
The Role of Rab Proteins in Parkinson’s Disease Synaptopathy



Abstract
1. Introduction
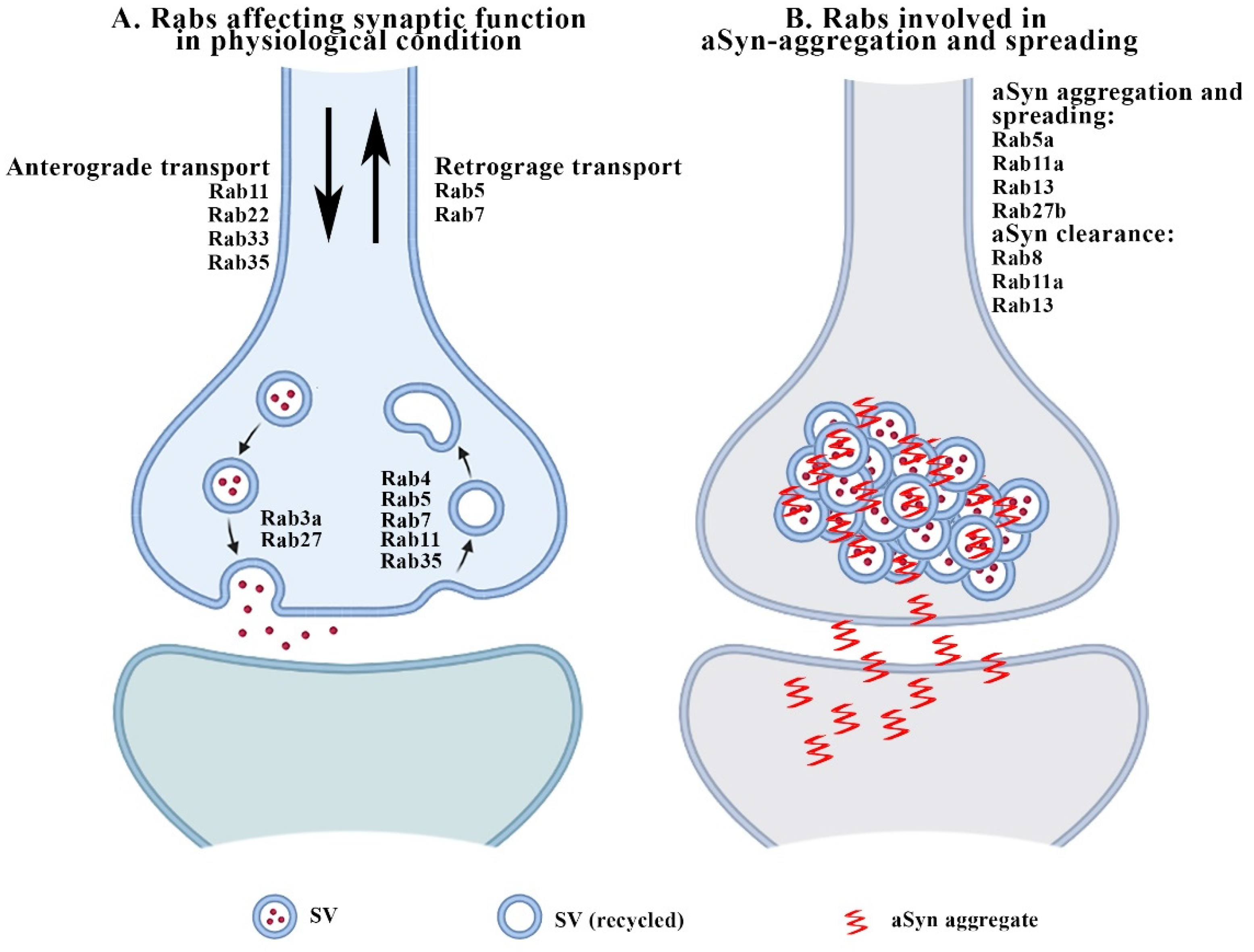
2. Rabs and Synaptic Function
3. Major Rab Alterations in PD and Other Forms of Parkinsonism: A Focus on aSyn-, GBA1- and LRRK2-Associated Synaptopathy
3.1. Rabs and aSyn-Related Synaptopathy
3.2. Critical Involvement of Rabs in LRRK2-Associated Parkinsonism
3.3. Involvement of Rabs in Other Genetic Forms of Parkinsonism
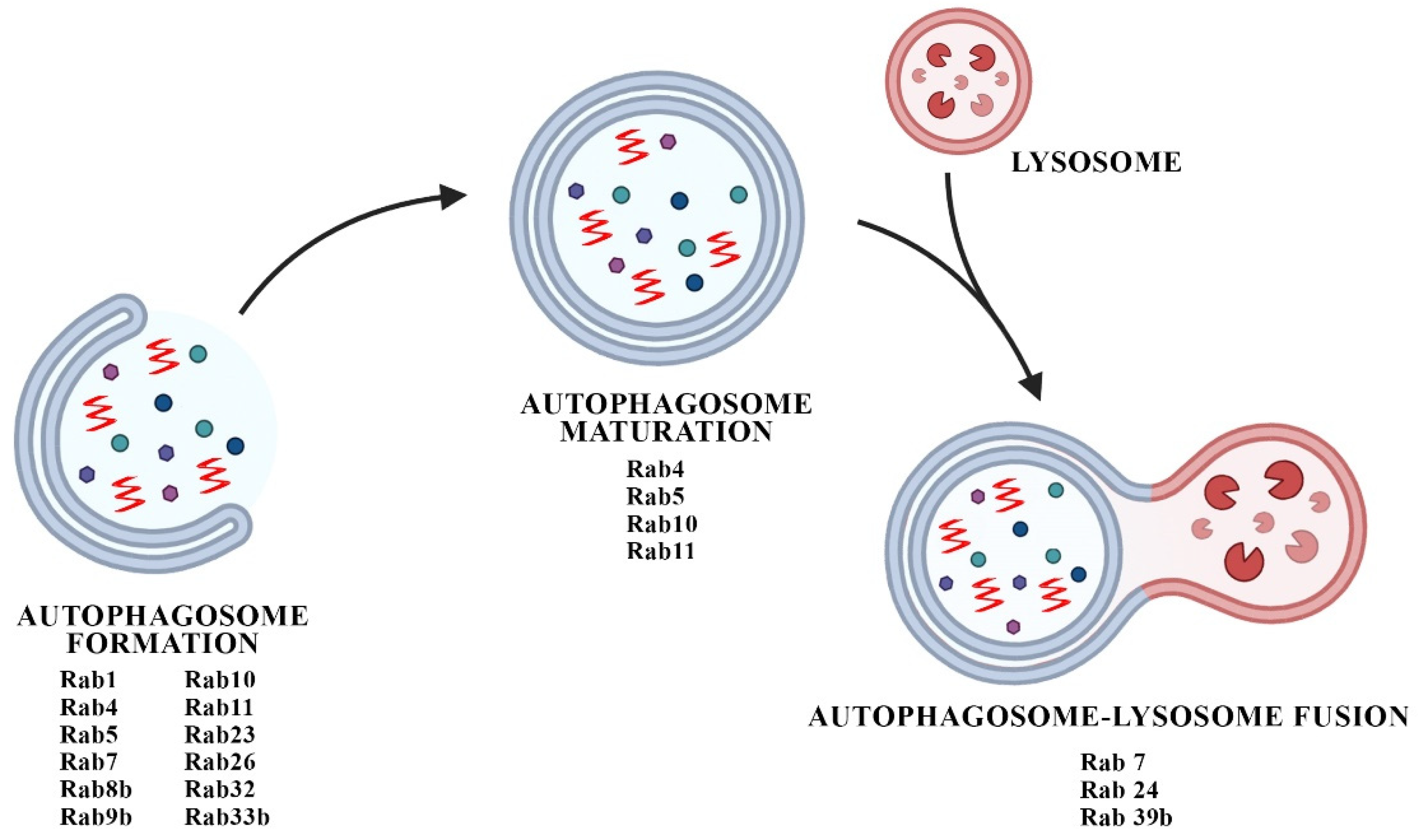
4. Interplay between Rabs and Autophagic Defects in PD Synaptopathy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bologna, M.; Truong, D.; Jankovic, J. The etiopathogenetic and pathophysiological spectrum of parkinsonism. J. Neurol. Sci. 2022, 433, 120012. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O. 50 years of levodopa. Mov. Disord. 2015, 30, 1008. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of alpha-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Definition of a Molecular Pathway Mediating -Synuclein Neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2017, 8, a024091. [Google Scholar] [CrossRef]
- Longhena, F.; Faustini, G.; Spillantini, M.G.; Bellucci, A. Living in Promiscuity: The Multiple Partners of Alpha-Synuclein at the Synapse in Physiology and Pathology. Int. J. Mol. Sci. 2019, 20, 141. [Google Scholar] [CrossRef]
- Reitboeck, P.G.; Anichtchik, O.; Bellucci, A.; Iovino, M.; Ballini, C.; Fineberg, E.; Ghetti, B.; Della Corte, L.; Spano, P.; Tofaris, G.K.; et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 2010, 133, 2032–2044. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Bellucci, A.; Antonini, A.; Pizzi, M.; Spano, P. The End Is the Beginning: Parkinson’s Disease in the Light of Brain Imaging. Front. Aging Neurosci. 2017, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Mercuri, N.B.; Venneri, A.; Faustini, G.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.; Bellucci, A.; Mercuri, N.B.; et al. Review: Parkinson’s disease: From synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 2016, 42, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Andica, C.; Kamagata, K.; Hatano, T.; Okuzumi, A.; Saito, A.; Nakazawa, M.; Ueda, R.; Motoi, Y.; Kamiya, K.; Suzuki, M.; et al. Neurite orientation dispersion and density imaging of the nigrostriatal pathway in Parkinson’s disease: Retrograde degeneration observed by tract-profile analysis. Park. Relat. Disord. 2018, 51, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef]
- Engelender, S.; Stefanis, L.; Oddo, S.; Bellucci, A. Can We Treat Neurodegenerative Proteinopathies by Enhancing Protein Degradation? Mov. Disord. 2022, 37, 1346–1359. [Google Scholar] [CrossRef]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Murphy, K.E.; Halliday, G.M. Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy 2014, 10, 1350–1351. [Google Scholar] [CrossRef]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.-W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef]
- Henderson, M.X.; Henrich, M.T.; Geibl, F.F.; Oertel, W.H.; Brundin, P.; Surmeier, D.J. The roles of connectivity and neuronal phenotype in determining the pattern of α-synuclein pathology in Parkinson’s disease. Neurobiol. Dis. 2022, 168, 105687. [Google Scholar] [CrossRef]
- Kim, C.Y.; Alcalay, R.N. Genetic Forms of Parkinson’s Disease. Semin. Neurol. 2017, 37, 135–146. [Google Scholar] [CrossRef]
- Lee, S.; Imai, Y.; Gehrke, S.; Liu, S.; Lu, B. The synaptic function of LRRK2. Biochem. Soc. Trans. 2012, 40, 1047–1051. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arranz, A.M.; Delbroek, L.; van Kolen, K.; Guimarães, M.R.; Mandemakers, W.; Daneels, G.; Matta, S.; Calafate, S.; Shaban, H.; Baatsen, P.; et al. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci. 2015, 128, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Beccano-Kelly, D.A.; Kuhlmann, N.; Tatarnikov, I.; Volta, M.; Munsie, L.N.; Chou, P.; Cao, L.-P.; Han, H.; Tapia, L.; Farrer, M.J.; et al. Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front. Cell. Neurosci. 2014, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, N.; Milnerwood, A.J. A Critical LRRK at the Synapse? The Neurobiological Function and Pathophysiological Dysfunction of LRRK2. Front. Mol. Neurosci. 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, E.; Gonnelli, A.; Cirnaru, M.-D.; Marte, A.; Plotegher, N.; Russo, I.; Civiero, L.; Cogo, S.; Carrion, M.P.; Franchin, C.; et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol. Neurodegener. 2016, 11, 1. [Google Scholar] [CrossRef]
- Cirnaru, M.D.; Marte, A.; Belluzzi, E.; Russo, I.; Gabrielli, M.; Longo, F.; Arcuri, L.; Murru, L.; Bubacco, L.; Matteoli, M.; et al. LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. Front. Mol. Neurosci. 2014, 7, 49. [Google Scholar] [CrossRef]
- Harris, T.W.; Hartwieg, E.; Horvitz, H.R.; Jorgensen, E.M. Mutations in Synaptojanin Disrupt Synaptic Vesicle Recycling. J. Cell Biol. 2000, 150, 589–600. [Google Scholar] [CrossRef]
- Geng, J.; Wang, L.; Lee, J.Y.; Chen, C.-K.; Chang, K.T. Phosphorylation of Synaptojanin Differentially Regulates Endocytosis of Functionally Distinct Synaptic Vesicle Pools. J. Neurosci. 2016, 36, 8882–8894. [Google Scholar] [CrossRef]
- Mignogna, M.L.; Giannandrea, M.; Gurgone, A.; Fanelli, F.; Raimondi, F.; Mapelli, L.; Bassani, S.; Fang, H.; van Anken, E.; Alessio, M.; et al. The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat. Commun. 2015, 6, 6504. [Google Scholar] [CrossRef]
- Niu, M.; Zheng, N.; Wang, Z.; Gao, Y.; Luo, X.; Chen, Z.; Fu, X.; Wang, Y.; Wang, T.; Liu, M.; et al. RAB39B Deficiency Impairs Learning and Memory Partially Through Compromising Autophagy. Front. Cell Dev. Biol. 2020, 8, 598622. [Google Scholar] [CrossRef]
- Koss, D.J.; Bondarevaite, O.; Adams, S.; Leite, M.; Giorgini, F.; Attems, J.; Outeiro, T.F. RAB39B is redistributed in dementia with Lewy bodies and is sequestered within aβ plaques and Lewy bodies. Brain Pathol. 2021, 31, 120–132. [Google Scholar] [CrossRef]
- Koss, D.J.; Campesan, S.; Giorgini, F.; Outeiro, T.F. Dysfunction of RAB39B-Mediated Vesicular Trafficking in Lewy Body Diseases. Mov. Disord. 2021, 36, 1744–1758. [Google Scholar] [CrossRef]
- Ribeiro, C.S.; Carneiro, K.; Ross, C.A.; Menezes, J.R.L.; Engelender, S. Synphilin-1 Is Developmentally Localized to Synaptic Terminals, and Its Association with Synaptic Vesicles Is Modulated by α-Synuclein. J. Biol. Chem. 2002, 277, 23927–23933. [Google Scholar] [CrossRef]
- Kim, M.J.; Deng, H.-X.; Wong, Y.C.; Siddique, T.; Krainc, D. The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum. Mol. Genet. 2017, 26, 729–741. [Google Scholar] [CrossRef]
- Tang, B.L. RAB39B’s role in membrane traffic, autophagy, and associated neuropathology. J. Cell. Physiol. 2021, 236, 1579–1592. [Google Scholar] [CrossRef]
- Casadei, N.; Pöhler, A.-M.; Tomás-Zapico, C.; Torres-Peraza, J.; Schwedhelm, I.; Witz, A.; Zamolo, I.; De Heer, R.; Spruijt, B.; Noldus, L.P.; et al. Overexpression of synphilin-1 promotes clearance of soluble and misfolded alpha-synuclein without restoring the motor phenotype in aged A30P transgenic mice. Hum. Mol. Genet. 2014, 23, 767–781. [Google Scholar] [CrossRef]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef]
- Zaarur, N.; Meriin, A.B.; Gabai, V.L.; Sherman, M.Y. Triggering Aggresome Formation. Dissecting aggresome-targeting and aggregation signals in synphilin 1. J. Biol. Chem. 2008, 283, 27575–27584. [Google Scholar] [CrossRef]
- Engelender, S. Ubiquitination of α-synuclein and autophagy in Parkinson’s disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef]
- Pan, P.-Y.; Zhu, J.; Rizvi, A.; Zhu, X.; Tanaka, H.; Dreyfus, C.F. Synaptojanin1 deficiency upregulates basal autophagosome formation in astrocytes. J. Biol. Chem. 2021, 297, 100873. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC 1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Lewis, P.A. LRRK2 and Autophagy. Adv. Neurobiol. 2017, 14, 89–105. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; Lopez de Maturana, R.; Sanchez-Pernaute, R.; Lopez de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Parganlija, D.; Klinkenberg, M.; Dominguez-Bautista, J.A.; Hetzel, M.; Gispert, S.; Chimi, M.A.; Dröse, S.; Mai, S.; Brandt, U.; Auburger, G.; et al. Loss of PINK1 Impairs Stress-Induced Autophagy and Cell Survival. PLoS ONE 2014, 9, e95288. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective Autophagy in Parkinson’s Disease: Role of Oxidative Stress. Mol. Neurobiol. 2012, 46, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef]
- Dasari, S.K.; Schejter, E.; Bialik, S.; Shkedy, A.; Levin-Salomon, V.; Levin-Zaidman, S.; Kimchi, A. Death by over-eating: The Gaucher disease associated gene GBA1, identified in a screen for mediators of autophagic cell death, is necessary for developmental cell death in Drosophila midgut. Cell Cycle 2017, 16, 2003–2010. [Google Scholar] [CrossRef]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef]
- Nelson, M.P.; Shacka, J.J. Autophagy Modulation in Disease Therapy: Where Do We Stand? Curr. Pathobiol. Rep. 2013, 1, 239–245. [Google Scholar] [CrossRef]
- Sesar, A.; Cacheiro, P.; López-López, M.; Camiña-Tato, M.; Quintáns, B.; Monroy-Jaramillo, N.; Alonso-Vilatela, M.-E.; Cebrián, E.; Yescas-Gómez, P.; Ares, B.; et al. Synaptotagmin XI in Parkinson’s disease: New evidence from an association study in Spain and Mexico. J. Neurol. Sci. 2016, 362, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Lin, Z.; Zheng, R.; Yan, Y.; Xue, N.; Yin, X.; Zhang, B. Association analysis of SYT11, FGF20, GCH1 rare variants in Parkinson’s disease. CNS Neurosci. Ther. 2021, 28, 175–177. [Google Scholar] [CrossRef]
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.P.; Goody, R.S. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Mignogna, M.L.; D’Adamo, P. Critical importance of RAB proteins for synaptic function. Small GTPases 2018, 9, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Ponce, L.; Cookson, M.R. The role of Rab GTPases in the pathobiology of Parkinson’ disease. Curr. Opin. Cell Biol. 2019, 59, 73–80. [Google Scholar] [CrossRef]
- Shi, M.-M.; Shi, C.-H.; Xu, Y.-M. Rab GTPases: The Key Players in the Molecular Pathway of Parkinson’s Disease. Front. Cell. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- Fukuda, M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol. Life Sci. 2008, 65, 2801–2813. [Google Scholar] [CrossRef]
- Kennedy, M.J.; Ehlers, M.D. Organelles and trafficking machinery for postsynaptic plasticity. Annu. Rev. Neurosci. 2006, 29, 325–362. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A. Review series: Rab GTPases and membrane identity: Causal or inconsequential? J. Cell Biol. 2013, 202, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Aivazian, D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004, 5, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The Guanine Nucleotide-Binding Switch in Three Dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Binotti, B.; Jahn, R.; Chua, J.J.E. Functions of Rab Proteins at Presynaptic Sites. Cells 2016, 5, 7. [Google Scholar] [CrossRef]
- Veleri, S.; Punnakkal, P.; Dunbar, G.L.; Maiti, P. Molecular Insights into the Roles of Rab Proteins in Intracellular Dynamics and Neurodegenerative Diseases. NeuroMolecular Med. 2018, 20, 18–36. [Google Scholar] [CrossRef]
- Fischer Von Mollard, G.; Südhof, T.C.; Jahn, R. A small GTP-binding protein dissociates from synaptic vesicles during exocytosis. Nature 1991, 349, 79–81. [Google Scholar] [CrossRef]
- Stahl, B.; von Mollard, G.F.; Walch-Solimena, C.; Jahn, R. GTP cleavage by the small GTP-binding protein Rab3A is associated with exocytosis of synaptic vesicles induced by alpha-latrotoxin. J. Biol. Chem. 1994, 269, 24770–24776. [Google Scholar] [CrossRef]
- Fukuda, M. Distinct Rab Binding Specificity of Rim1, Rim2, Rabphilin, and Noc2. Identification of a critical determinant of Rab3A/Rab27A recognition by Rim2. J. Biol. Chem. 2003, 278, 15373–15380. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Kanno, E.; Choi, S.; Sugimori, M.; Moreira, J.E.; Llinás, R.R.; Fukuda, M. Role of Rab27 in synaptic transmission at the squid giant synapse. Proc. Natl. Acad. Sci. USA 2008, 105, 16003–16008. [Google Scholar] [CrossRef] [PubMed]
- Pavlos, N.J.; Grønborg, M.; Riedel, D.; Chua, J.J.E.; Boyken, J.; Kloepper, T.H.; Urlaub, H.; Rizzoli, S.O.; Jahn, R. Quantitative Analysis of Synaptic Vesicle Rabs Uncovers Distinct Yet Overlapping Roles for Rab3a and Rab27b in Ca2+-Triggered Exocytosis. J. Neurosci. 2010, 30, 13441–13453. [Google Scholar] [CrossRef] [PubMed]
- Pavlos, N.J.; Jahn, R. Distinct yet overlapping roles of Rab GTPases on synaptic vesicles. Small GTPases 2011, 2, 77–81. [Google Scholar] [CrossRef][Green Version]
- Arias-Hervert, E.R.; Xu, N.; Njus, M.; Murphy, G.G.; Hou, Y.; Williams, J.A.; Lentz, S.I.; Ernst, S.A.; Stuenkel, E.L. Actions of Rab27B-GTPase on mammalian central excitatory synaptic transmission. Physiol. Rep. 2020, 8, e14428. [Google Scholar] [CrossRef]
- Schoch, S.; Castillo, P.E.; Jo, T.; Mukherjee, K.; Geppert, M.; Wang, Y.; Schmitz, F.; Malenka, R.C.; Südhof, T.C. RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 2002, 415, 321–326. [Google Scholar] [CrossRef]
- Schoch, S.; Mittelstaedt, T.; Kaeser, P.S.; Padgett, D.; Feldmann, N.; Chevaleyre, V.; Castillo, P.; Hammer, R.E.; Han, W.; Schmitz, F.; et al. Redundant functions of RIM1α and RIM2α in Ca2+-triggered neurotransmitter release. EMBO J. 2006, 25, 5852–5863. [Google Scholar] [CrossRef]
- Schlüter, O.M.; Schnell, E.; Verhage, M.; Tzonopoulos, T.; Nicoll, R.A.; Janz, R.; Malenka, R.C.; Geppert, M.; Südhof, T.C. Rabphilin Knock-Out Mice Reveal That Rabphilin Is Not Required for Rab3 Function in Regulating Neurotransmitter Release. J. Neurosci. 1999, 19, 5834–5846. [Google Scholar] [CrossRef]
- Deng, L.; Kaeser, P.S.; Xu, W.; Südhof, T.C. RIM Proteins Activate Vesicle Priming by Reversing Autoinhibitory Homodimerization of Munc13. Neuron 2011, 69, 317–331. [Google Scholar] [CrossRef]
- Dulubova, I.; Lou, X.; Lu, J.; Huryeva, I.; Alam, A.; Schneggenburger, R.; Südhof, T.C.; Rizo, J. A Munc13/RIM/Rab3 tripartite complex: From priming to plasticity? EMBO J. 2005, 24, 2839–2850. [Google Scholar] [CrossRef]
- Zarebidaki, F.; Camacho, M.; Brockmann, M.M.; Trimbuch, T.; Herman, M.A.; Rosenmund, C. Disentangling the Roles of RIM and Munc13 in Synaptic Vesicle Localization and Neurotransmission. J. Neurosci. 2020, 40, 9372–9385. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Kawamura, S.; Ozaki, K. An essential role of Rab5 in uniformity of synaptic vesicle size. J. Cell Sci. 2003, 116, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, T.; Wilsch-Bräuninger, M.; González-Gaitán, M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol. 2003, 161, 609–624. [Google Scholar] [CrossRef] [PubMed]
- De Hoop, M.J.; Huber, L.A.; Stenmark, H.; Williamson, E.; Zerial, M.; Parton, R.G.; Dotti, C.G. The involvement of the small GTP-binding protein Rab5a in neuronal endocytosis. Neuron 1994, 13, 11–22. [Google Scholar] [CrossRef]
- Fischer von Mollard, G.; Stahl, B.; Walch-Solimena, C.; Takei, K.; Daniels, L.; Khoklatchev, A.; de Camilli, P.; Südhof, T.C.; Jahn, R. Localization of Rab5 to synaptic vesicles identifies endosomal intermediate in synaptic vesicle recycling pathway. Eur. J. Cell Biol. 1994, 65, 319–326. [Google Scholar]
- Franchini, L.; Stanic, J.; Barzasi, M.; Zianni, E.; Mauceri, D.; Diluca, M.; Gardoni, F. Rabphilin-3A Drives Structural Modifications of Dendritic Spines Induced by Long-Term Potentiation. Cells 2022, 11, 1616. [Google Scholar] [CrossRef]
- Stanic, J.; Carta, M.; Eberini, I.; Pelucchi, S.; Marcello, E.; Genazzani, A.A.; Racca, C.; Mulle, C.; Di Luca, M.; Gardoni, F. Rabphilin 3A retains NMDA receptors at synaptic sites through interaction with GluN2A/PSD-95 complex. Nat. Commun. 2015, 6, 10181. [Google Scholar] [CrossRef]
- Nishino, H.; Saito, T.; Wei, R.; Takano, T.; Tsutsumi, K.; Taniguchi, M.; Ando, K.; Tomomura, M.; Fukuda, M.; Hisanaga, S.-I. The LMTK1-TBC1D9B-Rab11A Cascade Regulates Dendritic Spine Formation via Endosome Trafficking. J. Neurosci. 2019, 39, 9491–9502. [Google Scholar] [CrossRef]
- Szatmári, Z.; Sass, M. The autophagic roles of Rab small GTPases and their upstream regulators: A review. Autophagy 2014, 10, 1154–1166. [Google Scholar] [CrossRef]
- Binotti, B.; Pavlos, N.J.; Riedel, D.; Wenzel, D.; Vorbrüggen, G.; Schalk, A.M.; Kühnel, K.; Boyken, J.; Erck, C.; Martens, H.; et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. eLife 2015, 4, e05597. [Google Scholar] [CrossRef]
- Nakazawa, H.; Sada, T.; Toriyama, M.; Tago, K.; Sugiura, T.; Fukuda, M.; Inagaki, N. Rab33a Mediates Anterograde Vesicular Transport for Membrane Exocytosis and Axon Outgrowth. J. Neurosci. 2012, 32, 12712–12725. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.R.; Sim, J.C.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B Cause X-Linked Intellectual Disability and Early-Onset Parkinson Disease with α-Synuclein Pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.P.; Freeman, M.R. Wallerian degeneration, wld(s), and nmnat. Annu Rev. Neurosci. 2010, 33, 245–267. [Google Scholar] [CrossRef]
- Lesage, S.; Bras, J.; Cormier-Dequaire, F.; Condroyer, C.; Nicolas, A.; Darwent, L.; Guerreiro, R.; Majounie, E.; Federoff, M.; Heutink, P.; et al. Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol. Genet. 2015, 1, e9. [Google Scholar] [CrossRef]
- Mata, I.F.; Jang, Y.; Kim, C.H.; Hanna, D.S.; Dorschner, M.O.; Samii, A.; Agarwal, P.; Roberts, J.W.; Klepitskaya, O.; Shprecher, D.R.; et al. The RAB39B p.G192R mutation causes X-linked dominant Parkinson’s disease. Mol. Neurodegener. 2015, 10, 50. [Google Scholar] [CrossRef]
- Güldner, M.; Schulte, C.; Hauser, A.-K.; Gasser, T.; Brockmann, K. Broad clinical phenotype in Parkinsonism associated with a base pair deletion in RAB39B and additional POLG variant. Park. Relat. Disord. 2016, 31, 148–150. [Google Scholar] [CrossRef]
- Shi, C.-H.; Zhang, S.-Y.; Yang, Z.-H.; Yang, J.; Shang, D.-D.; Mao, C.-Y.; Liu, H.; Hou, H.-M.; Shi, M.-M.; Wu, J.; et al. A novel RAB39B gene mutation in X-linked juvenile parkinsonism with basal ganglia calcification. Mov. Disord. 2016, 31, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Löchte, T.; Brüggemann, N.; Vollstedt, E.-J.; Krause, P.; Domingo, A.; Rosales, R.; Lee, L.V.; Hopfner, F.; Westenberger, A.; Kühn, A.; et al. RAB39B mutations are a rare finding in Parkinson disease patients. Park. Relat. Disord. 2016, 23, 116–117. [Google Scholar] [CrossRef]
- Ciammola, A.; Carrera, P.; Di Fonzo, A.; Sassone, J.; Villa, R.; Poletti, B.; Ferrari, M.; Girotti, F.; Monfrini, E.; Buongarzone, G.; et al. X-linked Parkinsonism with Intellectual Disability caused by novel mutations and somatic mosaicism in RAB39B gene. Park. Relat. Disord. 2017, 44, 142–146. [Google Scholar] [CrossRef]
- Dalfó, E.; Barrachina, M.; Rosa, J.L.; Ambrosio, S.; Ferrer, I. Abnormal α-synuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol. Dis. 2004, 16, 92–97. [Google Scholar] [CrossRef]
- Wang, H.-L.; Lu, C.-S.; Yeh, T.-H.; Shen, Y.-M.; Weng, Y.-H.; Huang, Y.-Z.; Chen, R.-S.; Liu, Y.-C.; Cheng, Y.-C.; Chang, H.-C.; et al. Combined Assessment of Serum Alpha-Synuclein and Rab35 is a Better Biomarker for Parkinson’s Disease. J. Clin. Neurol. 2019, 15, 488–495. [Google Scholar] [CrossRef]
- Nakamura, S.; Kawamoto, Y.; Nakano, S.; Akiguchi, I. Expression of the endocytosis regulatory proteins Rab5 and Rabaptin-5 in glial cytoplasmic inclusions from brains with multiple system atrophy. Clin. Neuropathol. 2000, 19, 51–56. [Google Scholar]
- Dalfó, E.; Ferrer, I. α-Synuclein binding to rab3a in multiple system atrophy. Neurosci. Lett. 2005, 380, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.-C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; McCloskey, A.; Cheng, S.; Wu, M.; Xue, C.; Yu, Z.; Fu, J.; Liu, Y.; Luo, Z.-Q.; Liu, X. Regulation of the small GTPase Rab1 function by a bacterial glucosyltransferase. Cell Discov. 2018, 4, 5. [Google Scholar] [CrossRef]
- Dalfó, E.; Gómez-Isla, T.; Rosa, J.L.; Nieto Bodelón, M.; Cuadrado Tejedor, M.; Barrachina, M.; Ambrosio, S.; Ferrer, I. Abnormal α-Synuclein Interactions with Rab Proteins in α-Synuclein A30P Transgenic Mice. J. Neuropathol. Exp. Neurol. 2004, 63, 302–313. [Google Scholar] [CrossRef]
- Gonçalves, S.A.; Macedo, D.; Raquel, H.; Simões, P.D.; Giorgini, F.; Ramalho, J.; Barral, D.-C.; Ferreira Moita, L.; Outeiro, T.F. shRNA-Based Screen Identifies Endocytic Recycling Pathway Components That Act as Genetic Modifiers of Alpha-Synuclein Aggregation, Secretion and Toxicity. PLoS Genet. 2016, 12, e1005995. [Google Scholar] [CrossRef]
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. α-Synuclein Membrane Association Is Regulated by the Rab3a Recycling Machinery and Presynaptic Activity*. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of Neuronal Cell Death by Rab5A-dependent Endocytosis of α-Synuclein. J. Biol. Chem. 2001, 276, 27441–27448. [Google Scholar] [CrossRef]
- Underwood, R.; Wang, B.; Carico, C.; Whitaker, R.H.; Placzek, W.J.; Yacoubian, T.A. The GTPase Rab27b regulates the release, autophagic clearance, and toxicity of α-synuclein. J. Biol. Chem. 2020, 295, 8005–8016. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef]
- Haugarvoll, K.; Rademakers, R.; Kachergus, J.M.; Nuytemans, K.; Ross, O.A.; Gibson, J.M.; Tan, E.-K.; Gaig, C.; Tolosa, E.; Goldwurm, S.; et al. Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 2008, 70, 1456–1460. [Google Scholar] [CrossRef]
- Healy, D.G.; Wood, N.W.; Schapira, A. Test for LRRK2 mutations in patients with Parkinson’s disease. Pract. Neurol. 2008, 8, 381–385. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Martí-Massó, J.; Sánchez-Mut, J.V.; Paisán-Ruíz, C.; Martínez-Gil, A.; Ruiz-Martínez, J.; Sáenz, A.; Singleton, A.B.; Lopez de Munain, A.; Pérez-Tur, J. Parkinson’s disease due to the R1441G mutation in Dardarin: A founder effect in the basques. Mov. Disord. 2006, 21, 1954–1959. [Google Scholar] [CrossRef]
- Ho, D.H.; Jang, J.; Joe, E.-H.; Son, I.; Seo, H.; Seol, W. G2385R and I2020T Mutations Increase LRRK2 GTPase Activity. BioMed Res. Int. 2016, 2016, 7917128. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 2017, 6, e31012. [Google Scholar] [CrossRef] [PubMed]
- Tucci, A.; Nalls, M.A.; Houlden, H.; Revesz, T.; Singleton, A.B.; Wood, N.W.; Hardy, J.; Paisán-Ruíz, C. Genetic variability at the PARK16 locus. Eur. J. Hum. Genet. 2010, 18, 1356–1359. [Google Scholar] [CrossRef]
- Madero-Pérez, J.; Fernández, B.; Ordóñez, A.J.L.; Fdez, E.; Lobbestael, E.; Baekelandt, V.; Hilfiker, S. RAB7L1-Mediated Relocalization of LRRK2 to the Golgi Complex Causes Centrosomal Deficits via RAB8A. Front. Mol. Neurosci. 2018, 11, 417. [Google Scholar] [CrossRef]
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet. 2018, 27, 385–395. [Google Scholar] [CrossRef]
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropulou, A.F.; Freemantle, J.B.; Lis, P.; Vides, E.G.; Polinski, N.K.; Alessi, D.R. Endogenous Rab29 does not impact basal or stimulated LRRK2 pathway activity. Biochem. J. 2020, 477, 4397–4423. [Google Scholar] [CrossRef] [PubMed]
- Iannotta, L.; Biosa, A.; Kluss, J.H.; Tombesi, G.; Kaganovich, A.; Cogo, S.; Plotegher, N.; Civiero, L.; Lobbestael, E.; Baekelandt, V.; et al. Divergent Effects of G2019S and R1441C LRRK2 Mutations on LRRK2 and Rab10 Phosphorylations in Mouse Tissues. Cells 2020, 9, 2344. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suaga, P.; Rivero-Ríos, P.; Fdez, E.; Blanca Ramírez, M.; Ferrer, I.; Aiastui, A.; Lopez de Munain, A.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef]
- Rivero-Ríos, P.; Romo-Lozano, M.; Madero-Pérez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef]
- Kouranti, I.; Sachse, M.; Arouche, N.; Goud, B.; Echard, A. Rab35 Regulates an Endocytic Recycling Pathway Essential for the Terminal Steps of Cytokinesis. Curr. Biol. 2006, 16, 1719–1725. [Google Scholar] [CrossRef]
- Bae, E.-J.; Kim, D.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef]
- Waschbüsch, D.; Hübel, N.; Ossendorf, E.; Lobbestael, E.; Baekelandt, V.; Lindsay, A.J.; McCaffrey, M.W.; Khan, A.R.; Barnekow, A. Rab32 interacts with SNX6 and affects retromer-dependent Golgi trafficking. PLoS ONE 2019, 14, e0208889. [Google Scholar] [CrossRef]
- Waschbüsch, D.; Michels, H.; Strassheim, S.; Ossendorf, E.; Kessler, D.; Gloeckner, C.J.; Barnekow, A. LRRK2 Transport Is Regulated by Its Novel Interacting Partner Rab32. PLoS ONE 2014, 9, e111632. [Google Scholar] [CrossRef]
- Inoshita, T.; Arano, T.; Hosaka, Y.; Meng, H.; Umezaki, Y.; Kosugi, S.; Morimoto, T.; Koike, M.; Chang, H.-Y.; Imai, Y.; et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum. Mol. Genet. 2017, 26, 2933–2948. [Google Scholar] [CrossRef] [PubMed]
- Albanese, F.; Domenicale, C.; Volta, M.; Morari, M. Modeling Parkinson’s disease in LRRK2 mice: Focus on synaptic dysfunction and the autophagy-lysosomal pathway. Biochem. Soc. Trans. 2022, 50, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.D.; Shin, J.-H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.-I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, C.F.; Hijaz, B.A.; Singh, V.; Gcwensa, N.Z.; Kelly, K.; Boyden, E.S.; West, A.B.; Sarkar, D.; Volpicelli-Daley, L.A. Inhibition of LRRK2 kinase activity promotes anterograde axonal transport and presynaptic targeting of α-synuclein. Acta Neuropathol. Commun. 2021, 9, 180. [Google Scholar] [CrossRef]
- Plowey, E.D.; Johnson, J.W.; Steer, E.; Zhu, W.; Eisenberg, D.A.; Valentino, N.M.; Liu, Y.-J.; Chu, C.T. Mutant LRRK2 enhances glutamatergic synapse activity and evokes excitotoxic dendrite degeneration. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1596–1603. [Google Scholar] [CrossRef]
- Chou, J.-S.; Chen, C.-Y.; Chen, Y.-L.; Weng, Y.-H.; Yeh, T.-H.; Lu, C.-S.; Chang, Y.-M.; Wang, H.-L. (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol. Dis. 2014, 68, 190–199. [Google Scholar] [CrossRef]
- Nguyen, A.P.T.; Daniel, G.; Valdés, P.; Islam, M.S.; Schneider, B.L.; Moore, D.J. G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum. Mol. Genet. 2018, 27, 120–134. [Google Scholar] [CrossRef]
- Qin, Q.; Zhi, L.-T.; Li, X.-T.; Yue, Z.-Y.; Li, G.; Zhang, H. Effects of LRRK2 Inhibitors on Nigrostriatal Dopaminergic Neurotransmission. CNS Neurosci. Ther. 2017, 23, 162–173. [Google Scholar] [CrossRef]
- Nirujogi, R.S.; Tonelli, F.; Taylor, M.; Lis, P.; Zimprich, A.; Sammler, E.; Alessi, D.R. Development of a multiplexed targeted mass spectrometry assay for LRRK2-phosphorylated Rabs and Ser910/Ser935 biomarker sites. Biochem. J. 2021, 478, 299–326. [Google Scholar] [CrossRef]
- Petropoulou-Vathi, L.; Simitsi, A.; Valkimadi, P.-E.; Kedariti, M.; Dimitrakopoulos, L.; Koros, C.; Papadimitriou, D.; Papadimitriou, A.; Stefanis, L.; Alcalay, R.N.; et al. Distinct profiles of LRRK2 activation and Rab GTPase phosphorylation in clinical samples from different PD cohorts. Npj Park. Dis. 2022, 8, 73. [Google Scholar] [CrossRef]
- Mir, R.; Tonelli, F.; Lis, P.; Macartney, T.; Polinski, N.K.; Martinez, T.N.; Chou, M.-Y.; Howden, A.J.M.; König, T.; Hotzy, C.; et al. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 2018, 475, 1861–1883. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Unnithan, S.; Bryant, N.; Chang, A.; Rosenthal, L.S.; Pantelyat, A.; Dawson, T.M.; Al-Khalidi, H.R.; West, A.B. Elevated Urinary Rab10 Phosphorylation in Idiopathic Parkinson Disease. Mov. Disord. 2022, 37, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Atashrazm, F.; Hammond, D.; Perera, G.; Bolliger, M.F.; Matar, E.; Halliday, G.M.; Schüle, B.; Lewis, S.J.G.; Nichols, R.J.; Dzamko, N. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov. Disord. 2019, 34, 406–415. [Google Scholar] [CrossRef] [PubMed]
- George, A.A.; Hayden, S.; Holzhausen, L.C.; Ma, E.Y.; Suzuki, S.C.; Brockerhoff, S.E. Synaptojanin 1 Is Required for Endolysosomal Trafficking of Synaptic Proteins in Cone Photoreceptor Inner Segments. PLoS ONE 2014, 9, e84394. [Google Scholar] [CrossRef]
- Deng, H.-X.; Shi, Y.; Yang, Y.; Ahmeti, K.B.; Miller, N.; Huang, C.; Cheng, L.; Zhai, H.; Deng, S.; Nuytemans, K.; et al. Identification of TMEM230 mutations in familial Parkinson’s disease. Nat. Genet. 2016, 48, 733–739. [Google Scholar] [CrossRef]
- Sidransky, E.; Samaddar, T.; Tayebi, N.; Nichols, W.C.; Pankratz, N.; Foroud, T. Mutations in gba are associated with FAMILIAL parkinson disease susceptibility and age at onset. Neurology 2009, 73, 1424–1426. [Google Scholar] [CrossRef]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Evans, J.; Breen, D.P.; Barker, R.A.; Williams-Gray, C.H. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 695–702. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef]
- Gündner, A.L.; Duran-Pacheco, G.; Zimmermann, S.; Ruf, I.; Moors, T.; Baumann, K.; Jagasia, R.; van de Berg, W.D.J.; Kremer, T. Path mediation analysis reveals GBA impacts Lewy body disease status by increasing α-synuclein levels. Neurobiol. Dis. 2019, 121, 205–213. [Google Scholar] [CrossRef]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.-H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid β-glucosidase mutants linked to gaucher disease, parkinson disease, and lewy body dementia alter α-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y.; Gegg, M.; Chau, D.; Schapira, A. Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 2020, 134, 104620. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Yang, L.; Tran, H.; Yu, W.; Sun, A.X.; Chang, Y.Y.; Jung, B.C.; Lee, S.; Saw, T.Y.; Xiao, B.; et al. Lewy Body–like Inclusions in Human Midbrain Organoids Carrying Glucocerebrosidase and α-Synuclein Mutations. Ann. Neurol. 2021, 90, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sedor, S.; McGeary, I.; Cornblath, E.J.; Peng, C.; Riddle, D.M.; Li, H.L.; Zhang, B.; Brown, H.J.; Olufemi, M.F.; et al. Glucocerebrosidase Activity Modulates Neuronal Susceptibility to Pathological α-Synuclein Insult. Neuron 2019, 105, 822–836. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson Risk in Ashkenazi Jewish Patients With Gaucher Disease and GBA Heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef]
- Fecchio, C.; Palazzi, L.; de Laureto, P.P. α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules 2018, 23, 1531. [Google Scholar] [CrossRef]
- Rivero-Ríos, P.; Romo-Lozano, M.; Fasiczka, R.; Naaldijk, Y.; Hilfiker, S. LRRK2-Related Parkinson’s Disease Due to Altered Endolysosomal Biology With Variable Lewy Body Pathology: A Hypothesis. Front. Neurosci. 2020, 14, 556. [Google Scholar] [CrossRef]
- Klein, A.D.; Mazzulli, J.R. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107. [Google Scholar] [CrossRef]
- Morén, C.; Juárez-Flores, D.L.; Chau, K.-Y.; Gegg, M.; Garrabou, G.; González-Casacuberta, I.; Guitart-Mampel, M.; Tolosa, E.; Martí, M.J.; Cardellach, F.; et al. GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging 2019, 11, 10338–10355. [Google Scholar] [CrossRef]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.-H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Arshad, M.; Wang, W.; Zhao, D.; Xu, L.; Zhou, L. LRRK2 mediated Rab8a phosphorylation promotes lipid storage. Lipids Health Dis. 2018, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, P.; Schirinzi, T.; Meringolo, M.; Mercuri, N.B.; Pisani, A. Centrality of Early Synaptopathy in Parkinson’s Disease. Front. Neurol. 2018, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Ginns, E.I.; Mak, S.K.-K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Pará, C.; Bose, P.; Pshezhetsky, A.V. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J. Clin. Med. 2020, 9, 616. [Google Scholar] [CrossRef]
- Higashi, S.; Moore, D.; Minegishi, M.; Kasanuki, K.; Fujishiro, H.; Kabuta, T.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Localization of MAP1-LC3 in Vulnerable Neurons and Lewy Bodies in Brains of Patients With Dementia With Lewy Bodies. J. Neuropathol. Exp. Neurol. 2011, 70, 264–280. [Google Scholar] [CrossRef]
- Szegö, E.M.; van den Haute, C.; Höfs, L.; Baekelandt, V.; van der Perren, A.; Falkenburger, B.H. Rab7 reduces α-synuclein toxicity in rats and primary neurons. Exp. Neurol. 2022, 347, 113900. [Google Scholar] [CrossRef]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef]
- Lai, Y.; Kondapalli, C.; Lehneck, R.; Procter, J.B.; Dill, B.D.; Woodroof, H.I.; Gourlay, R.; Peggie, M.; Macartney, T.J.; Corti, O.; et al. Phosphoproteomic screening identifies Rab GTP ases as novel downstream targets of PINK 1. EMBO J. 2015, 34, 2840–2861. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Muqit, M.M.K. PTEN-induced kinase 1 (PINK1) and Parkin: Unlocking a mitochondrial quality control pathway linked to Parkinson’s disease. Curr. Opin. Neurobiol. 2022, 72, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P. Managing risky assets–mitophagy in vivo. J. Cell Sci. 2021, 134, jcs240465. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, M.; Madara, J.; Pankow, S.; Liu, X.; Yates, J., 3rd; Südhof, T.C.; Maximov, A. Synaptotagmin-11 mediates a vesicle trafficking pathway that is essential for development and synaptic plasticity. Genes Dev. 2019, 33, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kang, X.; Zhou, L.; Chai, Z.; Wu, Q.; Huang, R.; Xu, H.; Hu, M.; Sun, X.; Sun, S.; et al. Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat. Commun. 2018, 9, 81. [Google Scholar] [CrossRef]
- Decet, M.; Verstreken, P. Presynaptic Autophagy and the Connection with Neurotransmission. Front. Cell Dev. Biol. 2021, 9, 790721. [Google Scholar] [CrossRef]
- Liang, Y. Emerging Concepts and Functions of Autophagy as a Regulator of Synaptic Components and Plasticity. Cells 2019, 8, 34. [Google Scholar] [CrossRef]
- Shen, D.-N.; Zhang, L.-H.; Wei, E.-Q.; Yang, Y. Autophagy in synaptic development, function, and pathology. Neurosci. Bull. 2015, 31, 416–426. [Google Scholar] [CrossRef]
- Hill, S.E.; Colón-Ramos, D.A. The Journey of the Synaptic Autophagosome: A Cell Biological Perspective. Neuron 2020, 105, 961–973. [Google Scholar] [CrossRef]
- Kulkarni, V.V.; Anand, A.; Herr, J.B.; Miranda, C.; Vogel, M.C.; Maday, S. Synaptic activity controls autophagic vacuole motility and function in dendrites. J. Cell Biol. 2021, 220, e202002084. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Ryskalin, L.; Fornai, F. Interdependency Between Autophagy and Synaptic Vesicle Trafficking: Implications for Dopamine Release. Front. Mol. Neurosci. 2018, 11, 299. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Rab Alterations and aSyn-Related Synaptopathy | ||
| Rab | Alteration Type | References |
| Mutations Identified in Rabs | ||
| RAB39B | c.503C > A missense mutation | [92] |
| RAB39B | c.557G > A missense mutation | [94] |
| RAB39B | c.574G > A missense mutation | [95] |
| RAB39B | c.432delA single base pair deletion | [96] |
| RAB39B | c. 536dupA duplication | [97] |
| RAB39B | c.543A > G; c.215 + 61G > ; c.215 + 39C > G | [98] |
| RAB39B | c.137dupT; c.371delA | [99] |
| Activity or pathological changes | ||
| Rab3a | Loss of coupling with rabphilin | [100] |
| Rab35 | Increased levels in PD patient serum | [101] |
| Rab5 | Accumulation in GCI | [102,103] |
| Rab1 | Increased levels protect against aSyn-mediated neuron loss | [104] |
| Rab3a | Protects against aSyn-mediated neuron loss | [105] |
| Rab8a | Protects against aSyn-mediated neuron loss | [105] |
| Rab3a | Aberrant interaction with A30P aSyn | [107] |
| Rab5 | Aberrant interaction with A30P aSyn | [107] |
| Rab8 | Aberrant interaction with A30P aSyn | [107] |
| Rab8b | Promotes aSyn aggregate clearance | [108] |
| Rab11a | Promotes aSyn aggregate clearance/aSyn secretion | [108] |
| Rab13 | Promotes aSyn aggregate clearance/aSyn secretion | [108] |
| Rab3a | Regulates aSyn binding to presynaptic membranes | [109] |
| Rab5a | Mediates aSyn endocytosis (spreading?) | [110] |
| Rab27b | Reduces aSyn spreading via nonexosomal pathways | [111] |
| Rab Alterations in LRRK2-Associated Parkinsonism | ||
| Rab | Alteration Type | References |
| Phosphorylation/activity changes | ||
| Rab8a | Aberrant phosphorylation by G2019S-LRRK2 | [119] |
| Rab7L1 | Aberrant phosphorylation by R1441C, Y1699C and G2019S-LRRK2 | [120] |
| Rab10 | Phosphorylation increase (R1441C)/decrease (G2019S) | [123] |
| Rab7 | Decrease in protein activity induced by expression of PD-associated LRRK2 mutants | [124] |
| Rab8a | Impaired function mediated by G2019S-LRRK2 | [126] |
| Rab35 | Increased phosphorylation mediated by G2019S-LRRK2 | [128] |
| Rab impact on physiological and pathological LRRK2 trafficking | ||
| Trafficking changes | ||
| Rab29 | Abnormal recruitment of R1441G/C and Y1699C-LRRK2 to the Golgi without affecting LRRK2 phosphorylation activity | [121,122] |
| Rab32 | Regulates LRRK2 late endosomal transport | [130] |
| Rabs in Autophagy and Autophagy-Related Synaptic Alterations | ||
|---|---|---|
| Rab | ROLE | Reference |
| Rab39b | Autophagy activation and fusion of autophagosomes with lysosomes | [30] |
| Rab26 | Directs SVs into pre-autophagosomal structures | [90] |
| Rab1 | Autophagosome formation | [163] |
| Rab5 | Autophagosome formation | [163] |
| Rab7 | Autophagosome formation | [163] |
| Rab7 | Fusion of autophagosomes with lysosomes | [163] |
| Rab8b | Autophagosome formation (non-canonical autophagy) | [163] |
| Rab8b | Autophagy-based unconventional secretory pathway | [163] |
| Rab9a | Autophagosome formation | [163] |
| Rab11 | Amphisome formation | [163] |
| Rab23 | Autophagosome formation | [163] |
| Rab24 | Fusion of autophagosomes with lysosomes | [163] |
| Rab32 | Autophagosome formation | [163] |
| Rab33b | Autophagosome formation | [163] |
| Rab7 | Involved in autophagosome formation during mitophagy | [170] |
| Rab8a | Downstream target of PINK1 | [171] |
| Rab8b | Downstream target of PINK1 | [166] |
| Rab13 | Downstream target of PINK1 | [171] |
| Rab5 | Directs SVs to autophagy | [181] |
| Rab35 | Directs SVs to autophagy | [181] |
| Rab4 | Autophagosome formation and maturation | [181] |
| Rab5 | Autophagosome formation and maturation | [181] |
| Rab10 | Autophagosome formation and maturation | [181] |
| Rab11 | Autophagosome formation and maturation | [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellucci, A.; Longhena, F.; Spillantini, M.G. The Role of Rab Proteins in Parkinson’s Disease Synaptopathy. Biomedicines 2022, 10, 1941. https://doi.org/10.3390/biomedicines10081941
Bellucci A, Longhena F, Spillantini MG. The Role of Rab Proteins in Parkinson’s Disease Synaptopathy. Biomedicines. 2022; 10(8):1941. https://doi.org/10.3390/biomedicines10081941
Chicago/Turabian StyleBellucci, Arianna, Francesca Longhena, and Maria Grazia Spillantini. 2022. "The Role of Rab Proteins in Parkinson’s Disease Synaptopathy" Biomedicines 10, no. 8: 1941. https://doi.org/10.3390/biomedicines10081941
APA StyleBellucci, A., Longhena, F., & Spillantini, M. G. (2022). The Role of Rab Proteins in Parkinson’s Disease Synaptopathy. Biomedicines, 10(8), 1941. https://doi.org/10.3390/biomedicines10081941