Cong, X.S. Altered Composition of Gut Microbiota in Depression: A Systematic Review. Front. Psychiatry 2020, 11, 541. [Google Scholar]
- Chen, Y.J.; Wu, H.; Wu, S.D.; Lu, N.; Wang, Y.T.; Liu, H.N.; Dong, L.; Liu, T.T.; Shen, X.Z. Parasutterella, in Association with Irritable Bowel Syndrome and Intestinal Chronic Inflammation. J. Gastroenterol. Hepatol. 2018, 33, 1844–1852. [Google Scholar] [CrossRef]
- Luna, R.A.; Oezguen, N.; Balderas, M.; Venkatachalam, A.; Runge, J.K.; Versalovic, J.; Veenstra-VanderWeele, J.; Anderson, G.M.; Savidge, T.; Williams, K.C. Distinct Microbiome-Neuroimmune Signatures Correlate with Functional Abdominal Pain in Children with Autism Spectrum Disorder. Cmgh 2017, 3, 218–230. [Google Scholar] [CrossRef]
- Bush, J.R.; Alfa, M.J. Increasing Levels of Parasutterella in the Gut Microbiome Correlate with Improving Low-Density Lipoprotein Levels in Healthy Adults Consuming Resistant Potato Starch during a Randomised Trial. BMC Nutr. 2020, 6, 72. [Google Scholar] [CrossRef]
- Danneskiold-Samsøe, N.B.; Andersen, D.; Radulescu, I.D.; Normann-Hansen, A.; Brejnrod, A.; Kragh, M.; Madsen, T.; Nielsen, C.; Josefsen, K.; Fretté, X.; et al. A Safflower Oil Based High-Fat/High-Sucrose Diet Modulates the Gut Microbiota and Liver Phospholipid Profiles Associated with Early Glucose Intolerance in the Absence of Tissue Inflammation. Mol. Nutr. Food Res. 2017, 61, 1600528. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, Tryptophan Metabolism and the Brain-Gut-Microbiome Axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Ju, T.; Kong, J.Y.; Stothard, P.; Willing, B.P. Defining the Role of Parasutterella, a Previously Uncharacterized Member of the Core Gut Microbiota. ISME J. 2019, 13, 1520–1534. [Google Scholar] [CrossRef]
- Sasaki-Imamura, T.; Yoshida, Y.; Suwabe, K.; Yoshimura, F.; Kato, H. Molecular Basis of Indole Production Catalyzed by Tryptophanase in the Genus Prevotella. FEMS Microbiol. Lett. 2011, 322, 51–59. [Google Scholar] [CrossRef]
- Dong, T.S.; Guan, M.; Mayer, E.A.; Stains, J.; Liu, C.; Vora, P.; Jacobs, J.P.; Lagishetty, V.; Chang, L.; Barry, R.L.; et al. Obesity Is Associated with a Distinct Brain-Gut Microbiome Signature That Connects Prevotella and Bacteroides to the Brain’s Reward Center. Gut Microbes 2022, 14, 2051999. [Google Scholar] [CrossRef]
- Zhou, H.; Tai, J.; Xu, H.; Lu, X.; Meng, D. Xanthoceraside Could Ameliorate Alzheimer’s Disease Symptoms of Rats by Affecting the Gut Microbiota Composition and Modulating the Endogenous Metabolite Levels. Front. Pharmacol. 2019, 10, 1035. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, H.; Yu, B.; Tao, H.; Li, J.; Wu, Z.; Liu, G.; Yuan, C.; Guo, L.; Cui, B. Lycium Barbarum Polysaccharide Attenuates Myocardial Injury in High-Fat Diet-Fed Mice through Manipulating the Gut Microbiome and Fecal Metabolome. Food Res. Int. 2020, 138, 109778. [Google Scholar] [CrossRef]
- Olsen, I.; Hicks, S.D. Oral Microbiota and Autism Spectrum Disorder (ASD). J. Oral Microbiol. 2020, 12, 1702806. [Google Scholar] [CrossRef]
- Behrouzi, A.; Nafari, A.H.; Siadat, S.D. The Significance of Microbiome in Personalized Medicine. Clin. Transl. Med. 2019, 8, 16. [Google Scholar] [CrossRef]
- Li, N.; Yang, J.; Zhang, J.; Liang, C.; Wang, Y.; Chen, B.; Zhao, C.; Wang, J.; Zhang, G.; Zhao, D.; et al. Correlation of Gut Microbiome Between ASD Children and Mothers and Potential Biomarkers for Risk Assessment. Genom. Proteom. Bioinform. 2019, 17, 26–38. [Google Scholar] [CrossRef]
- Jurburg, S.D.; Konzack, M.; Eisenhauer, N.; Heintz-Buschart, A. The Archives Are Half-Empty: An Assessment of the Availability of Microbial Community Sequencing Data. Commun. Biol. 2020, 3, 474. [Google Scholar] [CrossRef]
- Klie, A.; Tsui, B.Y.; Mollah, S.; Skola, D.; Dow, M.; Hsu, C.N.; Carter, H. Increasing Metadata Coverage of SRA BioSample Entries Using Deep Learning-Based Named Entity Recognition. Database 2021, 2021, baab021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | ASD Samples | HC Samples | ASD Samples after Quality Filtering | HC Samples after Quality Filtering | Country | BioProject ID |
|---|---|---|---|---|---|---|
| Averina | 15 | 5 | 15 | 5 | Russia | PRJNA516054 |
| Coretti | 11 | 14 | 4 | 0 | Italy | PRJEB29421 |
| Dan | 142 | 143 | 142 | 143 | China | PRJNA453621 |
| Pulikkan | 30 | 24 | 30 | 24 | India | PRJNA355023 |
| Son | 59 | 44 | 59 | 44 | USA | PRJNA282013 |
| Zurita | 27 | 31 | 27 | 31 | Ecuador | PRJEB27306 |
| Vernocchi | 206 | 108 | 197 | 108 | Italy | PRJNA754695 |
| AGP | 50 | 50 | 47 | 47 | USA | - |
| Variable | Df | Sum | R2 | F | Pr (>F) |
|---|---|---|---|---|---|
| (A) Values prior to batch effect removal | |||||
| Phenotype | 1 | 0.0788 | 0.03050 | 39.354 | 0.001 |
| ìStudy ID | 6 | 74.081 | 0.28705 | 616.687 | 0.001 |
| Residual | 911 | 182.396 | 0.70676 | ||
| Total | 918 | 328.85 | 1.0000 | ||
| (B) Values after to batch effect removal | |||||
| Phenotype | 1 | 0.0782 | 0.02508 | 27.416 | 0.001 |
| Study ID | 6 | 42.040 | 0.13874 | 255.534 | 0.001 |
| Residual | 911 | 259.968 | 0.85795 | ||
| Total | 918 | 303.012 | 1.000 | ||
| Dataset | Algorithm Parameters | Threshold | Accuracy | Precision | Recall (TPR) & Specificity (TNR) | F-Score |
|---|---|---|---|---|---|---|
| Dan [12] | ntree = 1500, | 0.4540 | 0.85 | 0.85 | 0.86 | 0.85 |
| mtry = 10.21 | ||||||
| Vernocchi [18] | ntree = 1000, | 0.6190 | 0.72 | 0.82 | 0.72 | 0.77 |
| mtry = 10.21 | ||||||
| Son [15] | ntree = 1500, | 0.5640 | 0.41 | 0.49 | 0.41 | 0.44 |
| mtry = 6.21 |
| Algorithm | Strategy | Algorithm Parameters | Threshold | Accuracy | Precision | Recall (TPR) & Specificity (TNR) | F-Score |
|---|---|---|---|---|---|---|---|
| RF | 1 | ntree = 500, | 0.5580 | 0.67 | 0.71 | 0.67 | 0.70 |
| mtry = 7.21 | |||||||
| RF | 2 | ntree = 2000, | 0.5570 | 0.70 | 0.76 | 0.70 | 0.72 |
| mtry = 12.27 | |||||||
| RF | 3 | ntree = 500, | 0.5640 | 0.49 | 0.54 | 0.54 | 0.54 |
| mtry = 8.21 | |||||||
| GBM | 1 | n.trees = 1000, | 0.6545 | 0.62 | 0.68 | 0.62 | 0.65 |
| interaction.depth = 1, | |||||||
| n.minobsinnod = 1, | |||||||
| shrinkage = 0.1 | |||||||
| GBM | 2 | n.trees = 1000, | 0.6053 | 0.69 | 0.73 | 0.69 | 0.71 |
| interaction.depth = 1, | |||||||
| n.minobsinnod = 5, | |||||||
| shrinkage = 0.1 | |||||||
| GBM | 3 | n.trees = 2500, | 0.9853 | 0.48 | 0.54 | 0.49 | 0.47 |
| interaction.depth = 1, | |||||||
| n.minobsinnod = 0.1, | |||||||
| shrinkage = 20 | |||||||
| SVM | 1 | C = 1, sigma = 2.9802 × 10−8 | 0.5966 | 0.65 | 0.70 | 0.65 | 0.67 |
| SVM | 2 | C = 246, sigma = 3.1250 × 10−2 | 0.6025 | 0.69 | 0.74 | 0.70 | 0.72 |
| SVM | 3 | C = 81, sigma = 9.7656 × 10−4 | 0.5632 | 0.45 | 0.53 | 0.49 | 0.50 |
| Bacterial Taxa | Importance “RF” Algorithm | Importance “GBM” Algorithm | Importance “SVM” Algorithm |
|---|---|---|---|
| Alloprevotella | 1 | 1 | 5 |
| Clostridia UCG-014 | 6 | 6 | 13 |
| Faecalibacterium | 15 | 9 | 11 |
| Haemophilus | 13 | 4 | 12 |
| Parasutterella | 10 | 10 | 4 |
| [Eubacterium] siraeum group | 8 | 5 | |
| Actinomyces | 9 | 3 | |
| Bacteroides | 13 | 1 | |
| Gastranaerophilales | 7 | 12 | |
| Megamonas | 11 | 7 | |
| Muribaculaceae | 5 | 3 | |
| Negativibacillus | 4 | 2 | |
| Subdoligranulum | 15 | 6 | |
| [Eubacterium] xylanophilum group | 2 | ||
| Agathobacter | 15 | ||
| Alistipes | 10 | ||
| Blautia | 14 | ||
| Butyricicoccus | 7 | ||
| Enterococcus | 9 | ||
| Holdemanella | 12 | ||
| Lachnospira | 14 | ||
| RF39 | |||
| Romboutsia | 14 | 8 | |
| TM7x | 2 | ||
| Turicibacter | |||
| Tyzzerella | 3 | 11 | |
| Veillonella | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrucci, D.; Teofani, A.; Milanesi, M.; Fosso, B.; Putignani, L.; Messina, F.; Pesole, G.; Desideri, A.; Chillemi, G. Machine Learning Data Analysis Highlights the Role of Parasutterella and Alloprevotella in Autism Spectrum Disorders. Biomedicines 2022, 10, 2028. https://doi.org/10.3390/biomedicines10082028
Pietrucci D, Teofani A, Milanesi M, Fosso B, Putignani L, Messina F, Pesole G, Desideri A, Chillemi G. Machine Learning Data Analysis Highlights the Role of Parasutterella and Alloprevotella in Autism Spectrum Disorders. Biomedicines. 2022; 10(8):2028. https://doi.org/10.3390/biomedicines10082028
Chicago/Turabian StylePietrucci, Daniele, Adelaide Teofani, Marco Milanesi, Bruno Fosso, Lorenza Putignani, Francesco Messina, Graziano Pesole, Alessandro Desideri, and Giovanni Chillemi. 2022. "Machine Learning Data Analysis Highlights the Role of Parasutterella and Alloprevotella in Autism Spectrum Disorders" Biomedicines 10, no. 8: 2028. https://doi.org/10.3390/biomedicines10082028
APA StylePietrucci, D., Teofani, A., Milanesi, M., Fosso, B., Putignani, L., Messina, F., Pesole, G., Desideri, A., & Chillemi, G. (2022). Machine Learning Data Analysis Highlights the Role of Parasutterella and Alloprevotella in Autism Spectrum Disorders. Biomedicines, 10(8), 2028. https://doi.org/10.3390/biomedicines10082028