
Gene-Based Therapeutics for Parkinson’s Disease


Abstract
:1. Features of Parkinson’s Disease: From Theory to Therapy
1.1. Prevalence, Biology and Pathophysiology
1.2. Genetic Architecture of PD
1.3. Current Treatment Strategies
2. Gene Therapy for PD: Principles and Practicalities
2.1. Gene Therapy Trials for PD
2.1.1. Restoring the Physiological Balance of the Basal Ganglia
GABA
2.1.2. Enhancing Dopamine Synthesis
AADC
TH, AADC and GCH1
2.1.3. Neuroprotection and Regeneration
GDNF, NRTN
Neural Regeneration
2.1.4. Targeting Disease Genes
SNCA, LRRK2 and GBA
3. Prospective and Conclusive Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Kowal, S.L.; Ms, T.M.D.; Chakrabarti, R.; Bs, M.V.S.; Jain, A. The current and projected economic burden of Parkinson’s disease in the United States. Mov. Disord. 2013, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Dorsey, E.R.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. npj Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. Description of Parkinson’s Disease as a Clinical Syndrome. Ann. N. Y. Acad. Sci. 2006, 991, 1–14. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.V. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.; Roy, S. α-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Burré, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Südhof, T.C.; Brunger, A.T. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2013, 2, e00592. [Google Scholar] [CrossRef]
- Olanow, C.W.; Brundin, P. Parkinson’s Disease and Alpha Synuclein: Is Parkinson’s Disease a Prion-Like Disorder? Mov. Disord. 2013, 28, 31–40. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease and Related Neurodegenerative Disorders. Mol. Neurobiol. 2012, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.J.; Bs, J.K.; Forno, L.S.; Lincoln, S.; Wang, D.-S.; Hulihan, M.M.; Maraganore, D.M.; Gwinn, K.; Wszolek, Z.K.; Dickson, D.W.; et al. Comparison of kindreds with parkinsonism and ?-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Olanow, C.; Freeman, T.B. Transplanted dopaminergic neurons develop PD pathologic changes: A second case report. Mov. Disord. 2008, 23, 2303–2306. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.-I.; et al. α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild Type α-Synuclein Is Degraded by Chaperone-mediated Autophagy and Macroautophagy in Neuronal Cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Valera, E.; Rockenstein, E.; Trejo-Morales, M.; Adame, A.; Masliah, E. A brain-targeted, modified neurosin (kallikrein-6) reduces α-synuclein accumulation in a mouse model of multiple system atrophy. Mol. Neurodegener. 2015, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Mishizen-Eberz, A.J.; Guttmann, R.P.; Giasson, B.I.; Day, G.A., 3rd; Hodara, R.; Ischiropoulos, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Lynch, D.R. Distinct cleavage patterns of normal and pathologic forms of α-synuclein by calpain I in vitro. J. Neurochem. 2003, 86, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, M.; Vieira, S.; Schapira, A. Genetic causes of PD: A pathway to disease modification. Neuropharmacology 2020, 170, 108022. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puschmann, A.; Ross, O.A.; Vilariño-Güell, C.; Lincoln, S.J.; Kachergus, J.M.; Cobb, S.A.; Lindquist, S.G.; Nielsen, J.E.; Wszolek, Z.K.; Farrer, M.; et al. A Swedish family with de novo α-synuclein A53T mutation: Evidence for early cortical dysfunction. Park. Relat. Disord. 2009, 15, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Ki, C.-S.; Stavrou, E.; Davanos, N.; Lee, W.; Chung, E.; Kim, J.-Y.; Athanassiadou, A. The Ala53Thr mutation in the α-synuclein gene in a Korean family with Parkinson disease. Clin. Genet. 2007, 71, 471–473. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.B.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Tompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, e2181–e2185. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Koros, C.; Strohäker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibáñez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinsonʼs Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Maraganore, D.M.; De Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Krüger, R.; Rocca, W.A.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Collaborative Analysis of α-Synuclein Gene Promoter Variability and Parkinson Disease. JAMA 2006, 296, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.-Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s Disease Linked to Pathological α-Synuclein: New Targets for Drug Discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Tofaris, G.; Spillantini, M.G. Physiological and pathological properties of α-synuclein. Experientia 2007, 64, 2194–2201. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Brockmann, K.; Schulte, C.; Hauser, A.-K.; Lichtner, P.; Huber, H.; Maetzler, W.; Berg, D.; Gasser, T. SNCA: Major genetic modifier of age at onset of Parkinson’s disease. Mov. Disord. 2013, 28, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Guella, I.; Evans, D.M.; Szu-Tu, C.; Nosova, E.; Bsc, S.F.B.; Goldman, J.G.; Dalrymple-Alford, J.C.; Geurtsen, G.; Litvan, I.; Ross, O.; et al. α-synuclein genetic variability: A biomarker for dementia in Parkinson disease. Ann. Neurol. 2016, 79, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruίz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; Van Der Brug, M.; De Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations that Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldwurm, S.; Zini, M.; Mariani, L.; Tesei, S.; Miceli, R.; Sironi, F.; Clementi, M.; Bonifati, V.; Pezzoli, G. Evaluation of LRRK2 G2019S penetrance: Relevance for genetic counseling in Parkinson disease. Neurology 2007, 68, 1141–1143. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- Ramiίrez-Valle, F.; Braunstein, S.; Zavadil, J.; Formenti, S.C.; Schneider, R. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008, 181, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Dhungel, N.; Eleuteri, S.; Li, L.-B.; Kramer, N.; Chartron, J.; Spencer, B.; Kosberg, K.; Fields, J.A.; Stafa, K.; Adame, A.; et al. Parkinson’s Disease Genes VPS35 and EIF4G1 Interact Genetically and Converge on α-Synuclein. Neuron 2015, 85, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Mizuta, I.; Toyoda, A.; Ashida, R.; Takahashi, Y.; Goto, J.; Fukuda, Y.; Date, H.; Iwata, A.; Yamamoto, M.; et al. Mutations for Gaucher Disease Confer High Susceptibility to Parkinson Disease. Arch. Neurol. 2009, 66, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaolo, J.; Goker-Alpan, O.; Samaddar, T.; Lopez, G.; Sidransky, E. The association between mutations in the lysosomal protein glucocerebrosidase and parkinsonism. Mov. Disord. 2009, 24, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, A.; Hardy, J. A generalizable hypothesis for the genetic architecture of disease: Pleomorphic risk loci. Hum. Mol. Genet. 2011, 20, R158–R162. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Aquino, C.; Krauss, J.K.; Honey, C.R.; Bloem, B.R. Axial disability and deep brain stimulation in patients with Parkinson disease. Nat. Rev. Neurol. 2015, 11, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Paleologou, K.E.; Greer, B.; Abogrein, A.M.; King, J.E.; Salem, S.A.; Fullwood, N.J.; Benson, F.E.; Hewitt, R.; Ford, K.J.; et al. A strategy for designing inhibitors of α-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004, 18, 1315–1317. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Emadi, S.; Desplats, P.; Eleuteri, S.; Michael, S.; Kosberg, K.; Shen, J.; Rockenstein, E.; Patrick, C.; Adame, A.; et al. ESCRT-mediated Uptake and Degradation of Brain-targeted α-synuclein Single Chain Antibody Attenuates Neuronal Degeneration In Vivo. Mol. Ther. 2014, 22, 1753–1767. [Google Scholar] [CrossRef] [Green Version]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [Green Version]
- Polissidis, A.; Petropoulou-Vathi, L.; Nakos-Bimpos, M.; Rideout, H.J. The Future of Targeted Gene-Based Treatment Strategies and Biomarkers in Parkinson’s Disease. Biomolecules 2020, 10, 912. [Google Scholar] [CrossRef]
- Amer, M.H. Gene therapy for cancer: Present status and future perspective. Mol. Cell. Ther. 2014, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Prakash, V.; Moore, M.; Yáñez-Muñoz, R.J. Current Progress in Therapeutic Gene Editing for Monogenic Diseases. Mol. Ther. 2016, 24, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, M.S.; Samulski, R.J.; McCown, T.J. Adeno-associated virus (AAV) gene therapy for neurological disease. Neuropharmacology 2013, 69, 82–88. [Google Scholar] [CrossRef]
- Nanou, A.; Azzouz, M. Gene therapy for neurodegenerative diseases based on lentiviral vectors. Prog. Brain Res. 2009, 175, 187–200. [Google Scholar] [CrossRef]
- Duque, S.; Joussemet, B.; Riviere, C.; Marais, T.; Dubreil, L.; Douar, A.-M.; Fyfe, J.; Moullier, P.; Colle, M.-A.; Barkats, M. Intravenous Administration of Self-complementary AAV9 Enables Transgene Delivery to Adult Motor Neurons. Mol. Ther. 2009, 17, 1187–1196. [Google Scholar] [CrossRef]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; et al. T Lymphocyte-Directed Gene Therapy for ADA− SCID: Initial Trial Results After 4 Years. Science 1995, 270, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Bordignon, C.; Notarangelo, L.D.; Nobili, N.; Ferrari, G.; Casorati, G.; Panina, P.; Mazzolari, E.; Maggioni, D.; Rossi, C.; Servida, P.; et al. Gene Therapy in Peripheral Blood Lymphocytes and Bone Marrow for ADA− Immunodeficient Patients. Science 1995, 270, 470–475. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; Basile, G.D.S.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.-L.; et al. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Herzog, R.W. Gene Therapy for SCID-X1: Round 2. Mol. Ther. 2010, 18, 1891. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hammer, M.J.; Eckardt, P.; Barton-Burke, M. Informed Consent: A Clinical Trials Perspective. Oncol. Nurs. Forum 2016, 43, 694–696. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, K.; Aouida, M.; El-Agnaf, O. Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors. Int. J. Mol. Sci. 2020, 21, 7353. [Google Scholar] [CrossRef]
- Javed, H.; Menon, S.A.; Al-Mansoori, K.M.; Al-Wandi, A.; Majbour, N.K.; Ardah, M.T.; Varghese, S.; Vaikath, N.N.; Haque, M.E.; Azzouz, M.; et al. Development of Nonviral Vectors Targeting the Brain as a Therapeutic Approach For Parkinson’s Disease and Other Brain Disorders. Mol. Ther. 2016, 24, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.W.; Son, S.; Jang, J.; Youn, H.; Lee, S.; Lee, D.; Lee, Y.-S.; Jeong, J.M.; Kim, W.J.; Lee, D.S. A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA. Biomaterials 2011, 32, 4968–4975. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; A Lawlor, P.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 2007, 369, 2097–2105. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef]
- Niethammer, M.; Tang, C.C.; LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Sarkar, A.; et al. Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson’s disease. JCI Insight 2017, 2, e90133. [Google Scholar] [CrossRef] [Green Version]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; VanBrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009, 73, 1662–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christine, C.W.; Bankiewicz, K.S.; Van Laar, A.D.; Richardson, R.M.; Ravina, B.; Kells, A.P.; Boot, B.; Martin, A.J.; Nutt, J.; Ms, M.E.T.; et al. Magnetic resonance imaging–guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann. Neurol. 2019, 85, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christine, C.W.; Richardson, R.M.; Van Laar, A.D.; Thompson, M.E.; Fine, E.M.; Khwaja, O.S.; Li, C.; Liang, G.S.; Meier, A.; Roberts, E.W.; et al. Safety of AADC Gene Therapy for Moderately Advanced Parkinson Disease. Neurology 2022, 98, e40–e50. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Prakash, N.; Simuni, T. Clinical trial highlights: 1. gene therapy for parkinson’s, 2. phase 3 study in focus—intec pharma’s accordion pill, 3. clinical trials resources. J. Park. Dis. 2019, 9, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Mittermeyer, G.; Christine, C.W.; Rosenbluth, K.H.; Baker, S.L.; Starr, P.; Larson, P.; Kaplan, P.L.; Forsayeth, J.; Aminoff, M.J.; Bankiewicz, K.S. Long-Term Evaluation of a Phase 1 Study of AADC Gene Therapy for Parkinson’s Disease. Hum. Gene Ther. 2012, 23, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Azzouz, M.; Martin-Rendon, E.; Barber, R.D.; Mitrophanous, K.A.; Carter, E.E.; Rohll, J.B.; Kingsman, S.M.; Kingsman, A.J.; Mazarakis, N.D. Multicistronic Lentiviral Vector-Mediated Striatal Gene Transfer of Aromatic l-Amino Acid Decarboxylase, Tyrosine Hydroxylase, and GTP Cyclohydrolase I Induces Sustained Transgene Expression, Dopamine Production, and Functional Improvement in a Rat Model of Parkinson’s Disease. J. Neurosci. 2002, 22, 10302–10312. [Google Scholar] [CrossRef] [Green Version]
- Palfi, S.; Gurruchaga, J.M.; Ralph, G.S.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef]
- Marks, W.J.; Ostrem, J.L.; Verhagen, L.; A Starr, P.; Larson, P.S.; Bakay, R.A.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2–neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Baron, M.S.; Wichmann, T.; Ma, D.; Delong, M.R. Effects of Transient Focal Inactivation of the Basal Ganglia in Parkinsonian Primates. J. Neurosci. 2002, 22, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Emborg, M.E.; Carbon, M.; Holden, J.E.; During, M.J.; Ma, Y.; Tang, C.; Moirano, J.M.; Fitzsimons, H.; Roitberg, B.Z.; Tüccar, E.; et al. Subthalamic Glutamic Acid Decarboxylase Gene Therapy: Changes in Motor Function and Cortical Metabolism. J. Cereb. Blood Flow Metab. 2007, 27, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Kaplitt, M.G.; Fitzsimons, H.L.; Zuzga, D.S.; Liu, Y.; Oshinsky, M.L.; During, M.J. Subthalamic GAD Gene Therapy in a Parkinson’s Disease Rat Model. Science 2002, 298, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Leff, S.; Spratt, S.; Snyder, R.; Mandel, R. Long-term restoration of striatal l-aromatic amino acid decarboxylase activity using recombinant adeno-associated viral vector gene transfer in a rodent model of Parkinson’s disease. Neuroscience 1999, 92, 185–196. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Eberling, J.L.; Kohutnicka, M.; Jagust, W.; Pivirotto, P.; Bringas, J.; Cunningham, J.; Budinger, T.F.; Harvey-White, J. Convection-Enhanced Delivery of AAV Vector in Parkinsonian Monkeys; In Vivo Detection of Gene Expression and Restoration of Dopaminergic Function Using Pro-drug Approach. Exp. Neurol. 2000, 164, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankiewicz, K.S.; Forsayeth, J.; Eberling, J.L.; Sanchez-Pernaute, R.; Pivirotto, P.; Bringas, J.; Herscovitch, P.; Carson, R.E.; Eckelman, W.; Reutter, B.; et al. Long-Term Clinical Improvement in MPTP-Lesioned Primates after Gene Therapy with AAV-hAADC. Mol. Ther. 2006, 14, 564–570. [Google Scholar] [CrossRef]
- Muramatsu, S.-I.; Fujimoto, K.-I.; Kato, S.; Mizukami, H.; Asari, S.; Ikeguchi, K.; Kawakami, T.; Urabe, M.; Kume, A.; Sato, T.; et al. A Phase I Study of Aromatic L-Amino Acid Decarboxylase Gene Therapy for Parkinson’s Disease. Mol. Ther. 2010, 18, 1731–1735. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, W.S.; Richardson, R.M.; Kells, A.P.; Lamarre, C.; Bringas, J.; Pivirotto, P.; Salegio, E.A.; DeArmond, S.J.; Forsayeth, J.; Bankiewicz, K.S. Safety and Tolerability of Magnetic Resonance Imaging-Guided Convection-Enhanced Delivery of AAV2-hAADC with a Novel Delivery Platform in Nonhuman Primate Striatum. Hum. Gene Ther. 2012, 23, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Collier, T.J.; Sortwell, C.E. Therapeutic potential of nerve growth factors in Parkinson’s disease. Drugs Aging 1999, 14, 261–287. [Google Scholar] [CrossRef]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Bartus, R.T.; Herzog, C.D.; Chu, Y.; Wilson, A.; Brown, L.; Siffert, J.; Johnson, E.M., Jr.; Olanow, C.W.; Mufson, E.J.; Kordower, J.H. Bioactivity of AAV2-neurturin gene therapy (CERE-120): Differences between Parkinson’s disease and nonhuman primate brains. Mov. Disord. 2010, 26, 27–36. [Google Scholar] [CrossRef]
- Bartus, R.T.; Baumann, T.L.; Siffert, J.; Herzog, C.D.; Alterman, R.; Boulis, N.; Turner, D.A.; Stacy, M.; Lang, A.E.; Lozano, A.M.; et al. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 2013, 80, 1698–1701. [Google Scholar] [CrossRef] [Green Version]
- Georgievska, B. Aberrant Sprouting and Downregulation of Tyrosine Hydroxylase in Lesioned Nigrostriatal Dopamine Neurons Induced by Long-Lasting Overexpression of Glial Cell Line Derived Neurotrophic Factor in the Striatum by Lentiviral Gene Transfer. Exp. Neurol. 2002, 177, 461–474. [Google Scholar] [CrossRef]
- Rosenblad, C.; Georgievska, B.; Kirik, D. Long-term striatal overexpression of GDNF selectively downregulates tyrosine hydroxylase in the intact nigrostriatal dopamine system. Eur. J. Neurosci. 2003, 17, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Harvey, B.K.; Hoffman, A.; Wang, Y.; Chiang, Y.; Lupica, C. MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J. 2007, 22, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Kells, A.P.; Huang, E.; Lee, H.S.; Hadaczek, P.; Beyer, J.; Bringas, J.; Pivirotto, P.; Penticuff, J.; Eberling, J.; et al. Safety Evaluation of AAV2-GDNF Gene Transfer into the Dopaminergic Nigrostriatal Pathway in Aged and Parkinsonian Rhesus Monkeys. Hum. Gene Ther. 2009, 20, 1627–1640. [Google Scholar] [CrossRef]
- Quintino, L.; Manfré, G.; Wettergren, E.E.; Namislo, A.; Isaksson, C.; Lundberg, C. Functional Neuroprotection and Efficient Regulation of GDNF Using Destabilizing Domains in a Rodent Model of Parkinson’s Disease. Mol. Ther. 2013, 21, 2169–2180. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Björklund, T.; Lundberg, C.; Kirik, D.; Wandless, T.J. A General Chemical Method to Regulate Protein Stability in the Mammalian Central Nervous System. Chem. Biol. 2010, 17, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Decressac, M.; Kadkhodaei, B.; Mattsson, B.; Laguna, A.; Perlmann, T.; Björklund, A. α-Synuclein–Induced Down-Regulation of Nurr1 Disrupts GDNF Signaling in Nigral Dopamine Neurons. Sci. Transl. Med. 2012, 4, 163ra156. [Google Scholar] [CrossRef]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020, 181, 590–603.e16. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Choong, C.-J.; Nakamori, M.; Hayakawa, H.; Nishiyama, K.; Kasahara, Y.; Baba, K.; Nagata, T.; Yokota, T.; Tsuda, H.; et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting α-synuclein as a novel therapy for Parkinson’s disease. Sci. Rep. 2019, 9, 7567. [Google Scholar] [CrossRef]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [Green Version]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In Vivo RNAi-Mediated alpha-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.-J.; Choi, M.Y.; Im, H. Aggregation-defective α-synuclein mutants inhibit the fibrillation of Parkinson’s disease-linked α-synuclein variants. Biochem. Biophys. Res. Commun. 2009, 386, 165–169. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Bassett, A.R.; Wood, M.J.A. Precision Modulation of Neurodegenerative Disease-Related Gene Expression in Human iPSC-Derived Neurons. Sci. Rep. 2016, 6, 28420. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, P.N.; Chen, S.G.; Wilson-Delfosse, A.L. Leucine-rich repeat kinase 2 (LRRK2): A key player in the pathogenesis of Parkinson’s disease. J. Neurosci. Res. 2009, 87, 1283–1295. [Google Scholar] [CrossRef] [Green Version]
- Ness, D.; Ren, Z.; Gardai, S.; Sharpnack, D.; Johnson, V.J.; Brennan, R.J.; Brigham, E.F.; Olaharski, A.J. Leucine-Rich Repeat Kinase 2 (LRRK2)-Deficient Rats Exhibit Renal Tubule Injury and Perturbations in Metabolic and Immunological Homeostasis. PLoS ONE 2013, 8, e66164. [Google Scholar] [CrossRef] [Green Version]
- Fuji, R.N.; Flagella, M.; Baca, M.; Baptista, M.A.S.; Brodbeck, J.; Chan, B.K.; Fiske, B.K.; Honigberg, L.; Jubb, A.M.; Katavolos, P.; et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015, 7, 273ra15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.T.; John, N.; Delic, V.; Ikeda-Lee, K.; Kim, A.; Weihofen, A.; Swayze, E.; Kordasiewicz, H.B.; West, A.; Volpicelli-Daley, L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol. Ther.-Nucleic Acids 2017, 8, 508–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef] [Green Version]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.T.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef] [Green Version]
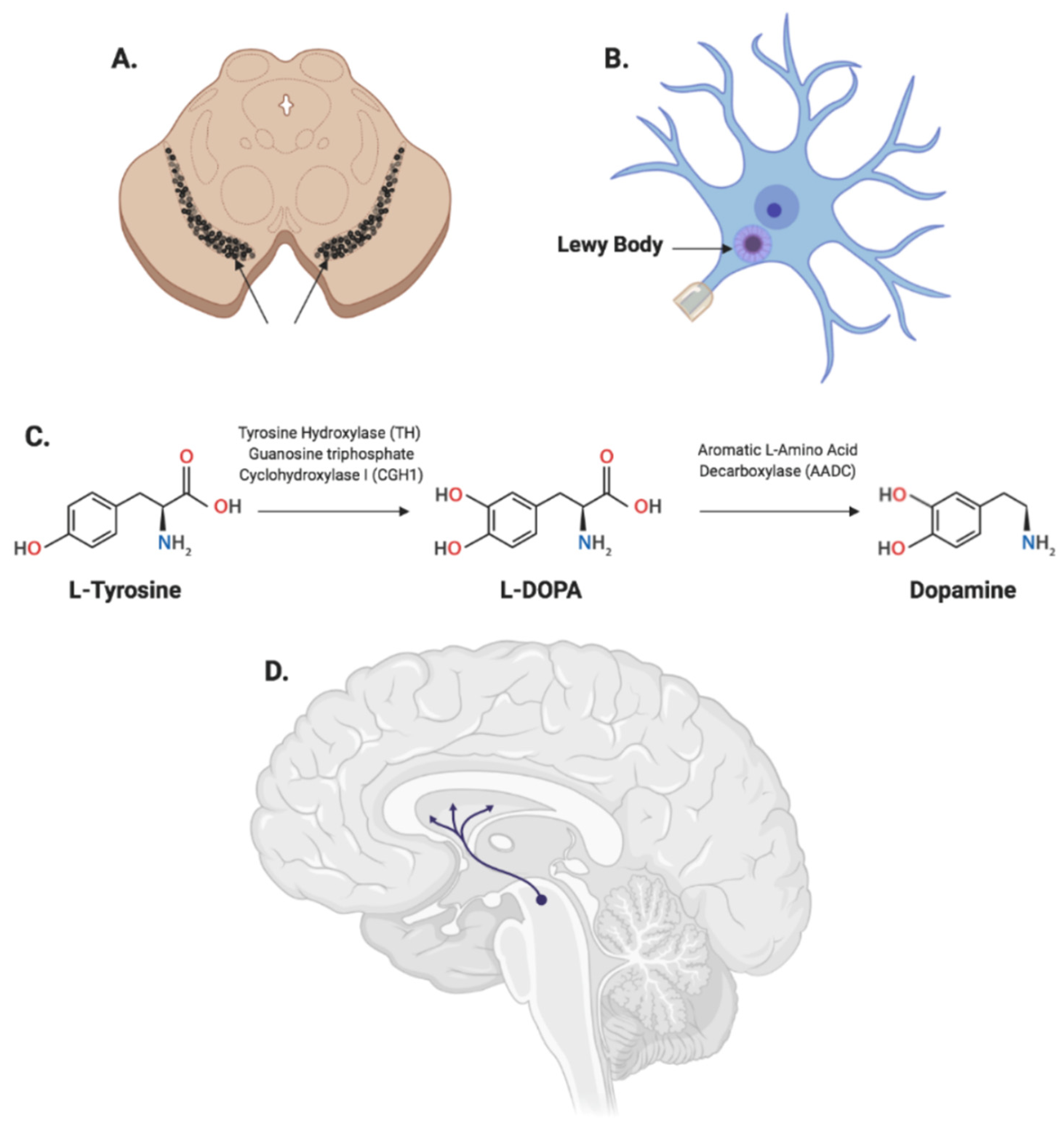

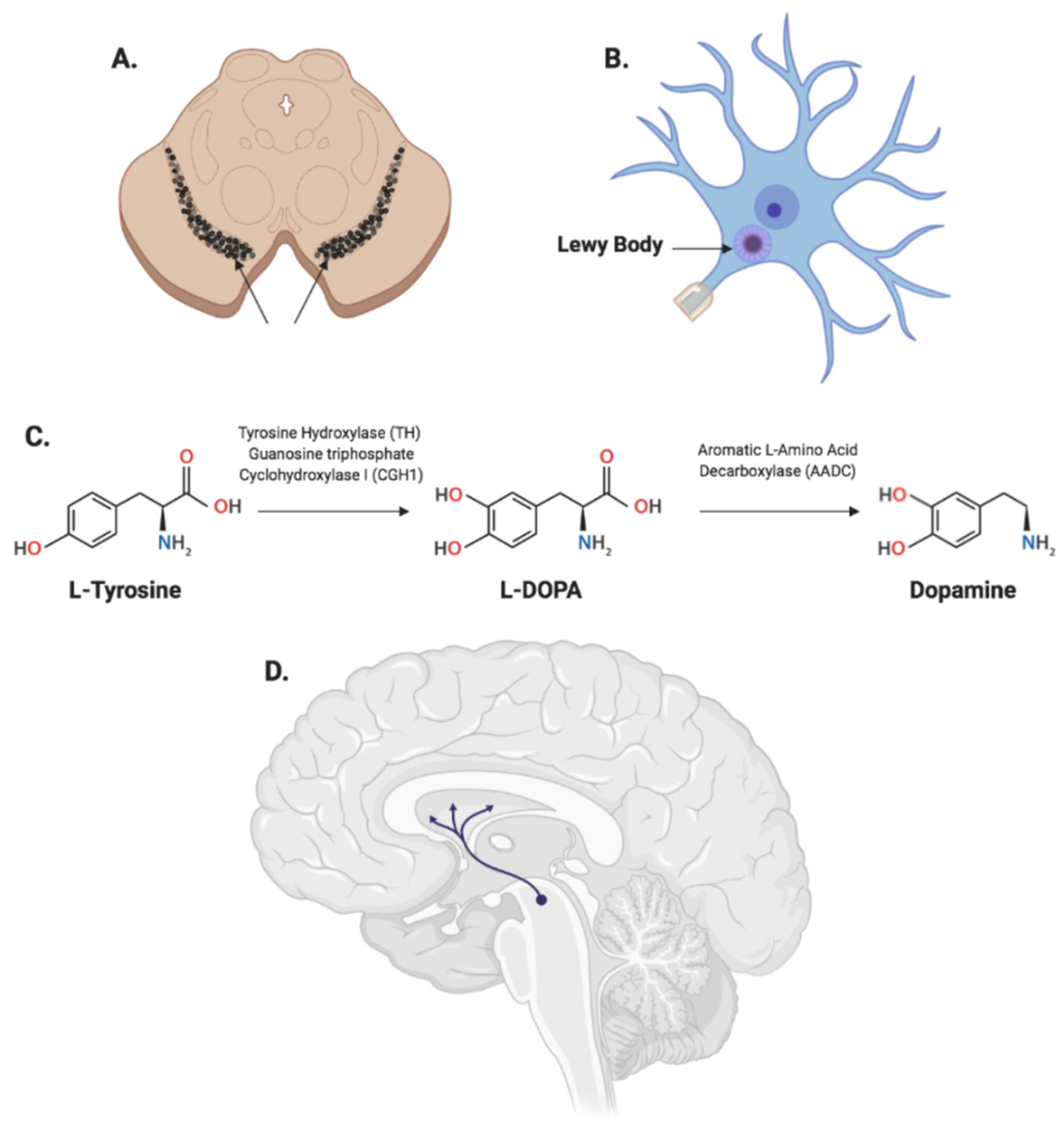{kind=link}
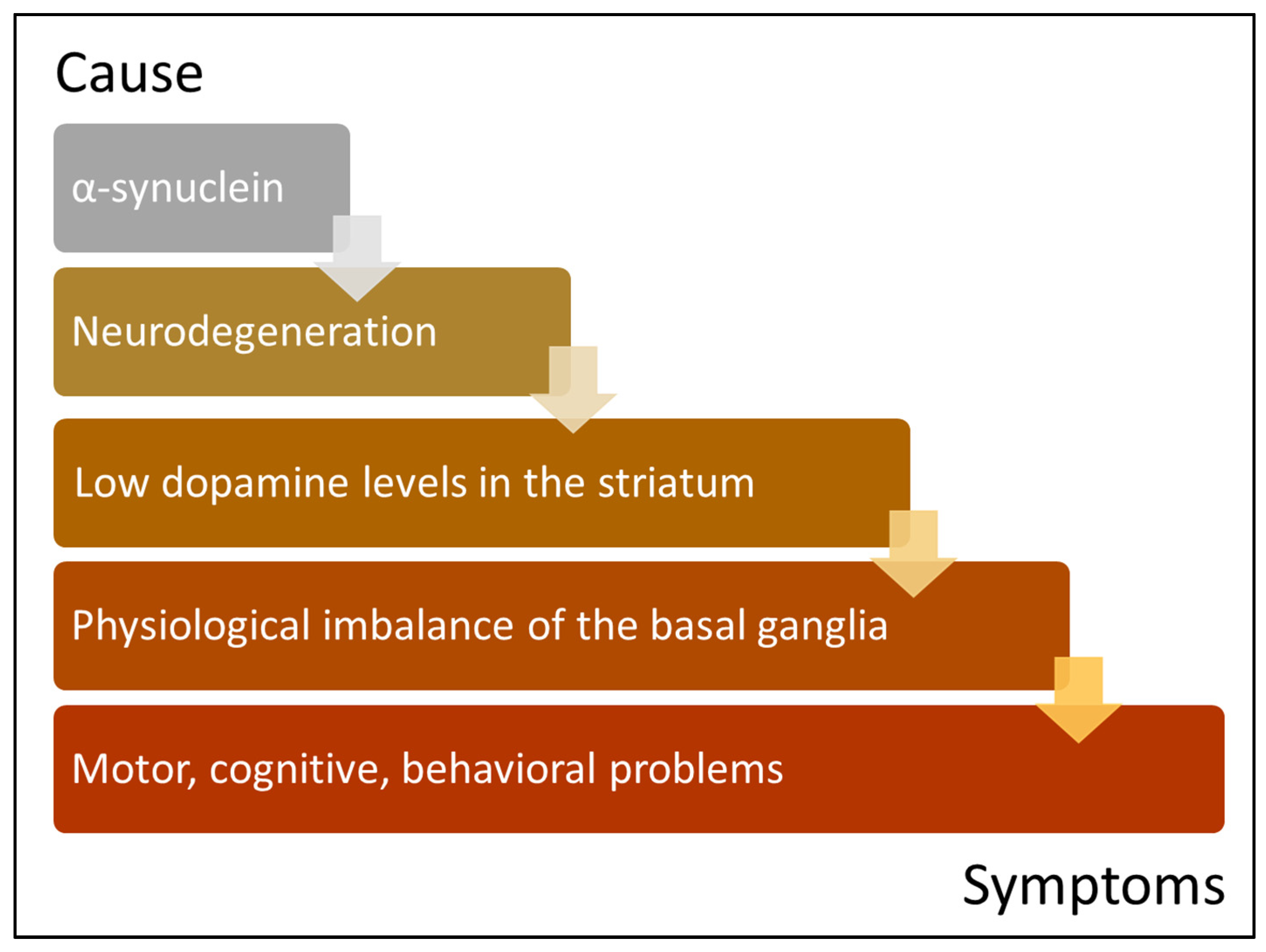
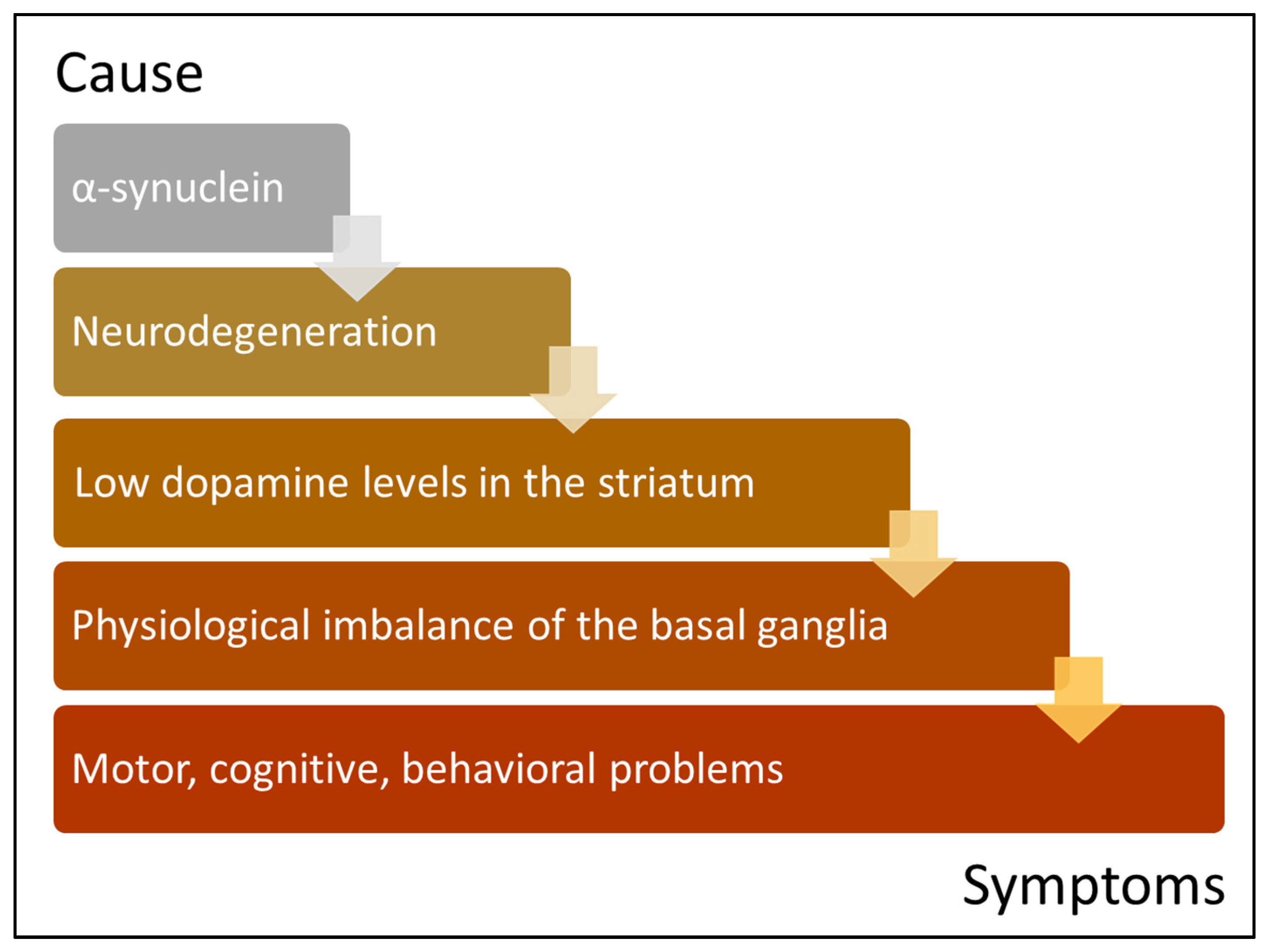{kind=link}
| Mode of Delivery | Phase | Duration | Primary Endpoint | Outcome(s) | Reference(s) | |
|---|---|---|---|---|---|---|
| Restoring the physiological balance of the basal ganglia | ||||||
| GABA | ||||||
| AAV2-GAD | IP (subthalamic nucleus) | I | 2003–2005 | Safety | UPDRS improvement persisted for 12 months, and reduced thalamic activity as assessed by 18F-FDG PET in all patients. | NCT00195143 [78] |
| AAV2-GAD | IP (subthalamic nucleus) | II | 2008–2010 | Disease severity and progression | UPDRS improvement over sham control group, diminished PD symptoms with no adverse events for 12 months in all patients. No improvement over conventional PD treatment. | NCT00643890 [79,80] |
| Enhancing dopamine synthesis | ||||||
| AADC | ||||||
| AAV2-AADC | IP (striatum) | I | 2004–2013 | Safety and tolerability | Clinical improvements in first 12 months in all patients followed by slow deterioration. One symptomatic and two asymptomatic intracranial hemorrhages followed. Increased ON time and reduced OFF time. Increased AADC activity as measured by 18FMT PET. | NCT00229736 [81] |
| AAV2-AADC | IP (striatum) | I | 2013–2020 | Safety and tolerability | Stable increase in AADC activity as measured by 18F-Levodopa PET at 6 months, and clinical improvements at 12 months with no serious adverse events in all patients. Improvements were stable or improved at 12, 24 and 36 months. | NCT01973543 [82,83] |
| AAV2-AADC | IP (putamen) | I/II | 2015–2018 | Safety | N/A | NCT02418598 |
| AAV2-AADC | IP (striatum) | I | 2017–2021 | Safety and suicide risk | Increase in AADC activity as measured by PET and increase in ON time without troublesome dyskinesia. | NCT03065192 |
| AAV2-AADC | IP (striatum) | II | 2018–2022 | Change in ON time without troublesome dyskinesia | N/A | NCT03562494 [84] |
| AADC, TH, CGH1 | ||||||
| Lentivirus-TH/AADC/CGH1 | IP (striatum) | I/II | 2008–2012 | Safety | Significant UPDRS improvement at 6 months. Mild to moderate but no serious drug-related adverse events were reported during the first 12 months. | NCT00627588 [85] |
| Lentivirus-TH/AADC/CGH1 | IP (striatum) | I/II | 2011–2022 | Long-term safety and tolerability | N/A | NCT01856439 [85] |
| Neuroprotection | ||||||
| GDNF | ||||||
| AAV2-GDNF | IP (putamen) | I | 2013–2022 | Safety and tolerability | Stable motor scores throughout study period. Increase in 18F-DOPA uptake as assessed by PET at 6 and 18 months in 10/13 and 12/13 patients, respectively. | NCT01621581 [84] |
| AAV2-GDNF | IP (putamen) | I | 2020–2026 | Safety and tolerability | N/A | NCT04167540 |
| NRTN | ||||||
| AAV2-NTN | IP (putamen) | I | 2005–2007 | Safety and tolerability | Improvement of 14 points in off-medication motor score of UPDRS and increase of 2.3 h in ON time without troublesome dyskinesia at 12 months. Non-significant improvements in several secondary measures. No change in 18F-levodopa uptake as assessed by PET | NCT00252850 [86] |
| AAV2-NTN | IP (putamen) | II | 2006–2008 | Disease severity and progression | No significant improvement in UPDRS over sham surgery control group at 12 months. Serious adverse events in 13/38 treated and in 4/20 control individuals. Three patients in the treated group and two in the sham surgery group developed tumors. | NCT00400634 [87] |
| AAV2-NTN | IP (substantia nigra + putamen) | I/II | 2009–2017 | Disease severity and progression | No serious adverse events in all patients. | NCT00985517 [88] |
| Targeting disease genes | ||||||
| AAV9-Gcase | IC | I/II | 2020–2027 | Safety and immunogenicity | N/A | NCT04127578 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalaby, K.E.; El-Agnaf, O.M.A. Gene-Based Therapeutics for Parkinson’s Disease. Biomedicines 2022, 10, 1790. https://doi.org/10.3390/biomedicines10081790
Shalaby KE, El-Agnaf OMA. Gene-Based Therapeutics for Parkinson’s Disease. Biomedicines. 2022; 10(8):1790. https://doi.org/10.3390/biomedicines10081790
Chicago/Turabian StyleShalaby, Karim E., and Omar M. A. El-Agnaf. 2022. "Gene-Based Therapeutics for Parkinson’s Disease" Biomedicines 10, no. 8: 1790. https://doi.org/10.3390/biomedicines10081790
APA StyleShalaby, K. E., & El-Agnaf, O. M. A. (2022). Gene-Based Therapeutics for Parkinson’s Disease. Biomedicines, 10(8), 1790. https://doi.org/10.3390/biomedicines10081790