
The Histone Deacetylase Inhibitor ITF2357 (Givinostat) Targets Oncogenic BRAF in Melanoma Cells and Promotes a Switch from Pro-Survival Autophagy to Apoptosis



 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Cultures
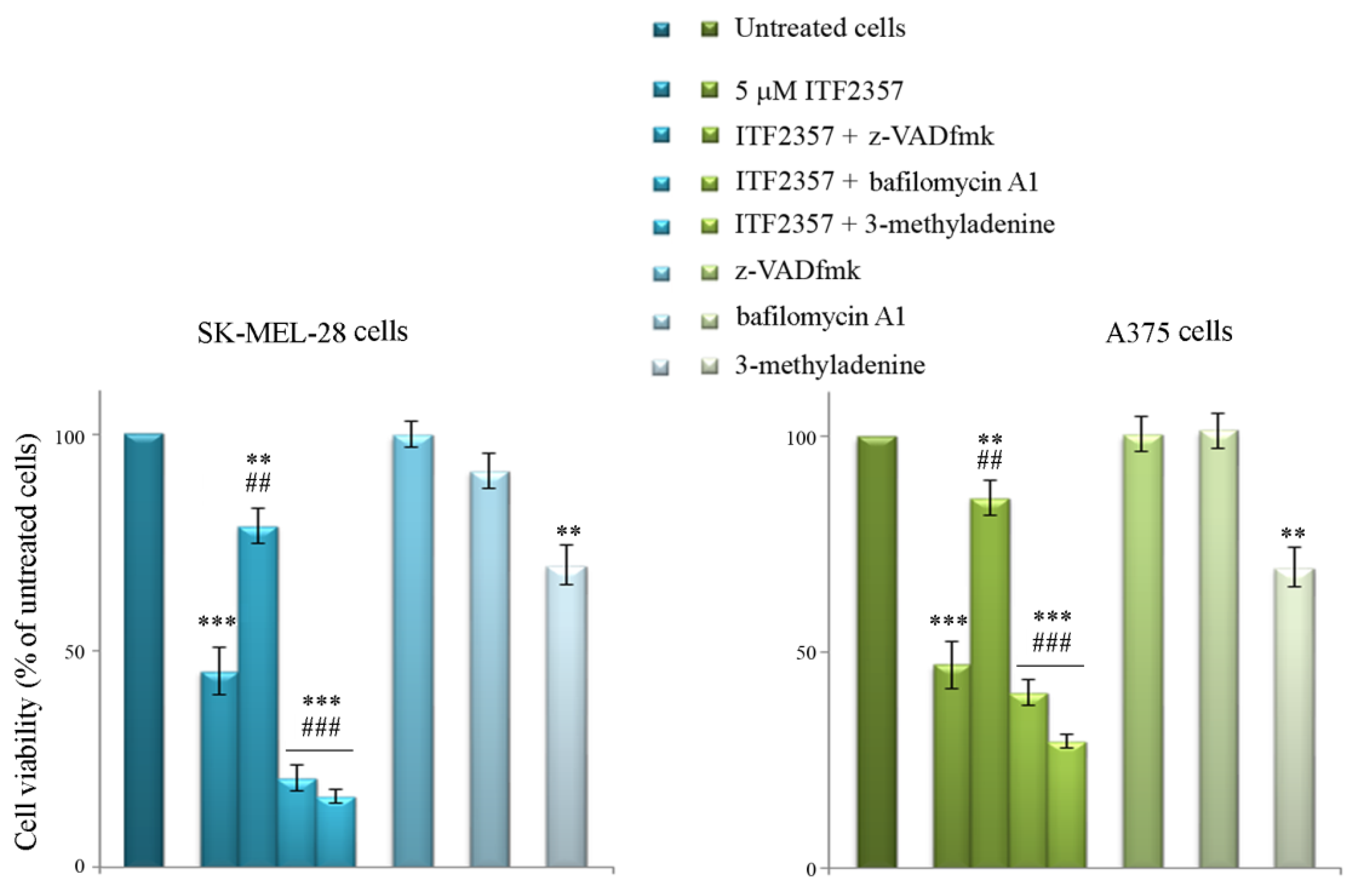
2.3. Evaluation of Cell Viability
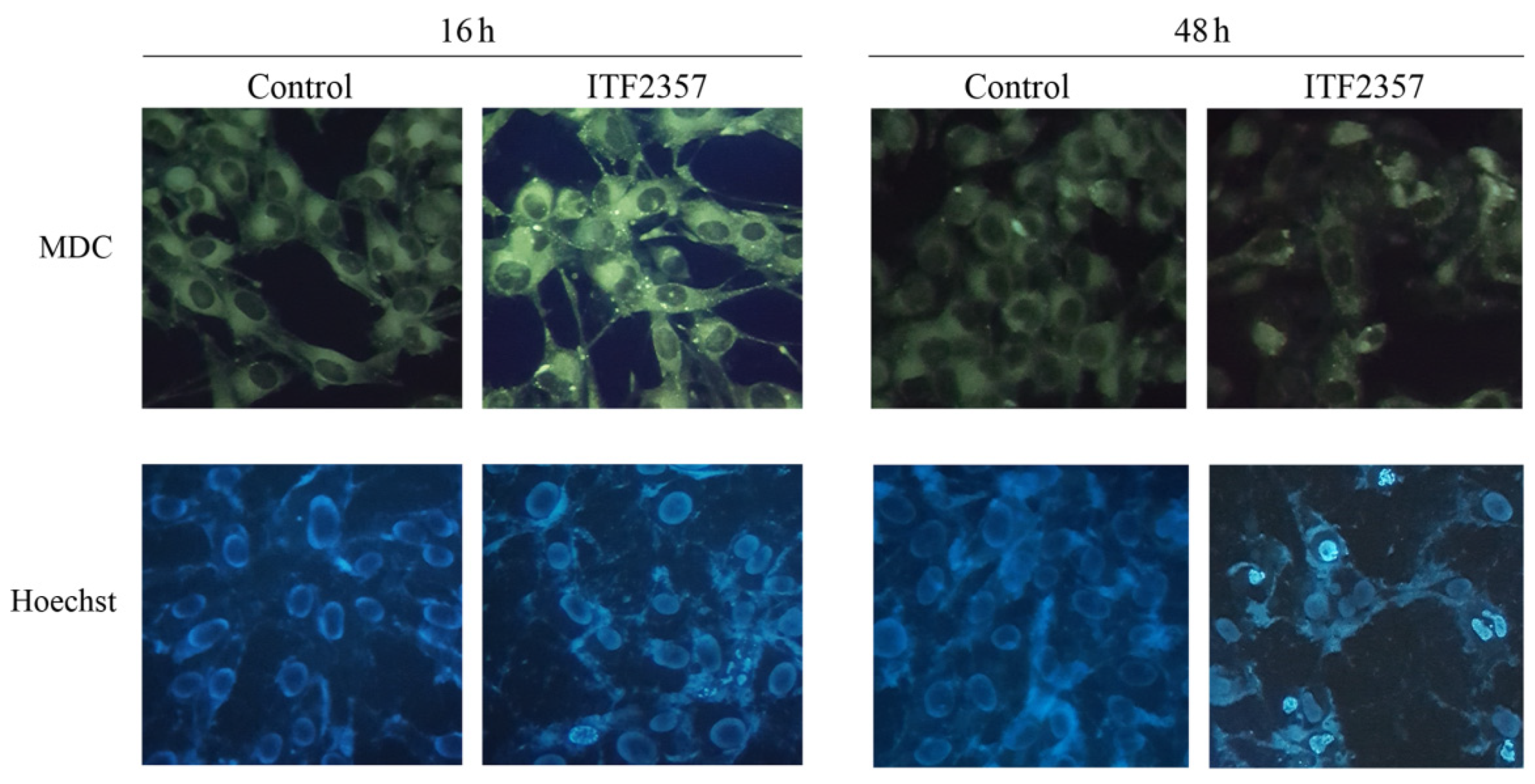
2.4. Evaluation of Autophagy by Monodansylcadaverine
2.5. Detection of Chromatin Condensation by Hoechst Staining
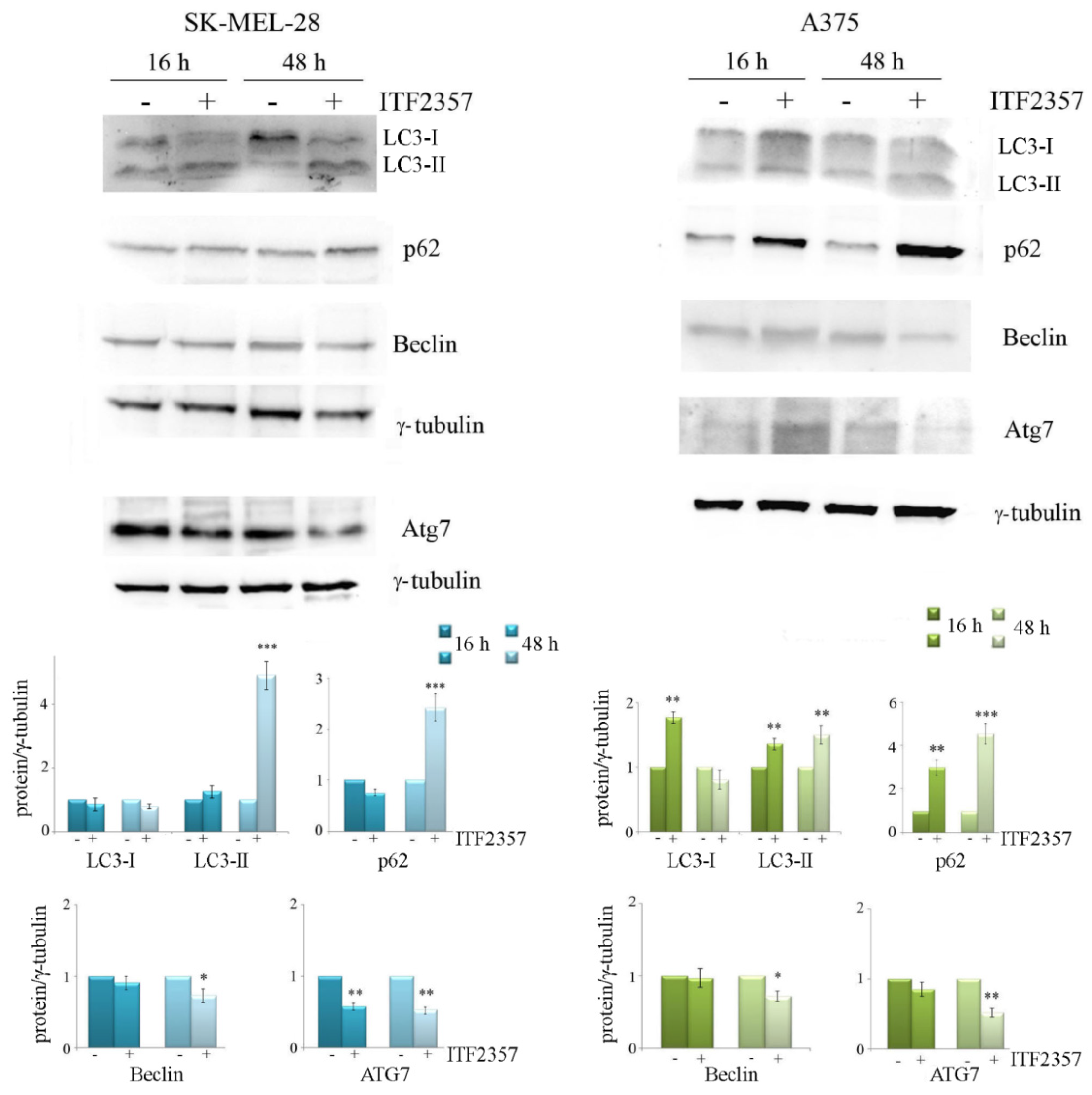
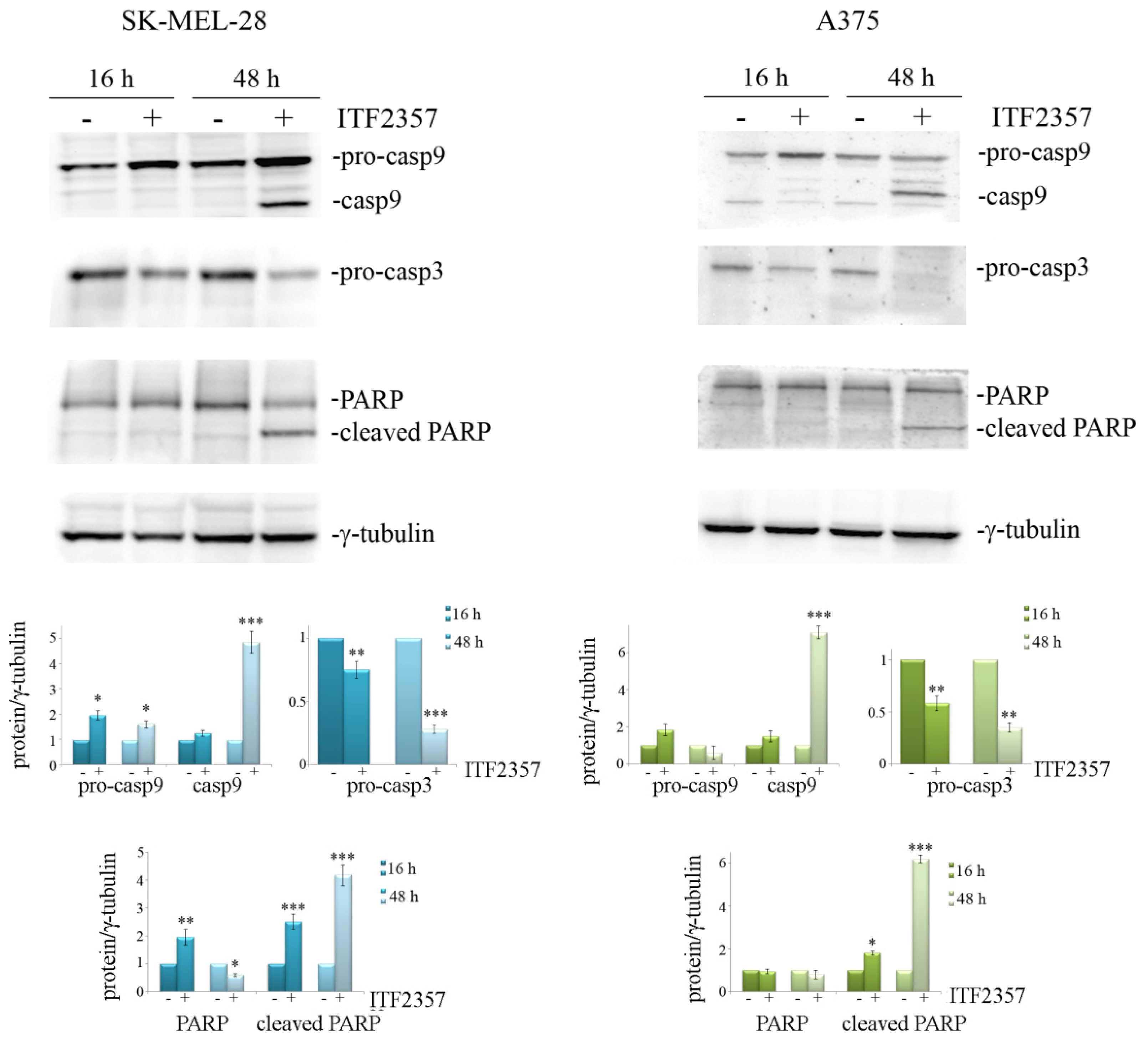
2.6. Western Blot Analysis
2.7. Semiquantitative RT-PCR
2.8. Statistical Analysis
3. Results
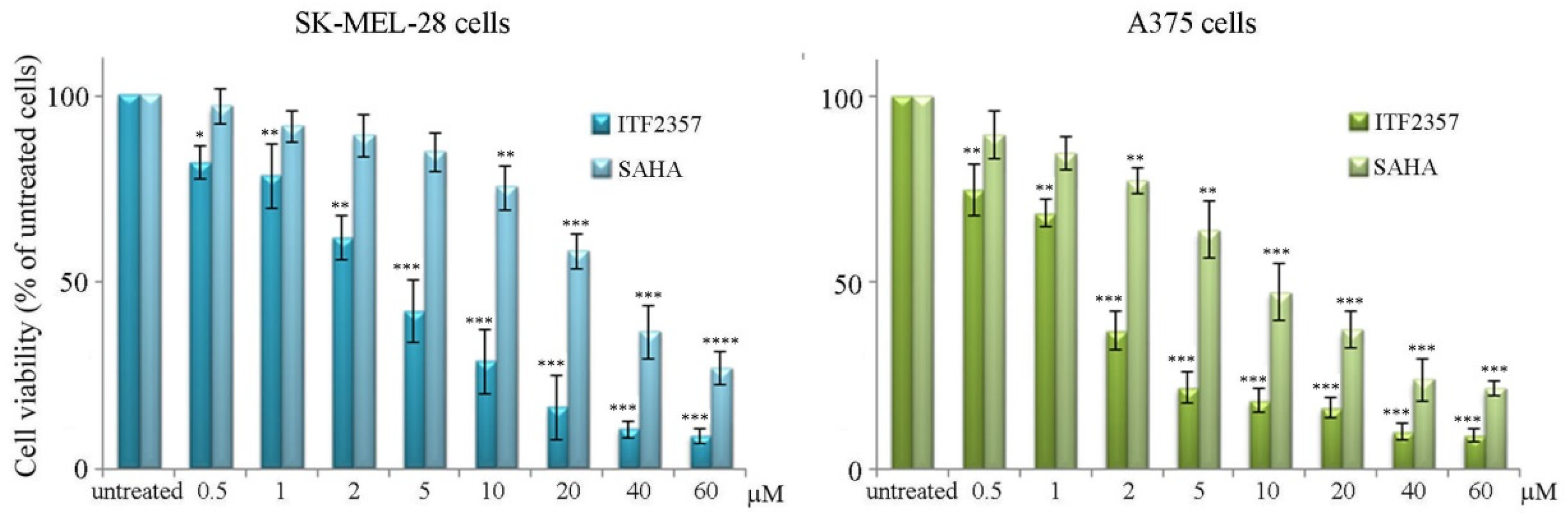
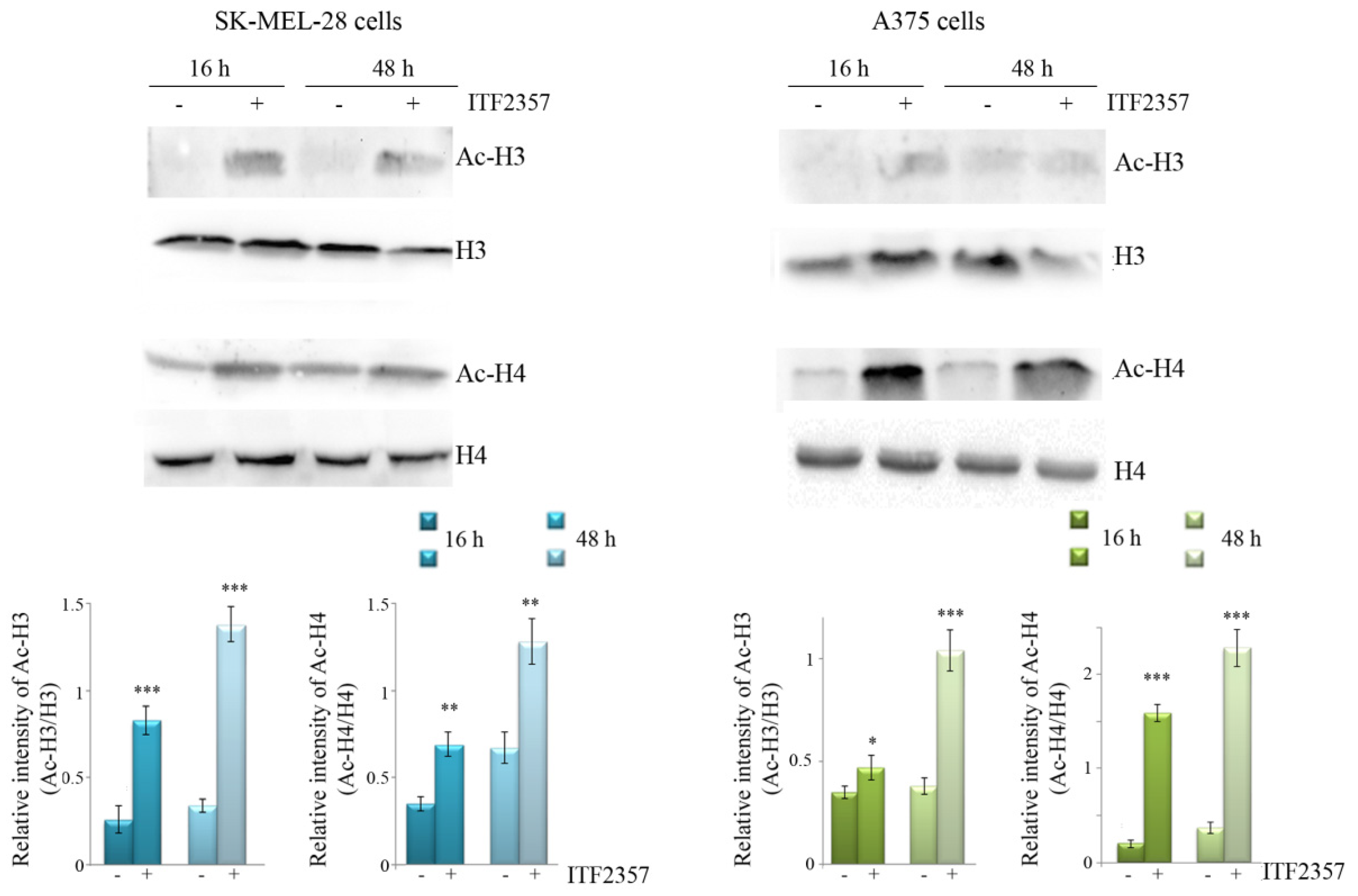
3.1. ITF2357 Potently Reduces Melanoma Cell Viability and Induces Histone Acetylation
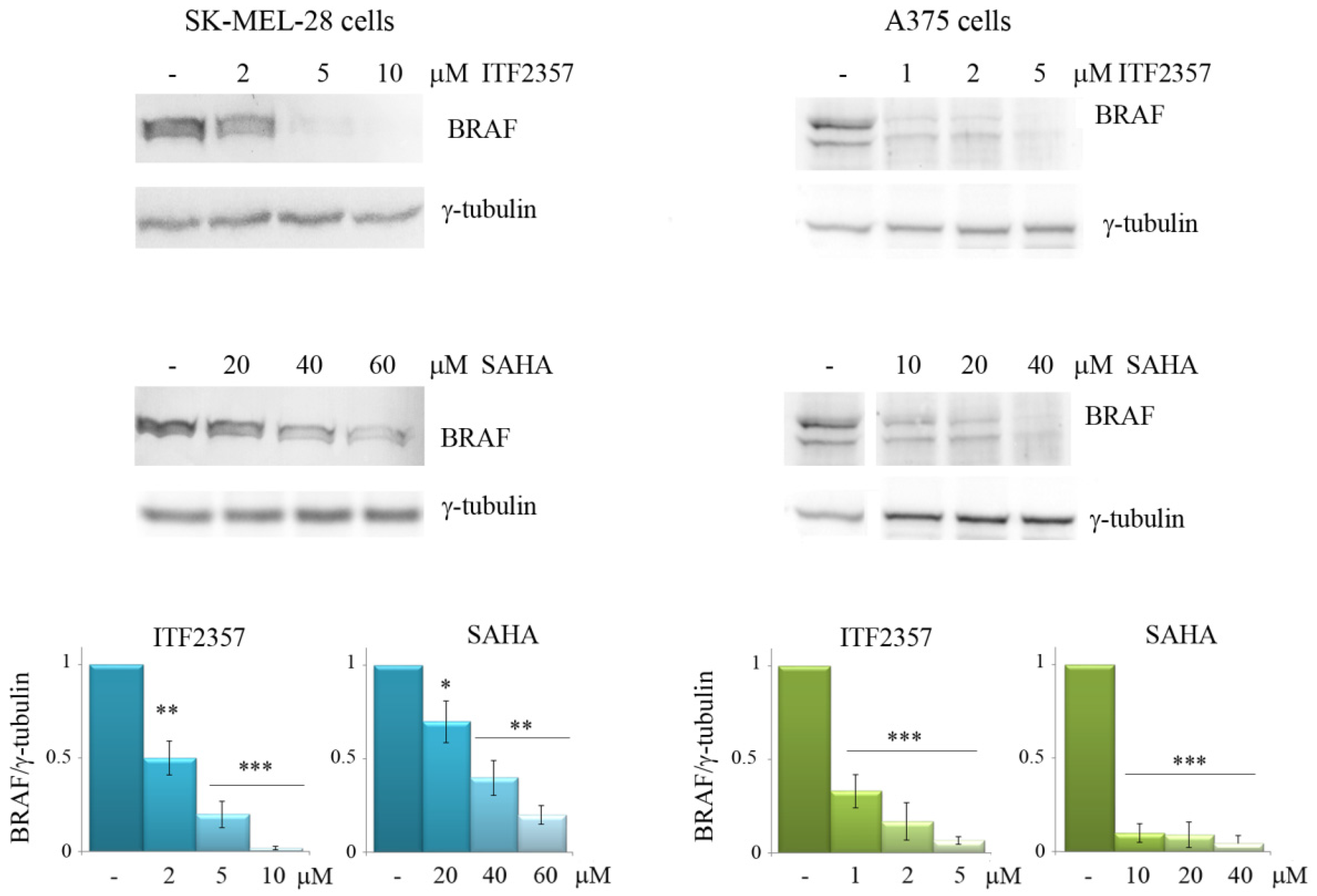
3.2. The Effects of ITF2357 on Oncogenic BRAF
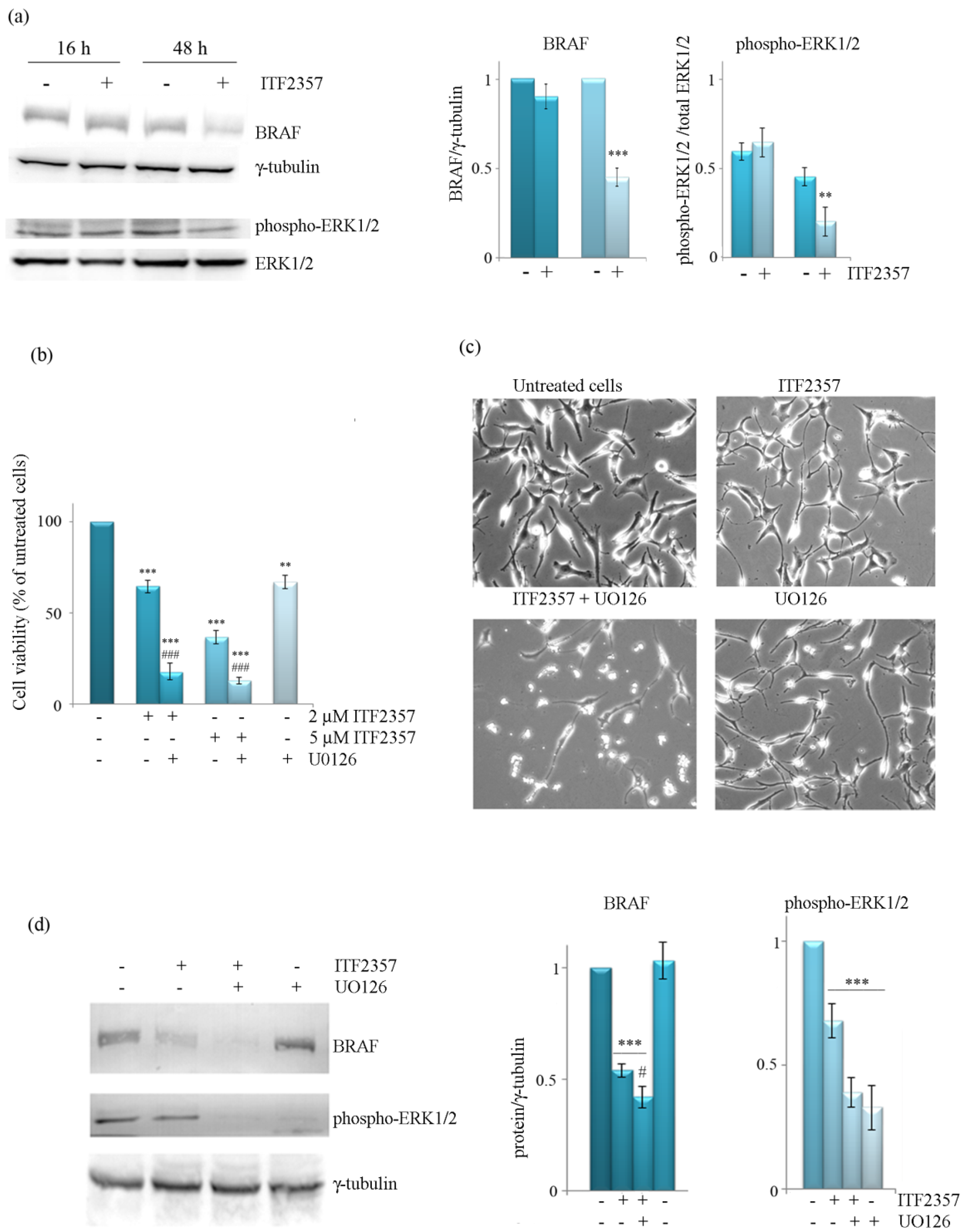
3.3. The Effect of ITF2357 on BRAF Mitogenic Signalling Cascade
3.4. ITF2357 Promotes a Switch from Autophagy to Caspase-Dependent Apoptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, J.M.; Tom, L.N.; Jagirdar, K.; Lambie, D.; Schaider, H.; Sturm, R.A.; Soyer, H.P.; Stark, M.S. The BRAF and NRAS Mutation Prevalence in Dermoscopic Subtypes of Acquired Naevi Reveals Constitutive Mitogen-activated Protein Kinase Pathway Activation. Br. J. Dermatol. 2018, 178, 191–197. [Google Scholar] [CrossRef]
- Valachis, A.; Ullenhag, G.J. Discrepancy in BRAF Status among Patients with Metastatic Malignant Melanoma: A Meta-Analysis. Eur. J. Cancer 2017, 81, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Erturk, K. BRAF V600E Mutation as a Prognostic Factor in Cutaneous Melanoma Patients. Derm. Ther. 2020, 33, e13270. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Flaherty, K.T. BRAF Targeted Therapy Changes the Treatment Paradigm in Melanoma. Nat. Rev. Clin. Oncol. 2011, 8, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Cohen, M.S. The Discovery of Vemurafenib for the Treatment of BRAF-Mutated Metastatic Melanoma. Exp. Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef]
- Dummer, R.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Kirkwood, J.M.; Chiarion Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; et al. Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N. Engl. J. Med. 2020, 383, 1139–1148. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and Adaptive Resistance to BRAF(V600E) Inhibition in Melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Tabolacci, C.; Cordella, M.; Mariotti, S.; Rossi, S.; Senatore, C.; Lintas, C.; Levati, L.; D’Arcangelo, D.; Facchiano, A.; D’Atri, S.; et al. Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines 2021, 9, 79. [Google Scholar] [CrossRef]
- Roelli, M.A.; Ruffieux-Daidié, D.; Stooss, A.; ElMokh, O.; Phillips, W.A.; Dettmer, M.S.; Charles, R.-P. PIK3CAH1047R-Induced Paradoxical ERK Activation Results in Resistance to BRAFV600E Specific Inhibitors in BRAFV600E PIK3CAH1047R Double Mutant Thyroid Tumors. Oncotarget 2017, 8, 103207–103222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, W.; Min, I.; Wyrwas, B.; Moore, M.; Zarnegar, R.; Fahey, T.J. BRAF V600E-Dependent Role of Autophagy in Uveal Melanoma. J. Cancer Res. Clin. Oncol. 2017, 143, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Bustos, S.O.; da Silva Pereira, G.J.; de Freitas Saito, R.; Gil, C.D.; Zanatta, D.B.; Smaili, S.S.; Chammas, R. Galectin-3 Sensitized Melanoma Cell Lines to Vemurafenib (PLX4032) Induced Cell Death through Prevention of Autophagy. Oncotarget 2018, 9, 14567–14579. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An Autophagy-Driven Pathway of ATP Secretion Supports the Aggressive Phenotype of BRAFV600E Inhibitor-Resistant Metastatic Melanoma Cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in Health and Disease: A Comprehensive Review. Biomed Pharm. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF Associated Autophagy Exploitation: BRAF and Autophagy Inhibitors Synergise to Efficiently Overcome Resistance of BRAF Mutant Colorectal Cancer Cells. Oncotarget 2016, 7, 9188–9221. [Google Scholar] [CrossRef]
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The Role of Autophagy in the Treatment of BRAF Mutant Colorectal Carcinomas Differs Based on Microsatellite Instability Status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Foreman, N.K.; Thorburn, A. Using BRAFV600E as a Marker of Autophagy Dependence in Pediatric Brain Tumors. Autophagy 2014, 10, 2077–2078. [Google Scholar] [CrossRef][Green Version]
- Wang, W.; Kang, H.; Zhao, Y.; Min, I.; Wyrwas, B.; Moore, M.; Teng, L.; Zarnegar, R.; Jiang, X.; Fahey, T.J. Targeting Autophagy Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. J. Clin. Endocrinol. Metab. 2017, 102, 634–643. [Google Scholar] [CrossRef]
- Run, L.; Wang, L.; Nong, X.; Li, N.; Huang, X.; Xiao, Y. Involvement of HMGB1 in Vemurafenib Resistance in Thyroid Cancer Cells Harboring BRAF (V6ooE) Mutation by Regulating Excessive Autophagy. Endocrine 2021, 71, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Goodall, M.L.; Wang, T.; Martin, K.R.; Kortus, M.G.; Kauffman, A.L.; Trent, J.M.; Gately, S.; MacKeigan, J.P. Development of Potent Autophagy Inhibitors That Sensitize Oncogenic BRAF V600E Mutant Melanoma Tumor Cells to Vemurafenib. Autophagy 2014, 10, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy Inhibition Overcomes Multiple Mechanisms of Resistance to BRAF Inhibition in Brain Tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef] [PubMed]
- Vagapova, E.; Kozlov, M.; Lebedev, T.; Ivanenko, K.; Leonova, O.; Popenko, V.; Spirin, P.; Kochetkov, S.; Prassolov, V. Selective Inhibition of HDAC Class I Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs. Biomedicines 2021, 9, 1846. [Google Scholar] [CrossRef] [PubMed]
- Daśko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Chen, Y.; Tsai, Y.-H.; Tseng, S.-H. HDAC Inhibitors and RECK Modulate Endoplasmic Reticulum Stress in Tumor Cells. Int. J. Mol. Sci. 2017, 18, 258. [Google Scholar] [CrossRef]
- Giuliano, M.; Pellerito, C.; Celesia, A.; Fiore, T.; Emanuele, S. Tributyltin(IV) Butyrate: A Novel Epigenetic Modifier with ER Stress- and Apoptosis-Inducing Properties in Colon Cancer Cells. Molecules 2021, 26, 5010. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for P53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef]
- López-Cobo, S.; Pieper, N.; Campos-Silva, C.; García-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Valés-Gómez, M. Impaired NK Cell Recognition of Vemurafenib-Treated Melanoma Cells Is Overcome by Simultaneous Application of Histone Deacetylase Inhibitors. OncoImmunology 2018, 7, e1392426. [Google Scholar] [CrossRef]
- Madorsky Rowdo, F.; Barón, A.; Gallagher, S.; Hersey, P.; Emran, A.; Von Euw, E.; Barrio, M.; Mordoh, J. Epigenetic Inhibitors Eliminate Senescent Melanoma BRAFV600E Cells That Survive Long-term BRAF Inhibition. Int. J. Oncol. 2020, 56, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Bettica, P.; Petrini, S.; D’Oria, V.; D’Amico, A.; Catteruccia, M.; Pane, M.; Sivo, S.; Magri, F.; Brajkovic, S.; Messina, S.; et al. Histological Effects of Givinostat in Boys with Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2016, 26, 643–649. [Google Scholar] [CrossRef]
- Savino, A.M.; Sarno, J.; Trentin, L.; Vieri, M.; Fazio, G.; Bardini, M.; Bugarin, C.; Fossati, G.; Davis, K.L.; Gaipa, G.; et al. The Histone Deacetylase Inhibitor Givinostat (ITF2357) Exhibits Potent Anti-Tumor Activity against CRLF2-Rearranged BCP-ALL. Leukemia 2017, 31, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Mensah, A.A.; Kwee, I.; Gaudio, E.; Rinaldi, A.; Ponzoni, M.; Cascione, L.; Fossati, G.; Stathis, A.; Zucca, E.; Caprini, G.; et al. Novel HDAC Inhibitors Exhibit Pre-Clinical Efficacy in Lymphoma Models and Point to the Importance of CDKN1A Expression Levels in Mediating Their Anti-Tumor Response. Oncotarget 2015, 6, 5059–5071. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, K.; Yao, C.; Kahwash, S.; Tang, Y.; Zhang, G.; Patterson, K.; Wang, Q.-E.; Zhao, W. Givinostat, a Type II Histone Deacetylase Inhibitor, Induces Potent Caspase-Dependent Apoptosis in Human Lymphoblastic Leukemia. Genes Cancer 2016, 7, 292–300. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.E21. [Google Scholar] [CrossRef]
- Del Bufalo, D.; Desideri, M.; De Luca, T.; Di Martile, M.; Gabellini, C.; Monica, V.; Busso, S.; Eramo, A.; De Maria, R.; Milella, M.; et al. Histone Deacetylase Inhibition Synergistically Enhances Pemetrexed Cytotoxicity through Induction of Apoptosis and Autophagy in Non-Small Cell Lung Cancer. Mol. Cancer 2014, 13, 230. [Google Scholar] [CrossRef]
- Di Martile, M.; Desideri, M.; Tupone, M.G.; Buglioni, S.; Antoniani, B.; Mastroiorio, C.; Falcioni, R.; Ferraresi, V.; Baldini, N.; Biagini, R.; et al. Histone Deacetylase Inhibitor ITF2357 Leads to Apoptosis and Enhances Doxorubicin Cytotoxicity in Preclinical Models of Human Sarcoma. Oncogenesis 2018, 7, 20. [Google Scholar] [CrossRef]
- Palamaris, K.; Moutafi, M.; Gakiopoulou, H.; Theocharis, S. Histone Deacetylase (HDAC) Inhibitors: A Promising Weapon to Tackle Therapy Resistance in Melanoma. IJMS 2022, 23, 3660. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC Inhibitors Enhance the Immunotherapy Response of Melanoma Cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef]
- Pellerito, C.; Emanuele, S.; Ferrante, F.; Celesia, A.; Giuliano, M.; Fiore, T. Tributyltin(IV) Ferulate, a Novel Synthetic Ferulic Acid Derivative, Induces Autophagic Cell Death in Colon Cancer Cells: From Chemical Synthesis to Biochemical Effects. J. Inorg. Biochem. 2020, 205, 110999. [Google Scholar] [CrossRef] [PubMed]
- Celesia, A.; Morana, O.; Fiore, T.; Pellerito, C.; D’Anneo, A.; Lauricella, M.; Carlisi, D.; De Blasio, A.; Calvaruso, G.; Giuliano, M.; et al. ROS-Dependent ER Stress and Autophagy Mediate the Anti-Tumor Effects of Tributyltin (IV) Ferulate in Colon Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8135. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, C.; Li, Z.; Xiao, J.; Li, C.; Wang, X.; Kong, P.; Cao, J.; Huang, F.; Li, Z.; et al. SOX2 Promotes Resistance of Melanoma with PD-L1 High Expression to T-Cell-Mediated Cytotoxicity That Can Be Reversed by SAHA. J. Immunother. Cancer 2020, 8, e001037. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone Deacetylase Inhibitor Selectively Induces P21WAF1 Expression and Gene-Associated Histone Acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef]
- Carlisi, D.; Vassallo, B.; Lauricella, M.; Emanuele, S.; D’Anneo, A.; Di Leonardo, E.; Di Fazio, P.; Vento, R.; Tesoriere, G. Histone Deacetylase Inhibitors Induce in Human Hepatoma HepG2 Cells Acetylation of P53 and Histones in Correlation with Apoptotic Effects. Int. J. Oncol. 2008, 32, 177–184. [Google Scholar] [CrossRef]
- Chiappetta, G.; Basile, A.; Arra, C.; Califano, D.; Pasquinelli, R.; Barbieri, A.; De Simone, V.; Rea, D.; Giudice, A.; Pezzullo, L.; et al. BAG3 Down-Modulation Reduces Anaplastic Thyroid Tumor Growth by Enhancing Proteasome-Mediated Degradation of BRAF Protein. J. Clin. Endocrinol. Metab. 2012, 97, E115–E120. [Google Scholar] [CrossRef]
- Kisselev, A.F. Site-Specific Proteasome Inhibitors. Biomolecules 2021, 12, 54. [Google Scholar] [CrossRef]
- Celesia, A.; Fiore, T.; Di Liberto, D.; Giuliano, M.; Pellerito, C.; Emanuele, S. Bortezomib Potentiates the Antitumor Effect of Tributyltin(IV) Ferulate in Colon Cancer Cells Exacerbating ER Stress and Promoting Apoptosis. Inorg. Chim. Acta 2022, 537, 120929. [Google Scholar] [CrossRef]
- Pitcher, D.S.; de Mattos-Shipley, K.; Tzortzis, K.; Auner, H.W.; Karadimitris, A.; Kleijnen, M.F. Bortezomib Amplifies Effect on Intracellular Proteasomes by Changing Proteasome Structure. EBioMedicine 2015, 2, 642–648. [Google Scholar] [CrossRef]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.-B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional Regulation of Autophagy-Lysosomal Function in BRAF-Driven Melanoma Progression and Chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef]
- Thorburn, A.; Morgan, M.J. Targeting Autophagy in BRAF -Mutant Tumors. Cancer Discov. 2015, 5, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, S.; Lauricella, M.; D’Anneo, A.; Carlisi, D.; De Blasio, A.; Di Liberto, D.; Giuliano, M. P62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles. Int. J. Mol. Sci. 2020, 21, 5029. [Google Scholar] [CrossRef] [PubMed]
- Chicote, J.; Yuste, V.J.; Boix, J.; Ribas, J. Cell Death Triggered by the Autophagy Inhibitory Drug 3-Methyladenine in Growing Conditions Proceeds With DNA Damage. Front. Pharmacol. 2020, 11, 580343. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, J.-Y.; Maehara, K.; Hayashi-Takanaka, Y.; Harada, A.; Fukuda, M.; Yamamoto, S.; Ichimaru, N.; Umehara, T.; Yokoyama, S.; Matsuda, R.; et al. Histone H4 Lysine 20 Acetylation Is Associated with Gene Repression in Human Cells. Sci. Rep. 2016, 6, 24318. [Google Scholar] [CrossRef]
- Hernandez, M.A.; Patel, B.; Hey, F.; Giblett, S.; Davis, H.; Pritchard, C. Regulation of BRAF Protein Stability by a Negative Feedback Loop Involving the MEK–ERK Pathway but Not the FBXW7 Tumour Suppressor. Cell. Signal. 2016, 28, 561–571. [Google Scholar] [CrossRef]
- Yamada, T.; Amann, J.M.; Tanimoto, A.; Taniguchi, H.; Shukuya, T.; Timmers, C.; Yano, S.; Shilo, K.; Carbone, D.P. Histone Deacetylase Inhibition Enhances the Antitumor Activity of a MEK Inhibitor in Lung Cancer Cells Harboring RAS Mutations. Mol. Cancer Ther. 2018, 17, 17–25. [Google Scholar] [CrossRef]
- Saei, A.; Palafox, M.; Benoukraf, T.; Kumari, N.; Jaynes, P.W.; Iyengar, P.V.; Muñoz-Couselo, E.; Nuciforo, P.; Cortés, J.; Nötzel, C.; et al. Loss of USP28-Mediated BRAF Degradation Drives Resistance to RAF Cancer Therapies. J. Exp. Med. 2018, 215, 1913–1928. [Google Scholar] [CrossRef]
- Angeletti, F.; Fossati, G.; Pattarozzi, A.; Würth, R.; Solari, A.; Daga, A.; Masiello, I.; Barbieri, F.; Florio, T.; Comincini, S. Inhibition of the Autophagy Pathway Synergistically Potentiates the Cytotoxic Activity of Givinostat (ITF2357) on Human Glioblastoma Cancer Stem Cells. Front. Mol. Neurosci. 2016, 9, 107. [Google Scholar] [CrossRef]
- Marampon, F.; Leoni, F.; Mancini, A.; Pietrantoni, I.; Codenotti, S.; Letizia, F.; Megiorni, F.; Porro, G.; Galbiati, E.; Pozzi, P.; et al. Histone Deacetylase Inhibitor ITF2357 (givinostat) Reverts Transformed Phenotype and Counteracts Stemness in In Vitro and In Vivo Models of Human Glioblastoma. J. Cancer Res. Clin. Oncol. 2019, 145, 393–409. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Value | ||
|---|---|---|
| SK-MEL-28 Cells | A375 Cells | |
| ITF2357 | 4.2 μM | 1.7 μM |
| SAHA | 26.9 μM | 9.2 μM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celesia, A.; Notaro, A.; Franzò, M.; Lauricella, M.; D’Anneo, A.; Carlisi, D.; Giuliano, M.; Emanuele, S. The Histone Deacetylase Inhibitor ITF2357 (Givinostat) Targets Oncogenic BRAF in Melanoma Cells and Promotes a Switch from Pro-Survival Autophagy to Apoptosis. Biomedicines 2022, 10, 1994. https://doi.org/10.3390/biomedicines10081994
Celesia A, Notaro A, Franzò M, Lauricella M, D’Anneo A, Carlisi D, Giuliano M, Emanuele S. The Histone Deacetylase Inhibitor ITF2357 (Givinostat) Targets Oncogenic BRAF in Melanoma Cells and Promotes a Switch from Pro-Survival Autophagy to Apoptosis. Biomedicines. 2022; 10(8):1994. https://doi.org/10.3390/biomedicines10081994
Chicago/Turabian StyleCelesia, Adriana, Antonietta Notaro, Marzia Franzò, Marianna Lauricella, Antonella D’Anneo, Daniela Carlisi, Michela Giuliano, and Sonia Emanuele. 2022. "The Histone Deacetylase Inhibitor ITF2357 (Givinostat) Targets Oncogenic BRAF in Melanoma Cells and Promotes a Switch from Pro-Survival Autophagy to Apoptosis" Biomedicines 10, no. 8: 1994. https://doi.org/10.3390/biomedicines10081994
APA StyleCelesia, A., Notaro, A., Franzò, M., Lauricella, M., D’Anneo, A., Carlisi, D., Giuliano, M., & Emanuele, S. (2022). The Histone Deacetylase Inhibitor ITF2357 (Givinostat) Targets Oncogenic BRAF in Melanoma Cells and Promotes a Switch from Pro-Survival Autophagy to Apoptosis. Biomedicines, 10(8), 1994. https://doi.org/10.3390/biomedicines10081994