
Tracking Clonal Evolution of Multiple Myeloma Using Targeted Next-Generation DNA Sequencing

 , , , , , , , , , , ,
, , , , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Targeted Sequencing
2.2. Cytogenetic Evaluation
2.3. Clinical Endpoints
2.4. Statistical Analysis
3. Results
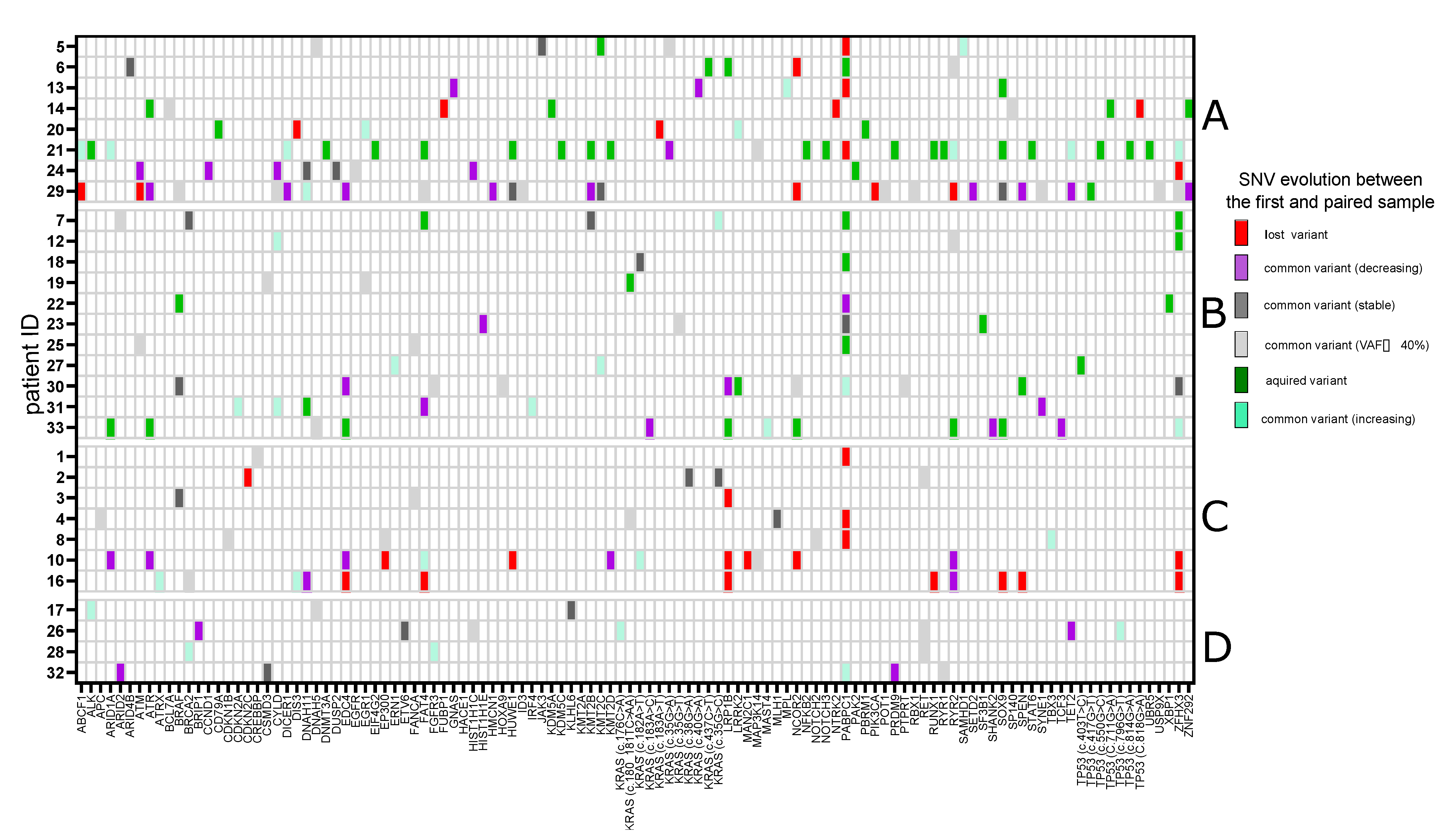
3.1. Single-Nucleotide Variants
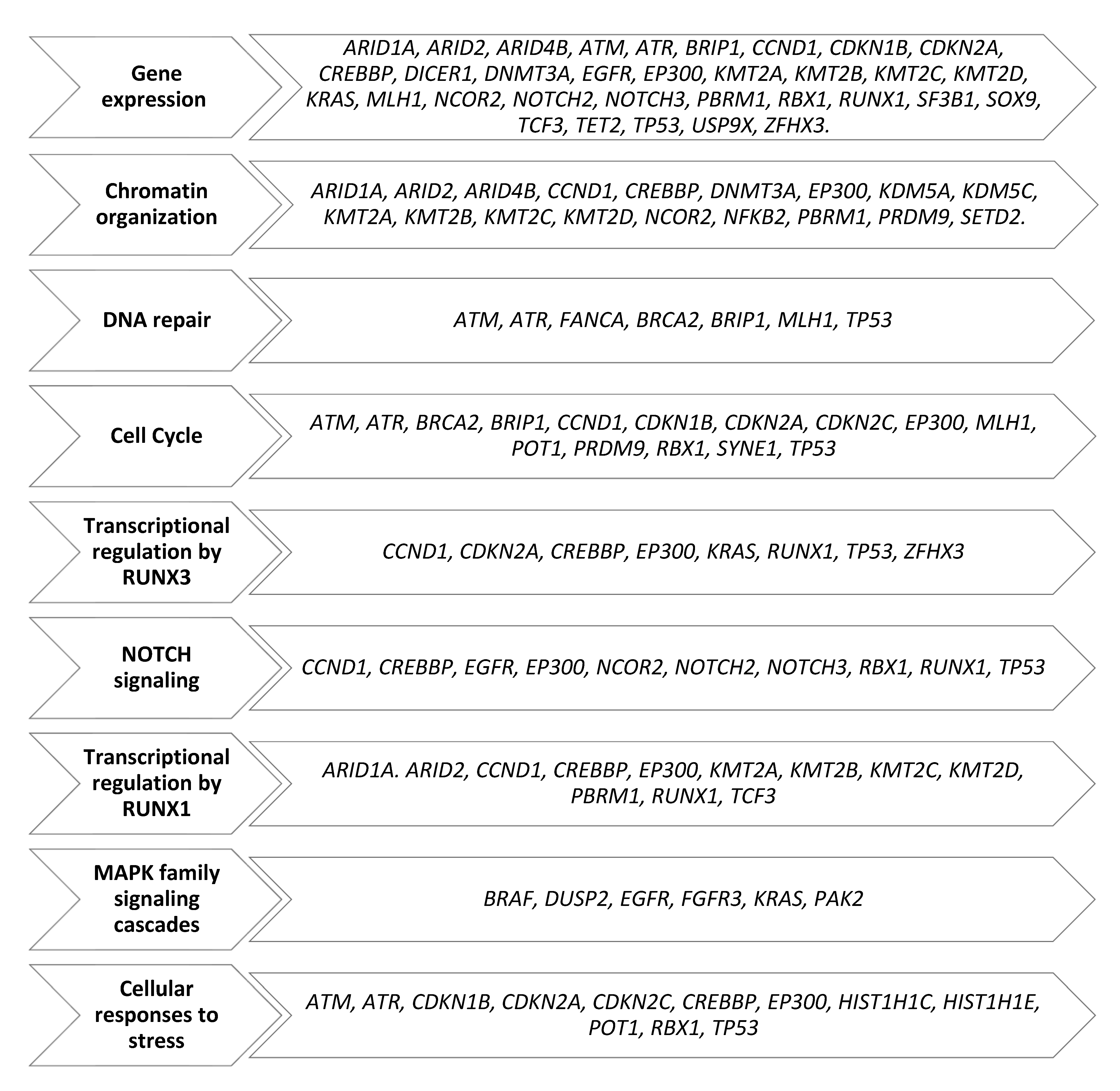
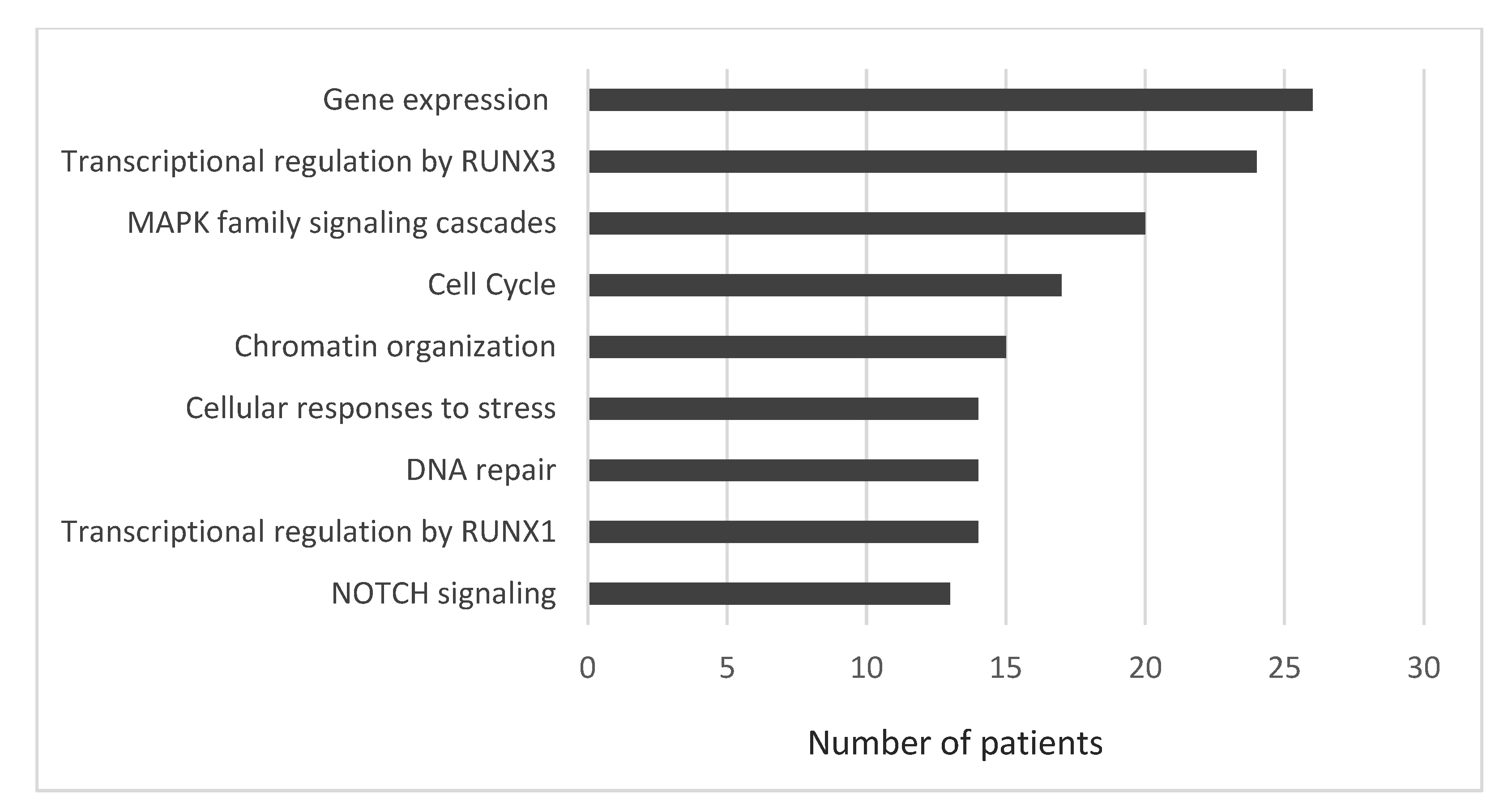
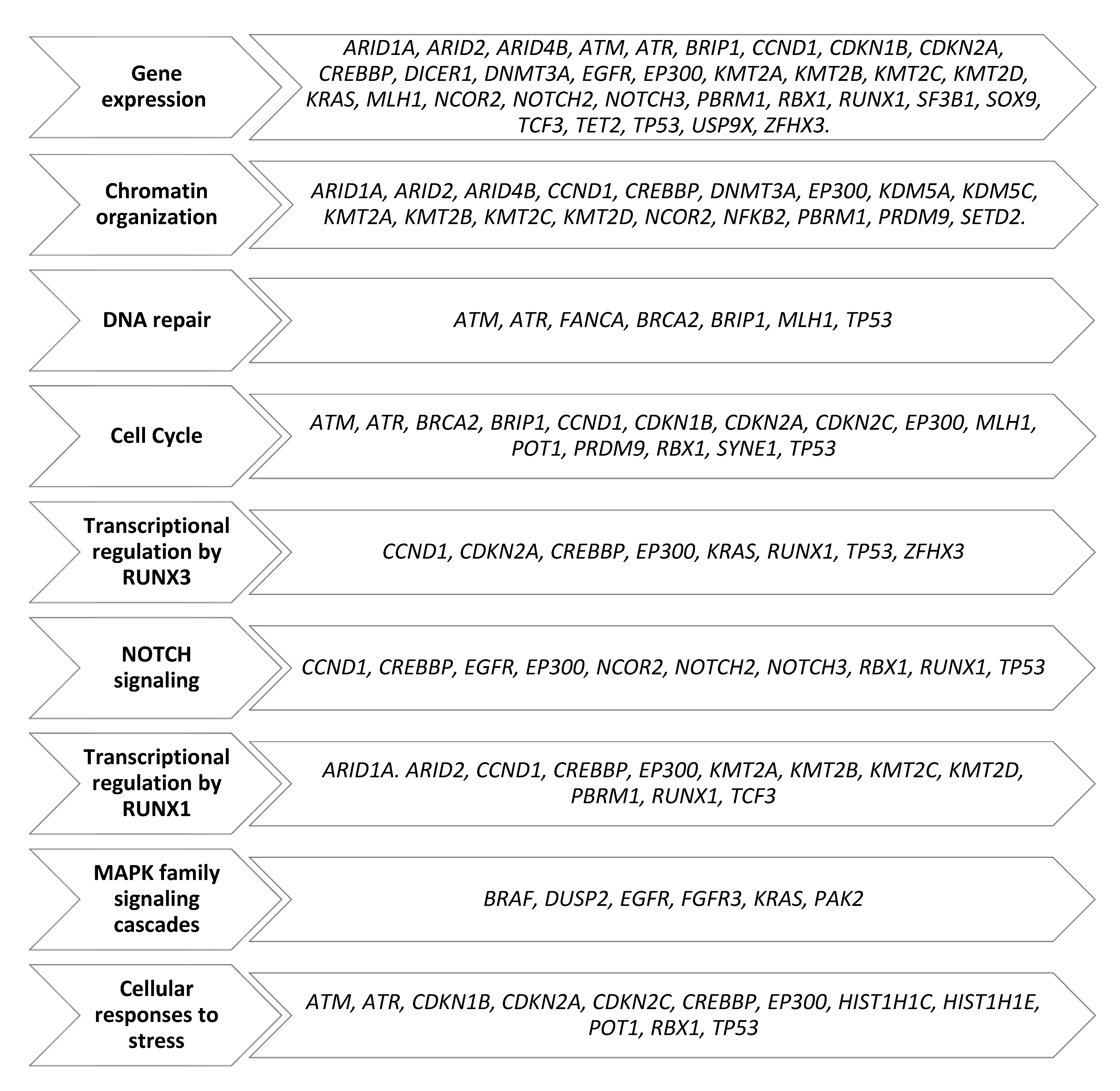
3.2. Single-Nucleotide Variants of Druggable Genes
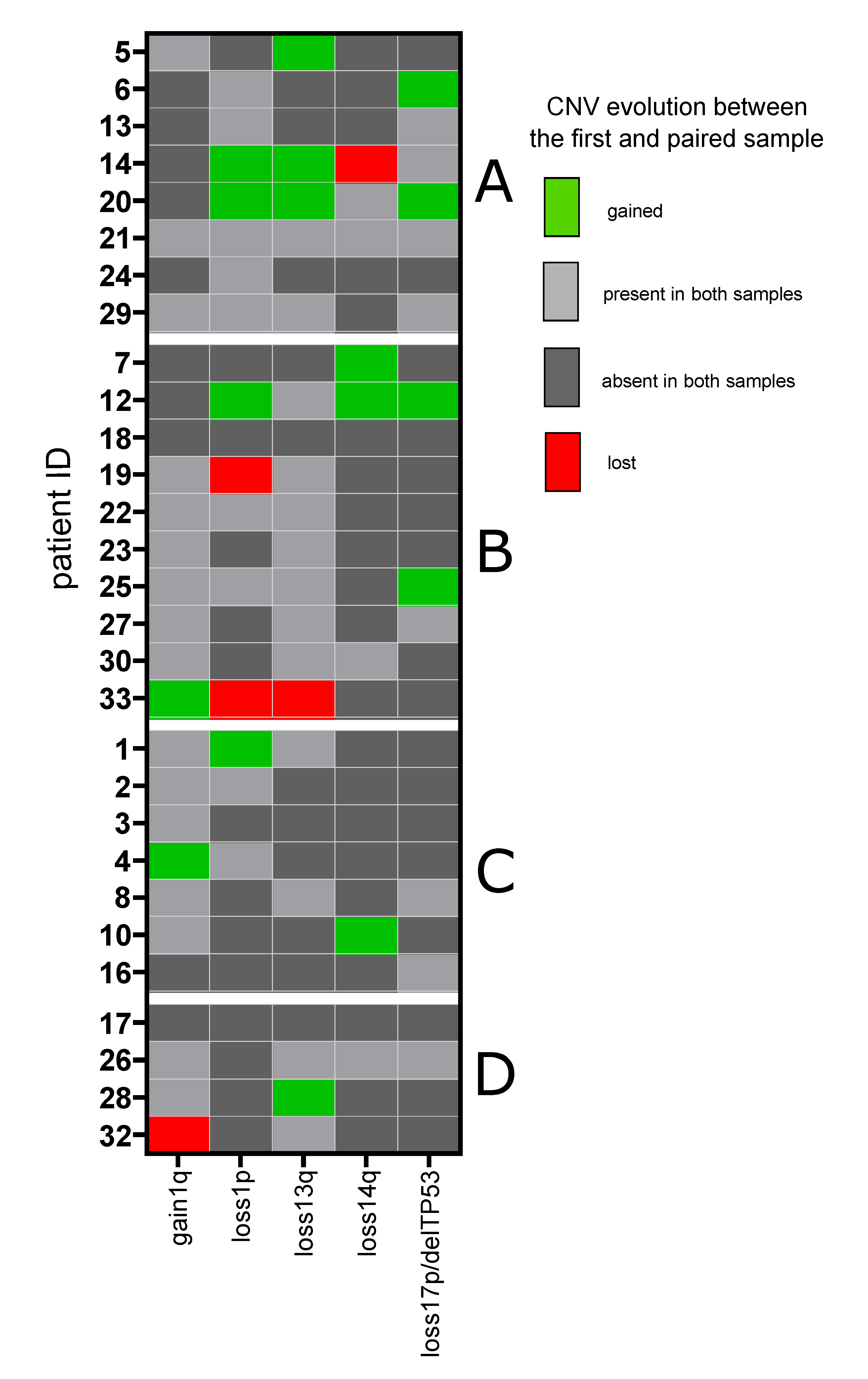
3.3. Copy Number Variants
3.4. IgH Translocations
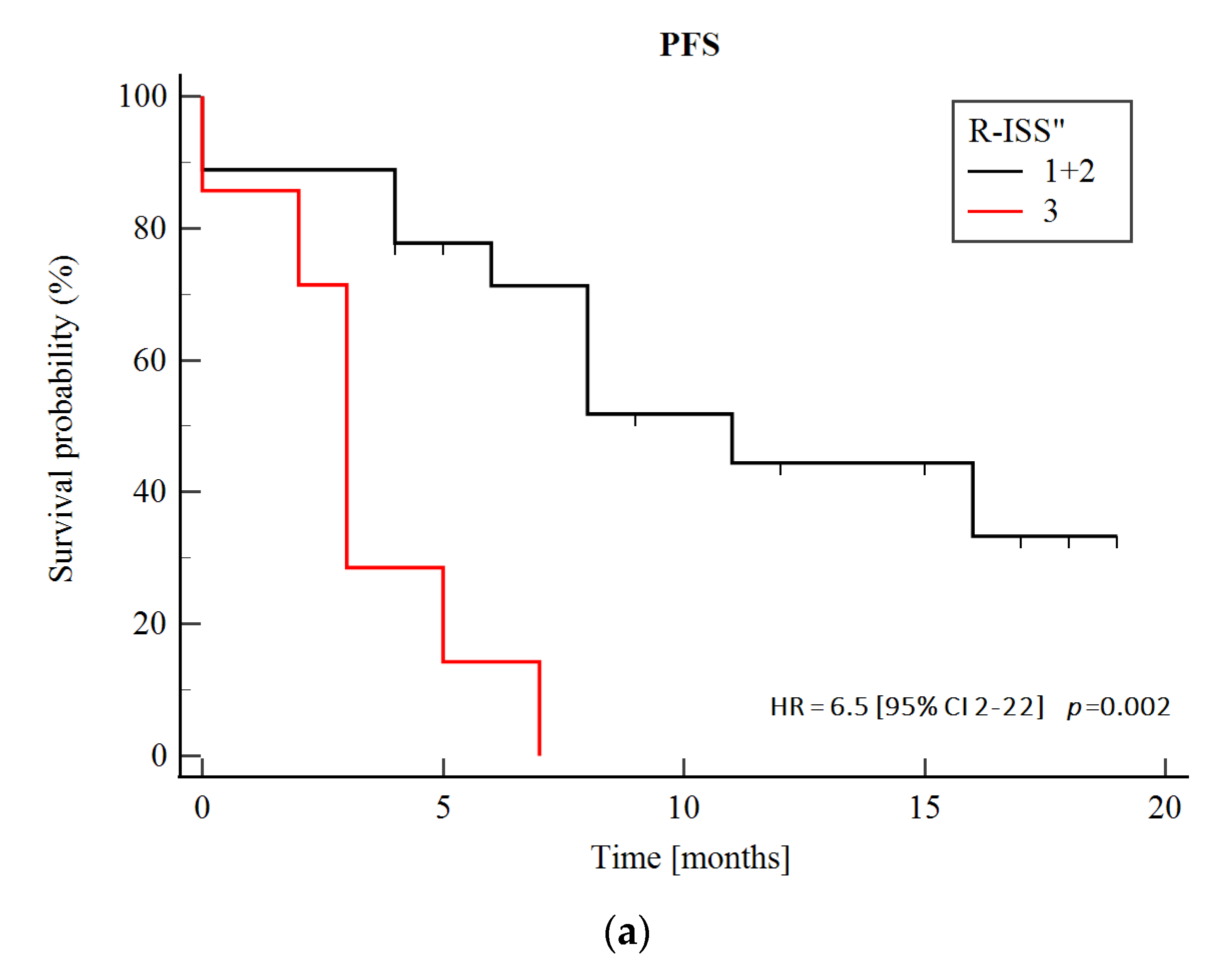
3.5. Prognostic Significance of Genetic Abnormalities
3.6. Biallelic Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V. Multiple Myeloma: 2020 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [Green Version]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial Genomic Heterogeneity in Multiple Myeloma Revealed by Multi-Region Sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Wilson, S.H.; et al. Single-Cell Genetic Analysis Reveals the Composition of Initiating Clones and Phylogenetic Patterns of Branching and Parallel Evolution in Myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal Selection and Double-Hit Events Involving Tumor Suppressor Genes Underlie Relapse in Myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal Heterogeneity is a Critical Early Event in the Development of Myeloma and Precedes the Development of Clinical Symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Cody Ashby, T.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Corre, J.; Cleynen, A.; Robiou du Pont, S.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple Myeloma Clonal Evolution in Homogeneously Treated Patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic Landscape and Chronological Reconstruction of Driver Events in Multiple Myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Perzyński, A.; Jamroziak, K.; Głodkowska-Mrówka, E. Clonal Evolution of Multiple Myeloma—Clinical and Diagnostic Implications. Diagnostics 2021, 11, 1534. [Google Scholar] [CrossRef]
- Farswan, A.; Jena, L.; Kaur, G.; Gupta, A.; Gupta, R.; Rani, L.; Sharma, A.; Kumar, L. Branching Clonal Evolution Patterns Predominate Mutational Landscape in Multiple Myeloma. Am. J. Cancer Res. 2021, 11, 5659. [Google Scholar] [PubMed]
- Ziccheddu, B.; Biancon, G.; Bagnoli, F.; de Philippis, C.; Maura, F.; Rustad, E.H.; Dugo, M.; Devecchi, A.; de Cecco, L.; Sensi, M.; et al. Integrative Analysis of the Genomic and Transcriptomic Landscape of Double-Refractory Multiple Myeloma. Blood Adv. 2020, 4, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Kortüm, K.M.; Langer, C.; Monge, J.; Bruins, L.; Zhu, Y.X.; Shi, C.X.; Jedlowski, P.; Egan, J.B.; Ojha, J.; Bullinger, L.; et al. Longitudinal Analysis of 25 Sequential Sample-Pairs Using a Custom Multiple Myeloma Mutation Sequencing Panel (M3P). Ann. Hematol. 2015, 94, 1205–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.R.; Weinhold, N.; Ashby, C.; Walker, B.A.; Wardell, C.; Pawlyn, C.; Rasche, L.; Melchor, L.; Cairns, D.A.; Gregory, W.M.; et al. Clonal Evolution in Myeloma: The Impact of Maintenance Lenalidomide and Depth of Response on the Genetics and Sub-Clonal Structure of Relapsed Disease in Uniformly Treated Newly Diagnosed Patients. Haematologica 2019, 104, 1440–1450. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Lenive, O.; Mitchell, J.; Jackson, G.; Owen, R.; Drayson, M.; Cook, G.; Jones, J.R.; Pawlyn, C.; Davies, F.E.; et al. Neutral Tumor Evolution in Myeloma is Associated with Poor Prognosis. Blood 2017, 130, 1639–1643. [Google Scholar] [CrossRef]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.J.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C.W. Subclonal Evolution in Disease Progression from MGUS/SMM to Multiple Myeloma Is Characterised by Clonal Stability. Leukemia 2018, 33, 457–468. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Greipp, P.R.; Miguel, J.S.; Dune, B.G.M.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Harousseau, J.L.; Kyle, R.A.; et al. International Staging System for Multiple Myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef]
- Oken, M.M.; Creech, R.H.; Tormey, D.C.; Horton, J.; Davis, T.E.; McFadden, E.T.; Carbone, P.P. Toxicity and Response Criteria of the Eastern Cooperative Oncology Group. Am. J. Clin. Oncol. 1982, 5, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; de Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.T.; Mudie, L.; et al. Analysis of the Genomic Landscape of Multiple Myeloma Highlights Novel Prognostic Markers and Disease Subgroups. Leukemia 2018, 32, 2604–2616. [Google Scholar] [CrossRef]
- Hoang, P.H.; Dobbins, S.E.; Cornish, A.J.; Chubb, D.; Law, P.J.; Kaiser, M.; Houlston, R.S. Whole-Genome Sequencing of Multiple Myeloma Reveals Oncogenic Pathways Are Targeted Somatically through Multiple Mechanisms. Leukemia 2018, 32, 2459–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Li, Y.; Sathiaseelan, V.; Raine, K.; Jones, D.; Ganly, P.; Cocito, F.; Bignell, G.; Chapman, M.A.; Sperling, A.S.; et al. A DNA Target-Enrichment Approach to Detect Mutations, Copy Number Changes and Immunoglobulin Translocations in Multiple Myeloma. Blood Cancer J. 2016, 6, e467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botta, C.; di Martino, M.T.; Ciliberto, D.; Cucè, M.; Correale, P.; Rossi, M.; Tagliaferri, P.; Tassone, P. A Gene Expression Inflammatory Signature Specifically Predicts Multiple Myeloma Evolution and Patients Survival. Blood Cancer J. 2016, 6, e511. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Douville, C.; Carter, H.; Kim, R.; Niknafs, N.; Diekhans, M.; Stenson, P.D.; Cooper, D.N.; Ryan, M.; Karchin, R. CRAVAT: Cancer-Related Analysis of Variants Toolkit. Bioinformatics 2013, 29, 647–648. [Google Scholar] [CrossRef]
- Masica, D.L.; Douville, C.; Tokheim, C.; Bhattacharya, R.; Kim, R.G.; Moad, K.; Ryan, M.C.; Karchin, R. CRAVAT 4: Cancer-Related Analysis of Variants Toolkit. Cancer Res. 2017, 77, e35–e38. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-Specific High-Throughput Annotation of Somatic Mutations: Computational Prediction of Driver Missense Mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.C.; Kim, D.; Carter, H.; Diekhans, M.; Ryan, M.C.; Karchin, R. CHASM and SNVBox: Toolkit for Detecting Biologically Important Single Nucleotide Mutations in Cancer. Bioinformatics 2011, 27, 2147–2148. [Google Scholar] [CrossRef] [PubMed]
- Darbyshire, M.; du Toit, Z.; Rogers, M.F.; Gaunt, T.R.; Campbell, C. Estimating the Frequency of Single Point Driver Mutations across Common Solid Tumours. Sci. Rep. 2019, 9, 13452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional Consequences of Cancer-Associated Amino Acid Substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, D.; Tanyalcin, I.; FertCrossed, J.S.D.; Gazzo, A.; Orlando, G.; Lenaerts, T.; Rooman, M.; Vranken, W. DEOGEN2: Prediction and Interactive Visualization of Single Amino Acid Variant Deleteriousness in Human Proteins. Nucleic Acids Res. 2017, 45, W201–W206. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the Clinical Impact of Human Mutation with Deep Neural Networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT Missense Predictions for Genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, S.M.; Lee, Y.; Jeong, D.; Yun, J.; Ryu, S.; Yoon, S.S.; Ahn, Y.O.; Hwang, S.M.; Lee, D.S. Prognostic Value of Integrated Cytogenetic, Somatic Variation, and Copy Number Variation Analyses in Korean Patients with Newly Diagnosed Multiple Myeloma. PLoS ONE 2021, 16, e0246322. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Aksenova, A.Y.; Zhuk, A.S.; Lada, A.G.; Zotova, I.V.; Stepchenkova, E.I.; Kostroma, I.I.; Gritsaev, S.V.; Pavlov, Y.I. Genome Instability in Multiple Myeloma: Facts and Factors. Cancers 2021, 13, 5949. [Google Scholar] [CrossRef]
- Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells 2020, 9, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 Are Exclusively Associated with Del(17p) in Multiple Myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corre, J.; Perrot, A.; Caillot, D.; Belhadj, K.; Hulin, C.; Leleu, X.; Mohty, M.; Facon, T.; Buisson, L.; do Souto, L.; et al. Del(17p) without TP53 Mutation Confers a Poor Prognosis in Intensively Treated Newly Diagnosed Patients with Multiple Myeloma. Blood 2021, 137, 1192–1195. [Google Scholar] [CrossRef]
- Caprio, C.; Sacco, A.; Giustini, V.; Roccaro, A.M. Epigenetic Aberrations in Multiple Myeloma. Cancers 2020, 12, 2996. [Google Scholar] [CrossRef]
- Barrio, S.; Munawar, U.; Zhu, Y.X.; Giesen, N.; Shi, C.X.; da Viá, M.; Sanchez, R.; Bruins, L.; Demler, T.; Müller, N.; et al. IKZF1/3 and CRL4CRBN E3 Ubiquitin Ligase Mutations and Resistance to Immunomodulatory Drugs in Multiple Myeloma. Haematologica 2020, 105, e237. [Google Scholar] [CrossRef] [Green Version]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.X.; Zhu, Y.X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted Sequencing of Refractory Myeloma Reveals a High Incidence of Mutations in CRBN and Ras Pathway Genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.; Rajkumar, S.V.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Hayman, S.R.; Hwa, Y.L.; Zeldenrust, S.R.; et al. Occurrence and Prognostic Significance of Cytogenetic Evolution in Patients with Multiple Myeloma. Blood Cancer J. 2016, 6, e401. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Painuly, U.; Vincent Rajkumar, S.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; et al. Impact of Acquired Del(17p) in Multiple Myeloma. Blood Adv. 2019, 3, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Perzyński, A.; Bluszcz, A.; Krzywdzińska, A.; Spyra-Górny, Z.; Jakacka, N.; Barankiewicz, J.; Borg, K.; Solarska, I.; Szpila, T.; Puła, B.; et al. The Impact of Cytogenetic Evolution and Acquisition of Del(17p) on the Prognosis of Patients with Multiple Myeloma. Pol. Arch. Intern. Med. 2020, 130, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Qin, X.; Liu, J.; Fan, H.; Yan, W.; Liu, L.; Du, C.; Yu, Z.; Xu, Y.; Hao, M.; et al. Clonal Phylogeny and Evolution of Critical Cytogenetic Aberrations in Multiple Myeloma at Single Cell Level by QM-FISH. Blood Adv. 2021, 6, 441–451. [Google Scholar] [CrossRef]
- George, A.; Turnbull, C. Tumor-Only Sequencing for Oncology Management: Germline-Focused Analysis and Implications. Genes Chromosomes Cancer 2021, 60, 352–357. [Google Scholar] [CrossRef]
- Montgomery, N.D.; Selitsky, S.R.; Patel, N.M.; Hayes, D.N.; Parker, J.S.; Weck, K.E. Identification of Germline Variants in Tumor Genomic Sequencing Analysis. J. Mol. Diagn. 2018, 20, 123–125. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | At the Time of the 1st Sample Collection (n = 30) | At the Time of the Paired Sample Collection (n = 30) | |
|---|---|---|---|
| Age (years), median (range) | 65 (50–77) | 67 (50–82) | |
| Female and male sex | 13 (43%), 17 (57%) | ||
| ECOG score | |||
| 0–1 | 23 (77%) | 23 (77%) | |
| ≥2 | 5 (16%) | 5 (16%) | |
| Not reported | 2 (7%) | 2 (7%) | |
| ISS staging | |||
| I | 7 (23%) | 8 (27%) | |
| II | 13 (43%) | 4 (13%) | |
| III | 9 (30%) | 14 (47%) | |
| Not reported | 1 (3%) | 4 (13%) | |
| R-ISS staging | |||
| I | 3 (10%) | 6 (20%) | |
| II | 23 (77%) | 13 (44%) | |
| III | 3 (10%) | 7 (23%) | |
| Not reported | 1 (3%) | 4 (13%) | |
| Cytogenetics/Copy number variants | |||
| t(4; 14) | 5 (17%) | ||
| 14q32 rearrangement, other a | 8 (27%) | ||
| 14q32 rearrangement, not specified | 1 (3%) | ||
| Detected in the 1st sample (n = 30) | Acquired in the paired sample (n = 30) | ||
| del1p | 10 (33%) | 4 (14%) | |
| gain1q21 | 19 (63%) | 4 (14%) | |
| del17p/17 monosomy | 8 (27%) | 4 (14%) | |
| del14q | 6 (20%) | 3 (10%) | |
| del13q | 13 (43%) | 5 (17%) | |
| Lines of therapy—in total, median (range) | 4 (1–8) | ||
| Lines of therapy, median (range) | Before the 1st sample | Between the 1st and paired samples | After the paired sample |
| 3 (1–6) b | 1 (1–3) | 1 (0–3) | |
| Multiple myeloma therapy | |||
| Exposure to PI (i.e., bortezomib, carfilzomib, ixazomib) | 8 (27%) | 24 (80%) | 12 (40%) |
| PI-refractoriness | 1 (3%) | 11 (37%) | 7 (23%) |
| Exposure to IMiD (i.e., thalidomide, lenalidomide, pomalidomide) | 10 (33%) | 15 (50%) | 10 (33%) |
| IMiD-refractoriness | 7 (23%) | 8 (27%) | 4 (13%) |
| Double-refractoriness to IMiD and PI | 0 | 4 (13%) | 1 (3%) |
| Exposure to cytotoxic agents (e.g., bendamustine, cyclophosphamide, doxorubicin, vincristine, melphalan) | 8 (27%) | 20 (67%) | 10 (33%) |
| Refractory to cytotoxic agents | 2 (7%) | 7 (23%) | 6 (20%) |
| Triple-refractoriness to IMiD, PI and alkylators | 0 | 2 (7%) | 0 |
| Autologous stem cell transplantation | 8 (27%) | 10 (33%) | 1 (3%) |
| Allogeneic stem cell transplantation | 1 (3%) | 1 (3%) | 1 (3%) |
| Patient ID | Sample Type | Gene | cDNA | ACMG Classification According to Varsome [36] |
|---|---|---|---|---|
| 8 | Dx and PD | EP300 | c.736A>G | Uncertain significance |
| 8 | Dx and PD | CDKN1B | c.180G>A | Pathogenic |
| 14 | PD | TP53 | c.711G>A | Pathogenic |
| 25 | PD and PD | ATM | c.6833T>A | Uncertain significance/minor pathogenic evidence |
| 28 | PD | BRCA2 | c.8182G>A | Uncertain significance |
| 29 | Dx and PD | USP9X | c.2939C>G | Uncertain significance/minor pathogenic evidence |
| 30 | Dx and PD | FGFR3 | c.2204G>A | Likely pathogenic |
| 7 | PD | FAT4 | c.10571G>A | Uncertain significance |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomon-Perzyński, A.; Barankiewicz, J.; Machnicki, M.; Misiewicz-Krzemińska, I.; Pawlak, M.; Radomska, S.; Krzywdzińska, A.; Bluszcz, A.; Stawiński, P.; Rydzanicz, M.; et al. Tracking Clonal Evolution of Multiple Myeloma Using Targeted Next-Generation DNA Sequencing. Biomedicines 2022, 10, 1674. https://doi.org/10.3390/biomedicines10071674
Salomon-Perzyński A, Barankiewicz J, Machnicki M, Misiewicz-Krzemińska I, Pawlak M, Radomska S, Krzywdzińska A, Bluszcz A, Stawiński P, Rydzanicz M, et al. Tracking Clonal Evolution of Multiple Myeloma Using Targeted Next-Generation DNA Sequencing. Biomedicines. 2022; 10(7):1674. https://doi.org/10.3390/biomedicines10071674
Chicago/Turabian StyleSalomon-Perzyński, Aleksander, Joanna Barankiewicz, Marcin Machnicki, Irena Misiewicz-Krzemińska, Michał Pawlak, Sylwia Radomska, Agnieszka Krzywdzińska, Aleksandra Bluszcz, Piotr Stawiński, Małgorzata Rydzanicz, and et al. 2022. "Tracking Clonal Evolution of Multiple Myeloma Using Targeted Next-Generation DNA Sequencing" Biomedicines 10, no. 7: 1674. https://doi.org/10.3390/biomedicines10071674
APA StyleSalomon-Perzyński, A., Barankiewicz, J., Machnicki, M., Misiewicz-Krzemińska, I., Pawlak, M., Radomska, S., Krzywdzińska, A., Bluszcz, A., Stawiński, P., Rydzanicz, M., Jakacka, N., Solarska, I., Borg, K., Spyra-Górny, Z., Szpila, T., Puła, B., Grosicki, S., Stokłosa, T., Płoski, R., ... Jamroziak, K. (2022). Tracking Clonal Evolution of Multiple Myeloma Using Targeted Next-Generation DNA Sequencing. Biomedicines, 10(7), 1674. https://doi.org/10.3390/biomedicines10071674