
β2-Adrenergic Receptor Expression and Intracellular Signaling in B Cells Are Highly Dynamic during Collagen-Induced Arthritis


Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Mice
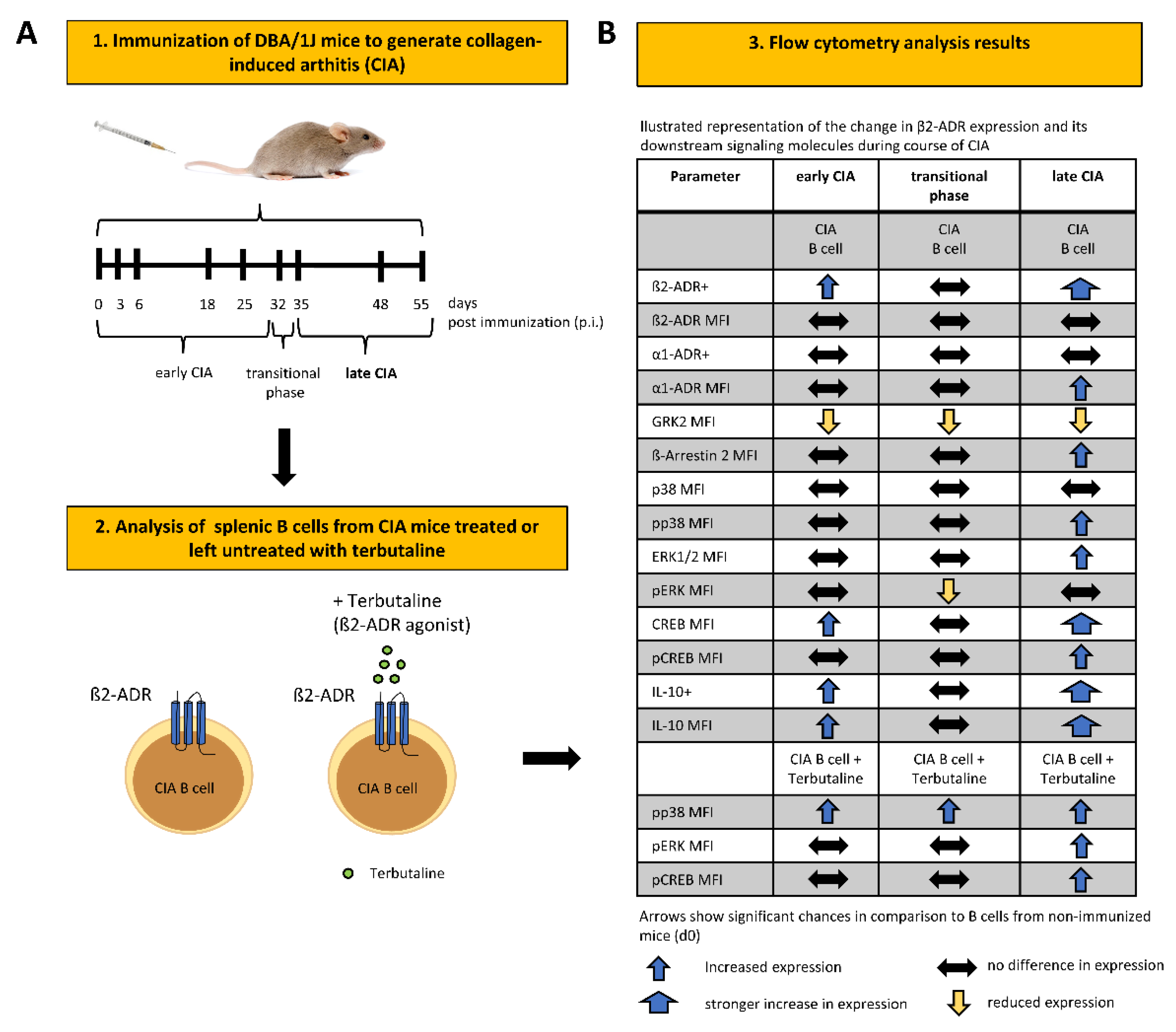
2.3. Collagen-Induced Arthritis (CIA)
2.4. B Cell Isolation
2.5. Stimulation of B Cells
2.6. Flow Cytometry
2.7. Statistics
3. Results
3.1. β2-ADR Positive B Cells Increase during Collagen-Induced Arthritis and Correlate with IL-10
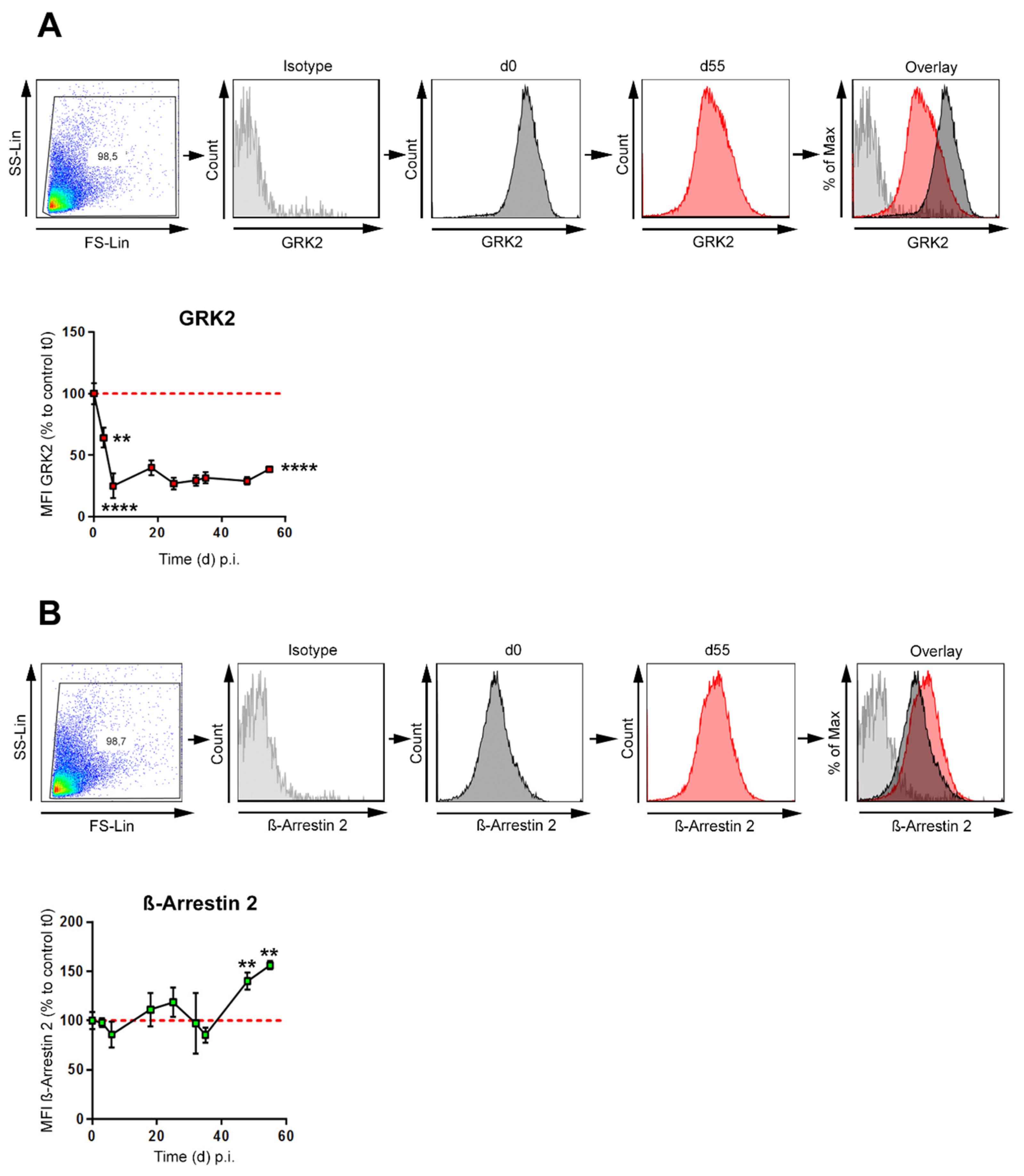
3.2. GRK-2 Is Downregulated during CIA, whereas β-Arrestin 2 Is Upregulated during Established CIA in B Cells
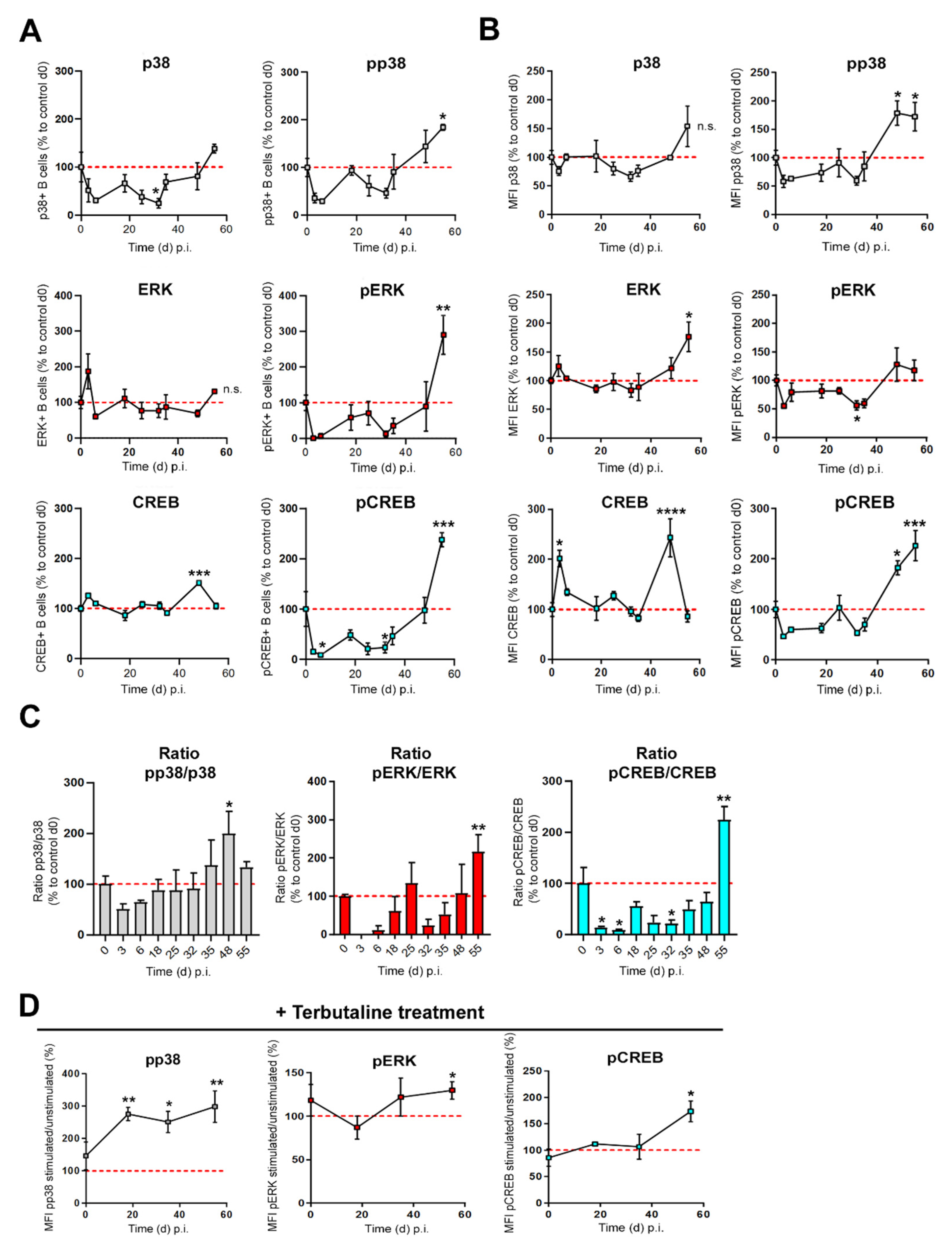
3.3. β2-ADR-Stimulated Increase of pp38, pERK and pCREB Was Associated with B Cell-Derived IL-10 Production in Established Collagen-Induced Arthritis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.C.; Geenen, R.; Godaert, G.L.; Bijlsma, J.W.; van Doornen, L.J. Elevated sympathetic nervous system activity in patients with recently diagnosed rheumatoid arthritis with active disease. Clin. Exp. Rheumatol. 2004, 22, 63–70. [Google Scholar]
- Kuis, W.; de Jong-de Vos van Steenwijk, C.; Sinnema, G.; Kavelaars, A.; Prakken, B.; Helders, P.J.; Heijnen, C.J. The autonomic nervous system and the immune system in juvenile rheumatoid arthritis. Brain Behav. Immun. 1996, 10, 387–398. [Google Scholar] [CrossRef]
- Tanaka, H.; Ueta, Y.; Yamashita, U.; Kannan, H.; Yamashita, H. Biphasic changes in behavioral, endocrine, and sympathetic systems in adjuvant arthritis in Lewis rats. Brain Res. Bull. 1996, 39, 33–37. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H. B-cell involvement in the pathogenesis of RA-is there a contribution of the sympathetic nervous system? Immunol. Res. 2008, 40, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Harle, P.; Mobius, D.; Carr, D.J.; Scholmerich, J.; Straub, R.H. An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum. 2005, 52, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.E.; Justen, H.P.; Scholmerich, J.; Straub, R.H. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J. 2000, 14, 2097–2107. [Google Scholar] [CrossRef]
- Jenei-Lanzl, Z.; Capellino, S.; Kees, F.; Fleck, M.; Lowin, T.; Straub, R.H. Anti-inflammatory effects of cell-based therapy with tyrosine hydroxylase-positive catecholaminergic cells in experimental arthritis. Ann. Rheum. Dis. 2015, 74, 444–451. [Google Scholar] [CrossRef]
- Miller, L.E.; Grifka, J.; Scholmerich, J.; Straub, R.H. Norepinephrine from synovial tyrosine hydroxylase positive cells is a strong indicator of synovial inflammation in rheumatoid arthritis. J. Rheumatol. 2002, 29, 427–435. [Google Scholar]
- Schorr, E.C.; Arnason, B.G. Interactions between the sympathetic nervous system and the immune system. Brain Behav. Immun. 1999, 13, 271–278. [Google Scholar] [CrossRef][Green Version]
- Kim, B.J.; Jones, H.P. Epinephrine-primed murine bone marrow-derived dendritic cells facilitate production of IL-17A and IL-4 but not IFN-gamma by CD4+ T cells. Brain Behav. Immun. 2010, 24, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Felten, D.L.; Ackerman, K.D.; Wiegand, S.J.; Felten, S.Y. Noradrenergic sympathetic innervation of the spleen: I. Nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J. Neurosci. Res. 1987, 18, 28–36. [Google Scholar] [CrossRef]
- Straub, R.H. Complexity of the bi-directional neuroimmune junction in the spleen. Trends Pharmacol. Sci. 2004, 25, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Emeny, R.T.; Gao, D.; Lawrence, D.A. Beta1-adrenergic receptors on immune cells impair innate defenses against Listeria. J. Immunol. 2007, 178, 4876–4884. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Medina, B.E.; Cadena-Medina, D.A.; Esparza, E.; Arrieta, A.J.; Kirken, R.A. Isoproterenol-induced beta-2 adrenergic receptor activation negatively regulates interleukin-2 signaling. Biochem. J. 2018, 475, 2907–2923. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, Y.; Matsumoto, M.; Togashi, H. Enhanced dendritic cell antigen uptake via alpha2 adrenoceptor-mediated PI3K activation following brief exposure to noradrenaline. J. Immunol. 2010, 185, 5762–5768. [Google Scholar] [CrossRef]
- Hasko, G. Receptor-mediated interaction between the sympathetic nervous system and immune system in inflammation. Neurochem. Res. 2001, 26, 1039–1044. [Google Scholar] [CrossRef]
- Honke, N.; Lowin, T.; Opgenoorth, B.; Shaabani, N.; Lautwein, A.; Teijaro, J.R.; Schneider, M.; Pongratz, G. Endogenously produced catecholamines improve the regulatory function of TLR9-activated B cells. PLoS Biol. 2022, 20, e3001513. [Google Scholar] [CrossRef]
- Harle, P.; Pongratz, G.; Albrecht, J.; Tarner, I.H.; Straub, R.H. An early sympathetic nervous system influence exacerbates collagen-induced arthritis via CD4+CD25+ cells. Arthritis Rheum. 2008, 58, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Klein, N.; Schaller, J.; Bellinger, D.L. Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant-induced arthritis. Brain Behav. Immun. 1999, 13, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Rauch, L.; Fassold, A.; Lowin, T.; Pongratz, G. Neuronally released sympathetic neurotransmitters stimulate splenic interferon-gamma secretion from T cells in early type II collagen-induced arthritis. Arthritis Rheum. 2008, 58, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, G.; Melzer, M.; Straub, R.H. The sympathetic nervous system stimulates anti-inflammatory B cells in collagen-type II-induced arthritis. Ann. Rheum. Dis. 2012, 71, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Backlund, J.; Vestberg, M.; Holmdahl, R. Collagen type II (CII)-specific antibodies induce arthritis in the absence of T or B cells but the arthritis progression is enhanced by CII-reactive T cells. Arthritis Res. Ther. 2004, 6, R544–R550. [Google Scholar] [CrossRef]
- Mauri, C.; Gray, D.; Mushtaq, N.; Londei, M. Prevention of arthritis by interleukin 10-producing B cells. J. Exp. Med. 2003, 197, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Fassold, A.; Falk, W.; Anders, S.; Hirsch, T.; Mirsky, V.M.; Straub, R.H. Soluble neuropilin-2, a nerve repellent receptor, is increased in rheumatoid arthritis synovium and aggravates sympathetic fiber repulsion and arthritis. Arthritis Rheum. 2009, 60, 2892–2901. [Google Scholar] [CrossRef] [PubMed]
- Candando, K.M.; Lykken, J.M.; Tedder, T.F. B10 cell regulation of health and disease. Immunol. Rev. 2014, 259, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Sanders, V.M. The beta2-adrenergic receptor on T and B lymphocytes: Do we understand it yet? Brain Behav. Immun. 2012, 26, 195–200. [Google Scholar] [CrossRef]
- Agac, D.; Estrada, L.D.; Maples, R.; Hooper, L.V.; Farrar, J.D. The beta2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav. Immun. 2018, 74, 176–185. [Google Scholar] [CrossRef]
- Kleibeuker, W.; Jurado-Pueyo, M.; Murga, C.; Eijkelkamp, N.; Mayor, F., Jr.; Heijnen, C.J.; Kavelaars, A. Physiological changes in GRK2 regulate CCL2-induced signaling to ERK1/2 and Akt but not to MEK1/2 and calcium. J. Neurochem. 2008, 104, 979–992. [Google Scholar] [CrossRef]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Banko, Z.; Pozsgay, J.; Szili, D.; Toth, M.; Gati, T.; Nagy, G.; Rojkovich, B.; Sarmay, G. Induction and Differentiation of IL-10-Producing Regulatory B Cells from Healthy Blood Donors and Rheumatoid Arthritis Patients. J. Immunol. 2017, 198, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H. Role of peripheral nerve fibres in acute and chronic inflammation in arthritis. Nat. Rev. Rheumatol. 2013, 9, 117–126. [Google Scholar] [CrossRef]
- Kohm, A.P.; Sanders, V.M. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol. Rev. 2001, 53, 487–525. [Google Scholar]
- Vazquez, A.; Auffredou, M.T.; Galanaud, P.; Leca, G. Modulation of IL-2- and IL-4-dependent human B cell proliferation by cyclic AMP. J. Immunol. 1991, 146, 4222–4227. [Google Scholar]
- Kavelaars, A. Regulated expression of alpha-1 adrenergic receptors in the immune system. Brain Behav. Immun. 2002, 16, 799–807. [Google Scholar] [CrossRef]
- Heijnen, C.J.; Rouppe van der Voort, C.; van de Pol, M.; Kavelaars, A. Cytokines regulate alpha(1)-adrenergic receptor mRNA expression in human monocytic cells and endothelial cells. J. Neuroimmunol. 2002, 125, 66–72. [Google Scholar] [CrossRef]
- Lubahn, C.L.; Lorton, D.; Schaller, J.A.; Sweeney, S.J.; Bellinger, D.L. Targeting alpha- and beta-Adrenergic Receptors Differentially Shifts Th1, Th2, and Inflammatory Cytokine Profiles in Immune Organs to Attenuate Adjuvant Arthritis. Front. Immunol. 2014, 5, 346. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, G.; Straub, R.H. The B cell, arthritis, and the sympathetic nervous system. Brain Behav. Immun. 2010, 24, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Platzer, C.; Meisel, C.; Vogt, K.; Platzer, M.; Volk, H.D. Up-regulation of monocytic IL-10 by tumor necrosis factor-alpha and cAMP elevating drugs. Int. Immunol. 1995, 7, 517–523. [Google Scholar] [CrossRef]
- Platzer, C.; Fritsch, E.; Elsner, T.; Lehmann, M.H.; Volk, H.D.; Prosch, S. Cyclic adenosine monophosphate-responsive elements are involved in the transcriptional activation of the human IL-10 gene in monocytic cells. Eur. J. Immunol. 1999, 29, 3098–3104. [Google Scholar] [CrossRef]
- Laukova, M.; Vargovic, P.; Vlcek, M.; Lejavova, K.; Hudecova, S.; Krizanova, O.; Kvetnansky, R. Catecholamine production is differently regulated in splenic T- and B-cells following stress exposure. Immunobiology 2013, 218, 780–789. [Google Scholar] [CrossRef]
- Capellino, S.; Weber, K.; Gelder, M.; Harle, P.; Straub, R.H. First appearance and location of catecholaminergic cells during experimental arthritis and elimination by chemical sympathectomy. Arthritis Rheum. 2012, 64, 1110–1118. [Google Scholar] [CrossRef]
- Lombardi, M.S.; Kavelaars, A.; Cobelens, P.M.; Schmidt, R.E.; Schedlowski, M.; Heijnen, C.J. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J. Immunol. 2001, 166, 1635–1640. [Google Scholar] [CrossRef]
- Lombardi, M.S.; Kavelaars, A.; Schedlowski, M.; Bijlsma, J.W.; Okihara, K.L.; Van de Pol, M.; Ochsmann, S.; Pawlak, C.; Schmidt, R.E.; Heijnen, C.J. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999, 13, 715–725. [Google Scholar] [CrossRef]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying beta-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef]
- Benovic, J.L. Novel beta2-adrenergic receptor signaling pathways. J. Allergy Clin. Immunol. 2002, 110, S229–S235. [Google Scholar] [CrossRef] [PubMed]
- Evron, T.; Daigle, T.L.; Caron, M.G. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 2012, 33, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Lindquist, C.A.; Schaller, J.; Washington, C.; Bellinger, D.L. Changes in the density and distribution of sympathetic nerves in spleens from Lewis rats with adjuvant-induced arthritis suggest that an injury and sprouting response occurs. J. Comp. Neurol. 2005, 489, 260–273. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.; Felten, S.Y.; Bellinger, D. Norepinephrine content in primary and secondary lymphoid organs is altered in rats with adjuvant-induced arthritis. Mech. Ageing Dev. 1997, 94, 145–163. [Google Scholar] [CrossRef]
- Han, C.C.; Ma, Y.; Li, Y.; Wang, Y.; Wei, W. Regulatory effects of GRK2 on GPCRs and non-GPCRs and possible use as a drug target (Review). Int. J. Mol. Med. 2016, 38, 987–994. [Google Scholar] [CrossRef]
- Han, C.; Li, Y.; Zhang, Y.; Wang, Y.; Cui, D.; Luo, T.; Zhang, Y.; Liu, Q.; Li, H.; Wang, C.; et al. Targeted inhibition of GRK2 kinase domain by CP-25 to reverse fibroblast-like synoviocytes dysfunction and improve collagen-induced arthritis in rats. Acta Pharm. Sin. B 2021, 11, 1835–1852. [Google Scholar] [CrossRef]
- Li, P.; Cook, J.A.; Gilkeson, G.S.; Luttrell, L.M.; Wang, L.; Borg, K.T.; Halushka, P.V.; Fan, H. Increased expression of beta-arrestin 1 and 2 in murine models of rheumatoid arthritis: Isoform specific regulation of inflammation. Mol. Immunol. 2011, 49, 64–74. [Google Scholar] [CrossRef]
- Li, H.; Hu, D.; Fan, H.; Zhang, Y.; LeSage, G.D.; Caudle, Y.; Stuart, C.; Liu, Z.; Yin, D. beta-Arrestin 2 negatively regulates Toll-like receptor 4 (TLR4)-triggered inflammatory signaling via targeting p38 MAPK and interleukin 10. J. Biol. Chem. 2014, 289, 23075–23085. [Google Scholar] [CrossRef]
- Gaffal, E.; Jakobs, M.; Glodde, N.; Schroder, R.; Kostenis, E.; Tuting, T. beta-arrestin 2 inhibits proinflammatory chemokine production and attenuates contact allergic inflammation in the skin. J. Investig. Derm. 2014, 134, 2131–2137. [Google Scholar] [CrossRef] [PubMed]
- Fan, H. beta-Arrestins 1 and 2 are critical regulators of inflammation. Innate Immun. 2014, 20, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Seregin, S.S.; Appledorn, D.M.; Patial, S.; Bujold, M.; Nance, W.; Godbehere, S.; Parameswaran, N.; Amalfitano, A. beta-Arrestins modulate Adenovirus-vector-induced innate immune responses: Differential regulation by beta-arrestin-1 and beta-arrestin-2. Virus Res. 2010, 147, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Li, X.G.; Xu, J. Anti-inflammatory activities of fenoterol through beta-arrestin-2 and inhibition of AMPK and NF-kappaB activation in AICAR-induced THP-1 cells. Biomed. Pharmacother. 2016, 84, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Capellino, S.; Cosentino, M.; Wolff, C.; Schmidt, M.; Grifka, J.; Straub, R.H. Catecholamine-producing cells in the synovial tissue during arthritis: Modulation of sympathetic neurotransmitters as new therapeutic target. Ann. Rheum. Dis. 2010, 69, 1853–1860. [Google Scholar] [CrossRef]
- Brenner, S.; Prosch, S.; Schenke-Layland, K.; Riese, U.; Gausmann, U.; Platzer, C. cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J. Biol. Chem. 2003, 278, 5597–5604. [Google Scholar] [CrossRef]
- Chung, E.Y.; Liu, J.; Homma, Y.; Zhang, Y.; Brendolan, A.; Saggese, M.; Han, J.; Silverstein, R.; Selleri, L.; Ma, X. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity 2007, 27, 952–964. [Google Scholar] [CrossRef]
- Ma, W.; Lim, W.; Gee, K.; Aucoin, S.; Nandan, D.; Kozlowski, M.; Diaz-Mitoma, F.; Kumar, A. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J. Biol. Chem. 2001, 276, 13664–13674. [Google Scholar] [CrossRef]
- Carlezon, W.A., Jr.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef]
- Steven, A.; Friedrich, M.; Jank, P.; Heimer, N.; Budczies, J.; Denkert, C.; Seliger, B. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 2020, 77, 4049–4067. [Google Scholar] [CrossRef] [PubMed]
- Baratki, B.L.; Huber, K.; Sarmay, G.; Matko, J.; Kovesdi, D. Inflammatory signal induced IL-10 production of marginal zone B-cells depends on CREB. Immunol. Lett. 2019, 212, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Krutzik, P.O.; Irish, J.M.; Nolan, G.P.; Perez, O.D. Analysis of protein phosphorylation and cellular signaling events by flow cytometry: Techniques and clinical applications. Clin. Immunol. 2004, 110, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.; Weckbecker, G.; Welzenbach, K. Intracellular Phospho-Flow cytometry reveals novel insights into TCR proximal signaling events. A comparison with Western blot. Cytom. Part A 2008, 73, 799–807. [Google Scholar] [CrossRef]
- Krutzik, P.O.; Trejo, A.; Schulz, K.R.; Nolan, G.P. Phospho flow cytometry methods for the analysis of kinase signaling in cell lines and primary human blood samples. Methods Mol. Biol. 2011, 699, 179–202. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (d) p.i | 0 | 3 | 6 | 18 | 25 | 32 | 35 | 48 |
|---|---|---|---|---|---|---|---|---|
| Arthritis score | 0 | 0 | 0.5 ± 0 | 3.17 ± 1.6 | 8.6 ± 1.6 | 20.1 ± 8.3 | 24.9 ± 13.1 | 29 ± 5.6 |
| Parameter | p38 | CREB | ERK | GRK2 | ß2-ADR | pp38 | pCREB | pERK | ß-Arrestin2 | 1-ADR | IL-10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Correlation | 0.5 | 0.21 | 0.44 | 0.32 | 0.25 | 0.38 | 0.43 | 0.48 | 0.42 | 0.54 | −0.244 |
| p-value | 0.0004 | 0.161 | 0.002 | 0.034 | 0.085 | 0.009 | 0.003 | 0.001 | 0.004 | 0.0002 | 0.141 |
| Parameter | p38 | CREB | ERK | GRK2 | β2-ADR | pp38 | pCREB | pERK | β-Arrestin2 | α1-ADR | IL-10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Correlation | −0.04 | −0.11 | −0.13 | −0.37 | −0.10 | −0.17 | −0.07 | −0.08 | −0.09 | −0.07 | 0.25 |
| p-value | 0.785 | 0.471 | 0.383 | 0.016 | 0.492 | 0.242 | 0.664 | 0.589 | 0.531 | 0.675 | 0.121 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honke, N.; Wiest, C.J.; Pongratz, G. β2-Adrenergic Receptor Expression and Intracellular Signaling in B Cells Are Highly Dynamic during Collagen-Induced Arthritis. Biomedicines 2022, 10, 1950. https://doi.org/10.3390/biomedicines10081950
Honke N, Wiest CJ, Pongratz G. β2-Adrenergic Receptor Expression and Intracellular Signaling in B Cells Are Highly Dynamic during Collagen-Induced Arthritis. Biomedicines. 2022; 10(8):1950. https://doi.org/10.3390/biomedicines10081950
Chicago/Turabian StyleHonke, Nadine, Clemens J. Wiest, and Georg Pongratz. 2022. "β2-Adrenergic Receptor Expression and Intracellular Signaling in B Cells Are Highly Dynamic during Collagen-Induced Arthritis" Biomedicines 10, no. 8: 1950. https://doi.org/10.3390/biomedicines10081950
APA StyleHonke, N., Wiest, C. J., & Pongratz, G. (2022). β2-Adrenergic Receptor Expression and Intracellular Signaling in B Cells Are Highly Dynamic during Collagen-Induced Arthritis. Biomedicines, 10(8), 1950. https://doi.org/10.3390/biomedicines10081950