
Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma



Abstract
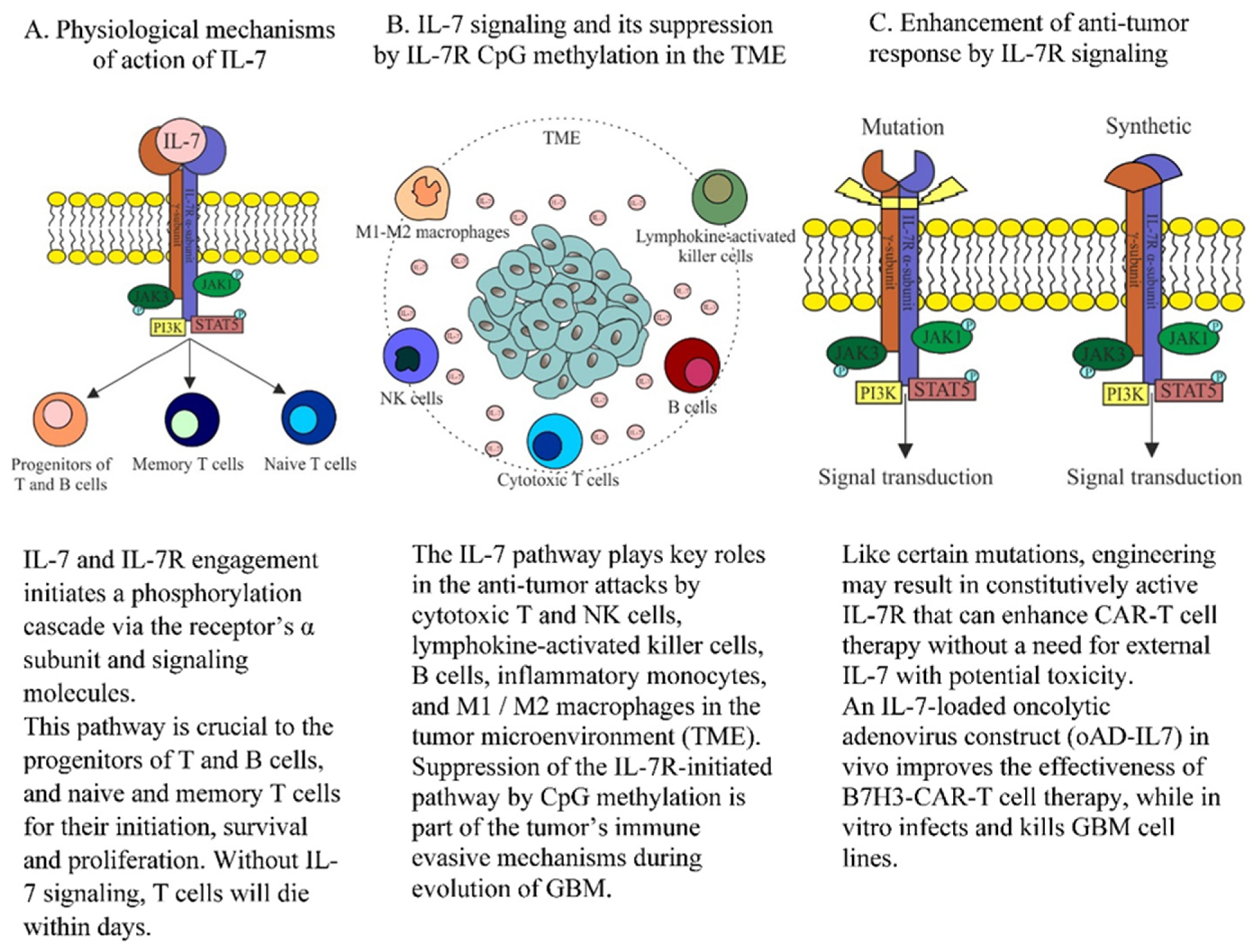
:1. Introduction
2. Materials and Methods
2.1. Subjects of the Study
2.2. GBM Cohort
2.3. Control Group
2.4. DNA Isolation and Bisulfite Sequencing
2.5. Validation Cohort-1
2.6. Validation Cohort-2
2.7. Bioinformatics and Statistics
3. Results
3.1. Differential DNA CpG Methylation of Immune Pathways in Paired GBM and Control Specimens (Table 1, Tables S2 and S3)

{kind=link}
| More Methylated Promoter and/or Gene within Pathways in the First Cohort Group Compared to the Second (Reference) Cohort Group | p-Value Range | Less Methylated Promoter and/or Gene within Pathways in the First Cohort Group Compared to the Second (Reference) Cohort Group | p-Value Range |
|---|---|---|---|
| GBMprim vs. CG | GBMprim vs. CG | ||
| Innate immune response (regulation of innate immune response, positive regulation of interleukin-6-mediated signaling pathway, positive regulation of macrophage cytokine production) | 0.006–0.0084 | Innate immune response (immune system development, regulation of cytokine-mediated signaling pathway) | 0.0032–0.0073 |
| Adaptive immune response (regulation of acute inflammatory response to antigenic stimulus) | 0.0084 | Adaptive immune response (positive regulation of CD8-positive-alpha-beta cytotoxic T cell extravasation, B and T cell receptor signaling pathway, T cell homeostasis, negative regulation of B cell activation, regulation of cytokine-mediated signaling) | 0.0019–0.0073 |
| Interleukin-7-mediated signaling pathway and response to interleukin-7 | 0.0001 | Interleukin-7-mediated signaling pathway and response to interleukin-7 | 0.0027 |
| GBMrec vs. CG | GBMrec vs. CG | ||
| Innate immune response (interleukin-6 mediated signaling pathway and response to interleukin 6, interleukin-11-mediated signaling pathway, positive regulation of NK T cell activation) | 0.0003–0.0092 | Innate immune response (positive regulation of myeloid leukocyte mediated immunity) | 0.0023 |
| Adaptive immune response (Interleukin-27-mediated signaling pathway, T cell differentiation in the thymus, Interleukin-11-mediated signaling pathway, positive regulation of T-helper 2 cell cytokine production) | 0.0009–0.0059 | Adaptive immune response (negative regulation of immature T-cell proliferation in thymus, positive regultation of immunoglobulin mediated immune response, immune response to tumor cells) | 0.0051–0.0082 |
| Interleukin-7-mediated signaling pathway and response to interleukin-7 | 0 | Interleukin-7-mediated signaling pathway and response to interleukin-7 | 0.0007 |
| GBMrec vs. GBMprim | GBMrec vs. GBMprim | ||
| Innate immune response (protection from natural killer cell mediated cytotoxicity, negative regulation of innate immune response and cytokine production) | 0.0069–0.0097 | Innate immune response (negative regulation of cytokine secretion, susceptibility to and positive regulation of natural killer cell mediated cytotoxicity, regulation of cell killing, positive regulation of leukocyte mediated immunity and cytotoxicity, negative regulation of interleukin-1 alpha production) | 0.0006–0.0056 |
| Adaptive immune response (antigen processing and presentation of endogeneous peptides, CD4-positive or CD8-positive alpha-beta T cell lineage commitment and proliferation, positive regulation of T cell mediated immunity, negative regulation of alpha-beta T cell proliferation) | 0.0011–0.0098 | Adaptive immune response (regulation of dendritic cell antigen processing and presentation, T cell homeostasis, regulation of lymphocyte mediated immunity, T cell mediated cytotoxicity, positive regulation of humoral immune response mediated by circulating immunoglobulin) | 0.0002–0.0093 |
| Interleukin-7-mediated signaling pathway and response to interleukin-7 | 0 | Interleukin-7-mediated signaling pathway and response to interleukin-7 | 0.0023 |
3.2. Differential Methylation of CpGs in Individual Promoter + Gene Regions of IL-7 and IL-7 Receptor in GBM Cohorts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Kraboth, Z.; Galik, B.; Tompa, M.; Kajtar, B.; Urban, P.; Gyenesei, A.; Miseta, A.; Kalman, B. DNA CpG methylation in sequential glioblastoma specimens. J. Cancer. Res. Clin. Oncol. 2020, 146, 2885–2896. [Google Scholar] [CrossRef]
- Kraboth, Z.; Kajtár, B.; Gálik, B.; Gyenesei, A.; Miseta, A.; Kalman, B. Involvement of the Catecholamine Pathway in Glioblastoma Development. Cells 2021, 10, 549. [Google Scholar] [CrossRef]
- Tompa, M.; Kajtar, B.; Galik, B.; Gyenesei, A.; Kalman, B. DNA methylation and protein expression of Wnt pathway markers in progressive glioblastoma. Pathol. Res. Pract. 2021, 222, 153429. [Google Scholar] [CrossRef]
- Najem, H.; Khasraw, M.; Heimberger, A.B. Immune Microenvironment Landscape in CNS Tumors and Role in Responses to Immunotherapy. Cells 2021, 10, 2032. [Google Scholar] [CrossRef]
- Liu, X.; Xing, H.; Liu, H.; Chen, J. Current status and future perspectives on immunotherapy in neoadjuvant therapy of resectable non-small cell lung cancer. Asia. Pac. J. Clin. Oncol. 2022, 18, 335–343. [Google Scholar] [CrossRef]
- Ossato, A.; Damuzzo, V.; Baldo, P.; Mengato, D.; Chiumente, M.; Messori, A. Immune checkpoint inhibitors as first line in advanced melanoma: Evaluating progression-free survival based on reconstructed individual patient data. Cancer Med. 2022. [Google Scholar] [CrossRef]
- Sanders, S.; Debinski, W. Challenges to successful implementation of the immune checkpoint inhibitors for treatment of glioblastoma. Int. J. Mol. Sci. 2020, 21, 2759. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta. Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Europian Nucleotide Archive; Primary Accession: PRJNA391429; EGAS00001002538. Available online: https://www.ebi.ac.uk/ena (accessed on 17 June 2022).
- National Cancer Institute, GDC Data Portal, TCGA-GBM database. Available online: https://portal.gdc.cancer.gov/repository?filters=%7B%22op%22%3A%22and%22%2C%22content%22%3A%5B%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.project.project_id%22%2C%22value%22%3A%5B%22TCGA-GBM%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22files.data_category%22%2C%22value%22%3A%5B%22DNA%20Methylation%22%5D%7D%7D%5D%7D&searchTableTab=cases (accessed on 18 December 2019).
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune escape in glioblastoma multiforme and the adaptation of immunotherapies for treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef]
- Mescher, M.F.; Curtsinger, J.M.; Agarwal, P.; Casey, K.A.; Gerner, M.; Hammerbeck, C.D.; Popescu, F.; Xiao, Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 2006, 211, 81–92. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.-A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470. [Google Scholar] [CrossRef]
- Sawada, M.; Itoh, Y.; Suzumura, A.; Marunouchi, T. Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci. Lett. 1993, 160, 131–134. [Google Scholar] [CrossRef]
- Mazzucchelli, R.; Durum, S.K. Interleukin-7 receptor expression: Intelligent design. Nat. Rev. Immunol. 2007, 7, 144–154. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, M.; Zhang, Z.; Tang, X.; Chen, Y.; Peng, A.; Peng, X.; Tong, A.; Zhou, L. Interleukin-7-loaded oncolytic adenovirus improves CAR-T cell therapy for glioblastoma. Cancer. Immunol. Immunother. 2021, 70, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Nakamura, Y.; Russell, S.M.; Ziegler, S.F.; Tsang, M.; Cao, X.; Leonard, W.J. Interleukin-2 receptor gamma chain: A functional component of the interleukin-7 receptor. Science 1993, 262, 1877–1880. [Google Scholar] [CrossRef] [PubMed]
- Perna, S.K.; Pagliara, D.; Mahendravada, A.; Liu, H.; Brenner, M.K.; Savoldo, B.; Dotti, G. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin. Cancer Res. 2014, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Baizan-Edge, A.; Stubbs, B.A.; Stubbington, M.J.T.; Bolland, D.J.; Tabbada, K.; Andrews, S.; Corcoran, A.E. IL-7R signaling activates widespread VH and DH gene usage to drive antibody diversity in bone marrow B cells. Cell Rep. 2021, 36, 109349. [Google Scholar] [CrossRef]
- Lin, K.; Gueble, S.E.; Sundaram, R.K.; Huseman, E.D.; Bindra, R.S.; Herzon, S.B. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 2022, 377, 502–511. [Google Scholar] [CrossRef]
- Cui, G.; Shimba, A.; Ma, G.; Takahara, K.; Tani-Ichi, S.; Zhu, Y.; Asahi, T.; Abe, A.; Miyachi, H.; Kitano, S.; et al. IL-7R-Dependent Phosphatidylinositol 3-Kinase Competes with the STAT5 Signal to Modulate T Cell Development and Homeostasis. J. Immunol. 2020, 204, 844–857. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S.; NABTT CNS Consortium. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Sportès, C.; Ahmadzadeh, M.; Fry, T.J.; Ngo, L.T.; Schwarz, S.L.; Stetler-Stevenson, M.; Morton, K.E.; Mavroukakis, S.A.; Morre, M.; et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J. Immunother. 2006, 29, 313–319. [Google Scholar] [CrossRef]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Kren, N.P.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 4, 448–459. [Google Scholar] [CrossRef]
- Khan, M.D.A.; Zheng, M.; Fu, J.; Tania, M.; Li, J.; Fu, J. Thymoquinone upregulates IL17RD in controlling the growth and metastasis of triple negative breast cancer cells in vitro. BMC Cancer 2022, 22, 707. [Google Scholar] [CrossRef]
- Mondal, P.; Natesh, J.; Penta, D.; Meeran, S.M. Progress and promises of epigenetic drugs and epigenetic diets in cancer prevention and therapy: A clinical update. Semin. Cancer Biol. 2022, 83, 503–522. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Huang, G.; Gao, T.; Huang, T.; Zou, M.; Zou, Y.; Duan, S. Epigenetic Changes Associated with Interleukin-10. Front. Immunol. 2020, 11, 1105. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.O.L.; Cramer, S.D.; Winer, H.Y.; Hixon, J.A.; Li, W.; Yunes, J.A.; Durum, S.K. Mutations that collaborate with IL-7Ra signaling pathways to drive ALL. Adv. Biol. Regul. 2021, 80, 100788. [Google Scholar] [CrossRef]
- Ronvaux, L.; Riva, M.; Coosemans, A.; Herzog, M.; Rommelaere, G.; Donis, N.; D’Hondt, L.; Douxfils, J. Liquid Biopsy in Glioblastoma. Cancers 2022, 14, 3394. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tompa, M.; Kraboth, Z.; Galik, B.; Kajtar, B.; Gyenesei, A.; Kalman, B. Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma. Biomedicines 2022, 10, 2174. https://doi.org/10.3390/biomedicines10092174
Tompa M, Kraboth Z, Galik B, Kajtar B, Gyenesei A, Kalman B. Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma. Biomedicines. 2022; 10(9):2174. https://doi.org/10.3390/biomedicines10092174
Chicago/Turabian StyleTompa, Marton, Zoltan Kraboth, Bence Galik, Bela Kajtar, Attila Gyenesei, and Bernadette Kalman. 2022. "Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma" Biomedicines 10, no. 9: 2174. https://doi.org/10.3390/biomedicines10092174
APA StyleTompa, M., Kraboth, Z., Galik, B., Kajtar, B., Gyenesei, A., & Kalman, B. (2022). Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma. Biomedicines, 10(9), 2174. https://doi.org/10.3390/biomedicines10092174