
The Alpha 7 Nicotinic Acetylcholine Receptor Does Not Affect Neonatal Brain Injury

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Experimental Design
2.2. In Vivo Procedures and Treatment
2.2.1. Animals
2.2.2. Hypoxia-Ischemia
2.2.3. Treatment
2.3. Behavioral Assessments of Motor Function
2.4. Sacrifice and Tissue Harvest
2.5. Protein and RNA Analysis
2.6. Caspase-3 Activity Assay
2.7. ELISA
2.8. QPCR
2.9. Immunohistochemistry
2.10. Neuron and Microglia Staining
2.11. Quantification of Brain Injury and Microglial Scoring
2.12. Statistical Analyses and Visualization
3. Results
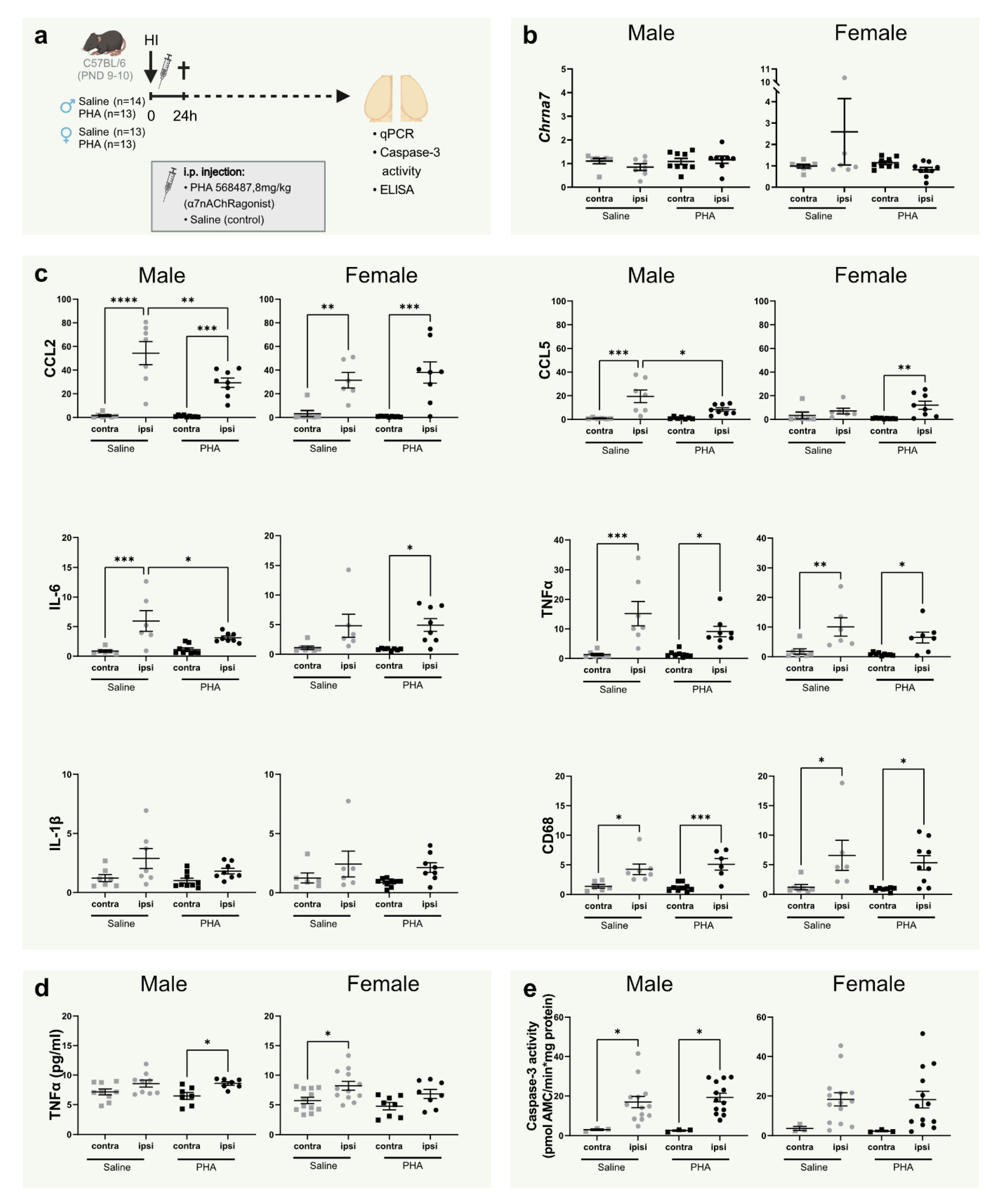
3.1. Activation of α7nAChR Does Not Alter α7nAChR mRNA Expression in the Brain
3.2. Activation of α7nAChR Decreases Gene Expression of CCL2, CCL5 and IL-6 24 h in the Brain 24 h after Hypoxia Ischemia in Male Pups
3.3. TNFα, Caspase-3 Activity and Tissue Loss Is Not Altered by α7nAChR Agonist Treatment at 24 h
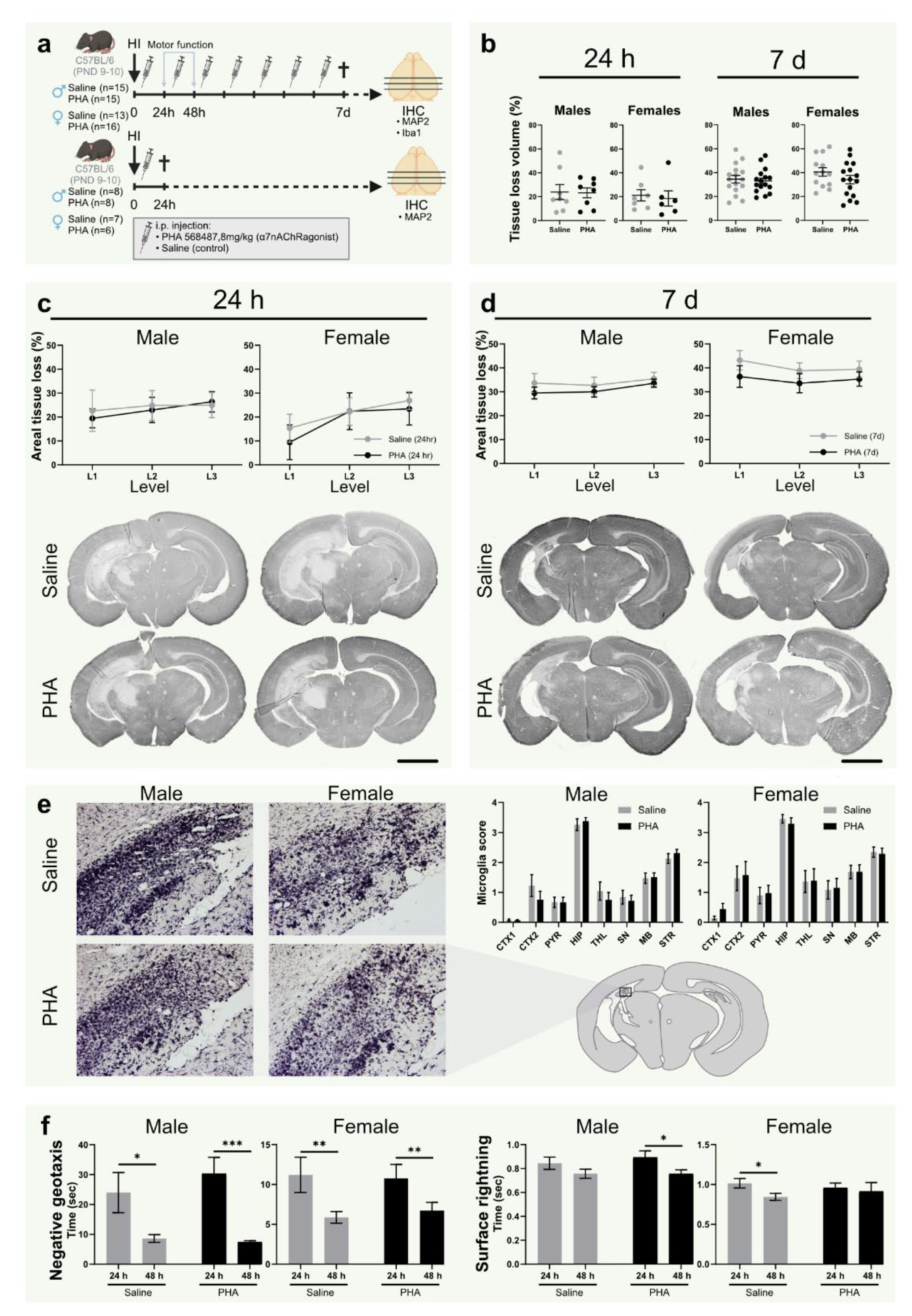
3.4. α7nAChR Activation Does Not Affect Tissue Loss or Microglial Activation in Neonatal Hypoxic Ischemic Brain Injury
3.5. Activation of α7nAChR Does Not Affect Motor Function or Weight Gain after HI
3.6. Knock-Out of α7nAChR Does Not Affect Tissue Loss in Neonatal Hypoxic-Ischemic Brain Injury
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.; Ritter, S.; Brotschi, B.; Werner, H.; Caflisch, J.; Martin, E.; Latal, B. Long-term neurodevelopmental outcome with hypoxic-ischemic encephalopathy. J. Pediatr. 2013, 163, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Simbruner, G.; Mittal, R.A.; Rohlmann, F.; Muche, R.; Participants, N.T. Systemic Hypothermia After Neonatal Encephalopathy: Outcomes of neo.nEURO.network RCT. Pediatrics 2010, 126, e771–e778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-H.; Cheng, G.-Q.; Shao, X.-M.; Liu, X.-Z.; Shan, R.-B.; Zhuang, D.-Y.; Zhou, C.-L.; Du, L.-Z.; Cao, Y.; Yang, Q.; et al. Selective Head Cooling with Mild Systemic Hypothermia after Neonatal Hypoxic-Ischemic Encephalopathy: A Multicenter Randomized Controlled Trial in China. J. Pediatr. 2010, 157, 367–372.e3. [Google Scholar] [CrossRef]
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010, 340, c363. [Google Scholar] [CrossRef]
- Dammann, O.; Leviton, A. Inflammatory brain damage in preterm newborns—Dry numbers, wet lab, and causal inferences. Early Hum. Dev. 2004, 79, 1–15. [Google Scholar] [CrossRef]
- Hedtjarn, M.; Mallard, C.; Hagberg, H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2004, 24, 1333–1351. [Google Scholar] [CrossRef]
- Vexler, Z.S.; Tang, X.N.; Yenari, M.A. Inflammation in adult and neonatal stroke. Clin. Neurosci. Res. 2006, 6, 293–313. [Google Scholar] [CrossRef]
- Concepcion, K.R.; Zhang, L. Corticosteroids and perinatal hypoxic-ischemic brain injury. Drug Discov. Today 2018, 23, 1718–1732. [Google Scholar] [CrossRef]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Alkondon, M.; Pereira, E.F.R.; Castro, N.G.; Schrattenholz, A.; Barbosa, C.T.F.; Bonfante-Cabarcas, R.; Aracava, Y.; Eisenberg, H.M.; Maelicke, A. Properties of neuronal nicotinic acetylcholine receptors: Pharmacological characterization and mod-ulation of synaptic function. J. Pharmacol. Exp. Ther. 1997, 280, 1117–1136. [Google Scholar]
- Kihara, T.; Shimohama, S.; Sawada, H.; Honda, K.; Nakamizo, T.; Shibasaki, H.; Toshiaki, K.; Akinori, A. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem. 2001, 276, 13541–13546. [Google Scholar] [CrossRef]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Jiang, T.; Wu, M.; Zhang, Z.; Yan, C.; Ma, Z.; He, S.; Wei, Y.; Pu, K.; Wang, Q. Electroacupuncture attenuated cerebral ischemic injury and neuroinflammation through al-pha7nAChR-mediated inhibition of NLRP3 inflammasome in stroke rats. Mol. Med. 2019, 25, 22. [Google Scholar] [CrossRef]
- Krafft, P.R.; McBride, D.; Rolland, W.B.; Lekic, T.; Flores, J.J.; Zhang, J.H. α7 Nicotinic Acetylcholine Receptor Stimulation Attenuates Neuroinflammation through JAK2-STAT3 Activation in Murine Models of Intracerebral Hemorrhage. BioMed Res. Int. 2017, 2017, 8134653. [Google Scholar] [CrossRef]
- Laudenbach, V.; Medja, F.; Zoli, M.; Rossi, F.M.; Evrard, P.; Changeux, J.-P.; Gressens, P. Selective activation of central subtypes of the nicotinic acetylcholine receptor has opposite effects on neonatal excitotoxic brain injuries. FASEB J. 2002, 16, 423–425. [Google Scholar] [CrossRef]
- Colas, L.; Domercq, M.; Ramos-Cabrer, P.; Palma, A.; Gómez-Vallejo, V.; Padro, D.; Plaza-García, S.; Pulagam, K.R.; Higuchi, M.; Matute, C.; et al. In vivo imaging of Alpha7 nicotinic receptors as a novel method to monitor neuroinflammation after cerebral ischemia. Glia 2018, 66, 1611–1624. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Fu, X.; Zhang, D.; Yu, L.; Li, N.; Gao, Y.; Liu, X.; Yin, C.; Ke, J.; et al. Alpha-7 Nicotinic Receptor Signaling Pathway Participates in the Neurogenesis Induced by ChAT-Positive Neurons in the Subventricular Zone. Transl. Stroke Res. 2017, 8, 484–493. [Google Scholar] [CrossRef]
- Han, Z.; Li, L.; Wang, L.; Degos, V.; Maze, M.; Su, H. Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J. Neurochem. 2014, 131, 498–508. [Google Scholar] [CrossRef]
- Krafft, P.R.; Altay, O.; Rolland, W.B.; Duris, K.; Lekic, T.; Tang, J.; Zhang, J.H. alpha7 nicotinic acetylcholine receptor agonism confers neuroprotection through GSK-3beta inhibition in a mouse model of intracerebral hemorrhage. Stroke 2012, 43, 844–850. [Google Scholar] [CrossRef]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Svedin, P.; Hagberg, H.; Sävman, K.; Zhu, C.; Mallard, C. Matrix Metalloproteinase-9 Gene Knock-out Protects the Immature Brain after Cerebral Hypoxia–Ischemia. J. Neurosci. 2007, 27, 1511–1518. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Walker, D.P.; Wishka, D.G.; Piotrowski, D.W.; Jia, S.; Reitz, S.C.; Yates, K.M.; Myers, J.K.; Vetman, T.N.; Margolis, B.J.; Jacobsen, E.J.; et al. Design, synthesis, structure-activity relationship, and in vivo activity of azabicyclic aryl amides as alpha7 nicotinic acetylcholine receptor agonists. Bioorg. Med. Chem. 2006, 14, 8219–8248. [Google Scholar] [CrossRef]
- Feather-Schussler, D.N.; Ferguson, T.S. A Battery of Motor Tests in a Neonatal Mouse Model of Cerebral Palsy. J. Vis. Exp. 2016, 117, e53569. [Google Scholar] [CrossRef]
- Li, T.; Li, K.; Zhang, S.; Wang, Y.; Xu, Y.; Cronin, S.J.F.; Sun, Y.; Zhang, Y.; Xie, C.; Rodriguez, J.I.; et al. Overexpression of apoptosis inducing factor aggravates hypoxic-ischemic brain injury in neonatal mice. Cell Death Dis. 2020, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Ek, C.J.; Mallard, C.; Johansson, M.E. Perinatal hypoxia-ischemia reduces alpha 7 nicotinic receptor expression and selective alpha 7 nicotinic receptor stimulation suppresses inflammation and promotes microglial Mox phenotype. Biomed. Res. Int. 2014, 2014, 718769. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.; Svedin, P.; Ardalan, M.; Levy, O.; Ek, C.J.; Mallard, C.; Lai, J. Staphylococcus epidermidis Sensitizes Perinatal Hypoxic-Ischemic Brain Injury in Male but Not Female Mice. Front. Immunol. 2020, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Ferreira, E.; Poupon, L.; Zelco, A.; Leverin, A.L.; Nair, S.; Jonsdotter, A.; Carlsson, Y.; Thornton, C.; Hagberg, H.; Rahim, A.A. Neuroprotective exendin-4 enhances hypothermia therapy in a model of hypoxic-ischaemic enceph-alopathy. Brain 2018, 141, 2925–2942. [Google Scholar] [CrossRef]
- Hammarlund, M.E.; Darsalia, V.; Mjörnstedt, F.; Pattanaik, B.; Mallard, C.; Rocha-Ferreira, E.; Patrone, C.; Johansson, M.E. The selective alpha7 nicotinic acetylcholine receptor agonist AR-R17779 does not affect ischemia–reperfusion brain injury in mice. Biosci. Rep. 2021, 41, BSR20210736. [Google Scholar] [CrossRef]
- Johansson, M.E.; Ulleryd, M.A.; Bernardi, A.; Lundberg, A.M.; Andersson, A.; Folkersen, L.; Fogelstrand, L.; Islander, U.; Yan, Z.-Q.; Hansson, G.K. α7 Nicotinic Acetylcholine Receptor Is Expressed in Human Atherosclerosis and Inhibits Disease in Mice—Brief Report. Arter. Thromb. Vasc. Biol. 2014, 34, 2632–2636. [Google Scholar] [CrossRef]
- Ulleryd, M.A.; Mjörnstedt, F.; Panagaki, D.; Yang, L.J.; Engevall, K.; Gutiérrez, S.; Wang, Y.; Gan, L.-M.; Nilsson, H.; Michaëlsson, E.; et al. Stimulation of alpha 7 nicotinic acetylcholine receptor (alpha7nAChR) inhibits atherosclerosis via im-munomodulatory effects on myeloid cells. Atherosclerosis 2019, 287, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Q.; Zhang, W.-J.; Su, D.-F.; Zhang, G.-Q.; Miao, C.-Y. Cellular responses and functions of α7 nicotinic acetylcholine receptor activation in the brain: A narrative review. Ann. Transl. Med. 2021, 9, 509. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, X.; Hui, Y.; Zhu, C.; Wu, J.; Taylor, D.H.; Ji, J.; Fan, W.; Huang, Z.; Hu, J. Activation of α7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: Implications for Parkinson’s disease. Neuropharmacology 2015, 91, 87–96. [Google Scholar] [CrossRef]
- Revathikumar, P.; Bergqvist, F.; Gopalakrishnan, S.; Korotkova, M.; Jakobsson, P.-J.; Lampa, J.; Le Maître, E. Immunomodulatory effects of nicotine on interleukin 1β activated human astrocytes and the role of cyclooxygenase 2 in the underlying mechanism. J. Neuroinflamm. 2016, 13, 256. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef]
- Suzuki, T.; Hide, I.; Matsubara, A.; Hama, C.; Harada, K.; Miyano, K.; Andrä, M.; Matsubayashi, H.; Sakai, N.; Kohsaka, S.; et al. Microglial α7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J. Neurosci. Res. 2006, 83, 1461–1470. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef]
- Rocha-Ferreira, E.; Vincent, A.; Bright, S.; Peebles, D.M.; Hristova, M. The duration of hypothermia affects short-term neuro-protection in a mouse model of neonatal hypoxic ischaemic injury. PLoS ONE 2018, 13, e0199890. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Aly, H.; Khashaba, M.T.; El-Ayouty, M.; El-Sayed, O.; Hasanein, B.M. IL-1β, IL-6 and TNF-α and outcomes of neonatal hypoxic ischemic encephalopathy. Brain Dev. 2006, 28, 178–182. [Google Scholar] [CrossRef]
- Szaflarski, J.; Burtrum, D.; Silverstein, F.S. Cerebral Hypoxia-Ischemia Stimulates Cytokine Gene Expression in Perinatal Rats. Stroke 1995, 26, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Bona, E.; Andersson, A.-L.; Blomgren, K.; Gilland, E.; Puka-Sundvall, M.; Gustafson, K.; Hagberg, H. Chemokine and Inflammatory Cell Response to Hypoxia-Ischemia in Immature Rats. Pediatr. Res. 1999, 45, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, K.; Llovera, G.; Recasens, M.; Chertoff, M.; Giménez-Llort, L.; Gonzalez, B.; Acarin, L. Temporal Expression of Cytokines and Signal Transducer and Activator of Transcription Factor 3 Acti-vation after Neonatal Hypoxia/Ischemia in Mice. Dev. Neurosci. 2013, 35, 212–225. [Google Scholar] [CrossRef]
- Hagberg, H.; Gilland, E.; Bona, E.; Hanson, L.-Å.; Hahn-Zoric, M.; Blennow, M.; Holst, M.; McRae, A.; Söder, O. Enhanced Expression of Interleukin (IL)-1 and IL-6 Messenger RNA and Bioactive Protein after Hypox-ia-Ischemia in Neonatal Rats. Pediatr. Res. 1996, 40, 603–609. [Google Scholar] [CrossRef]
- Andres, R.H.; Choi, R.; Pendharkar, A.V.; Gaeta, X.; Wang, N.; Nathan, J.K.; Chua, J.Y.; Lee, S.W.; Palmer, T.D.; Steinberg, G.K.; et al. The CCR2/CCL2 interaction mediates the transendothelial recruitment of intravascularly delivered neural stem cells to the ischemic brain. Stroke 2011, 42, 2923–2931. [Google Scholar] [CrossRef]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte Chemoattractant Protein-1 Deficiency is Protective in a Murine Stroke Model. J. Cereb. Blood Flow Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, K.; Suzuki, N. Ambivalent Aspects of Interleukin-6 in Cerebral Ischemia: Inflammatory versus Neurotrophic Aspects. J. Cereb. Blood Flow Metab. 2009, 29, 464–479. [Google Scholar] [CrossRef]
- Thornton, C.; Leaw, B.; Mallard, C.; Nair, S.; Jinnai, M.; Hagberg, H. Cell Death in the Developing Brain after Hypoxia-Ischemia. Front. Cell. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Xu, F.; Wang, X.; Shibata, M.; Uchiyama, Y.; Blomgren, K.; Hagberg, H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J. Neurochem. 2006, 96, 1016–1027. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Mottahedin, A.; Stridh, L.; Yellowhair, T.R.; Jantzie, L.L.; Northington, F.J.; Mallard, C. Evidence for Sexual Dimorphism in the Response to TLR3 Activation in the Developing Neonatal Mouse Brain: A Pilot Study. Front. Physiol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Hurn, P.D.; Vannucci, S.J.; Hagberg, H. Adult or Perinatal Brain Injury. Stroke 2005, 36, 193–195. [Google Scholar] [CrossRef]
- Cheng, J.; Hurn, P.D. Sex shapes experimental ischemic brain injury. Steroids 2010, 75, 754–759. [Google Scholar] [CrossRef]
- Skiöld, B.; Alexandrou, G.; Padilla, N.; Blennow, M.; Vollmer, B.; Ådén, U. Sex Differences in Outcome and Associations with Neonatal Brain Morphology in Extremely Preterm Children. J. Pediatr. 2014, 164, 1012–1018. [Google Scholar] [CrossRef]
- Otto, S.L.; Yakel, J.L. The α7 nicotinic acetylcholine receptors regulate hippocampal adult-neurogenesis in a sexually dimorphic fashion. Anat. Embryol. 2019, 224, 829–846. [Google Scholar] [CrossRef]
- Vexler, Z.S.; Yenari, M.A. Does inflammation after stroke affect the developing brain differently than adult brain? Dev. Neurosci. 2009, 31, 378–393. [Google Scholar] [CrossRef]
- Doverhag, C.; Hedtjärn, M.; Poirier, F.; Mallard, C.; Hagberg, H.; Karlsson, A.; Sävman, K. Galectin-3 contributes to neonatal hypoxic–ischemic brain injury. Neurobiol. Dis. 2010, 38, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Hunter, B.E.; de Fiebre, C.M.; Papke, R.L.; Kem, W.R.; Meyer, E.M. A novel nicotinic agonist facilitates induction of long-term potentiation in the rat hippocampus. Neurosci. Lett. 1994, 168, 130–134. [Google Scholar] [CrossRef]
- Meyer, E.M.; Kuryatov, A.; Gerzanich, V.; Lindstrom, J.; Papke, R.L. Analysis of 3-(4-hydroxy, 2-Methoxybenzylidene)anabaseine selectivity and activity at human and rat alpha-7 nicotinic receptors. J. Pharmacol. Exp. Ther. 1998, 287, 918–925. [Google Scholar] [PubMed]
- Garg, B.K.; Loring, R.H. GTS-21 has cell-specific anti-inflammatory effects independent of α7 nicotinic acetylcholine receptors. PLoS ONE 2019, 14, e0214942. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammarlund, M.E.; Ek, C.J.; Akar, S.; Karlsson, A.; Pattanaik, B.; Mjörnstedt, F.; Svedin, P.; Ardalan, M.; Rocha-Ferreira, E.; Mallard, C.; et al. The Alpha 7 Nicotinic Acetylcholine Receptor Does Not Affect Neonatal Brain Injury. Biomedicines 2022, 10, 2023. https://doi.org/10.3390/biomedicines10082023
Hammarlund ME, Ek CJ, Akar S, Karlsson A, Pattanaik B, Mjörnstedt F, Svedin P, Ardalan M, Rocha-Ferreira E, Mallard C, et al. The Alpha 7 Nicotinic Acetylcholine Receptor Does Not Affect Neonatal Brain Injury. Biomedicines. 2022; 10(8):2023. https://doi.org/10.3390/biomedicines10082023
Chicago/Turabian StyleHammarlund, Maria E., C. Joakim Ek, Sukaina Akar, Alma Karlsson, Bagmi Pattanaik, Filip Mjörnstedt, Pernilla Svedin, Maryam Ardalan, Eridan Rocha-Ferreira, Carina Mallard, and et al. 2022. "The Alpha 7 Nicotinic Acetylcholine Receptor Does Not Affect Neonatal Brain Injury" Biomedicines 10, no. 8: 2023. https://doi.org/10.3390/biomedicines10082023
APA StyleHammarlund, M. E., Ek, C. J., Akar, S., Karlsson, A., Pattanaik, B., Mjörnstedt, F., Svedin, P., Ardalan, M., Rocha-Ferreira, E., Mallard, C., & Johansson, M. E. (2022). The Alpha 7 Nicotinic Acetylcholine Receptor Does Not Affect Neonatal Brain Injury. Biomedicines, 10(8), 2023. https://doi.org/10.3390/biomedicines10082023