
Stage II of Chronic Kidney Disease—A Tipping Point in Disease Progression?

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
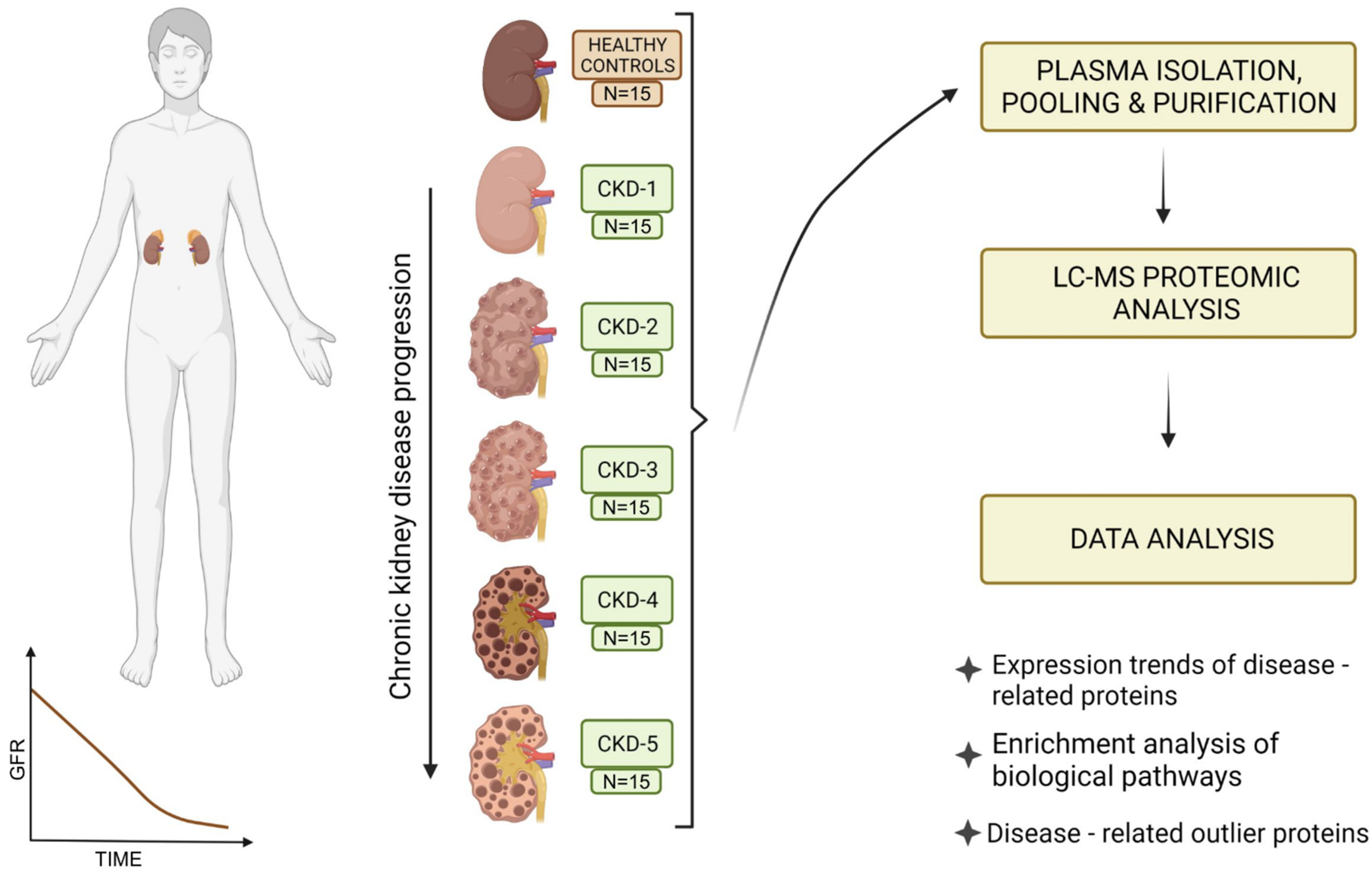
2.1. Study Participants and Study Outline
2.2. Plasma Sample Collection and Heparin Column Chromatography
2.3. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
2.4. Data Analysis
3. Results
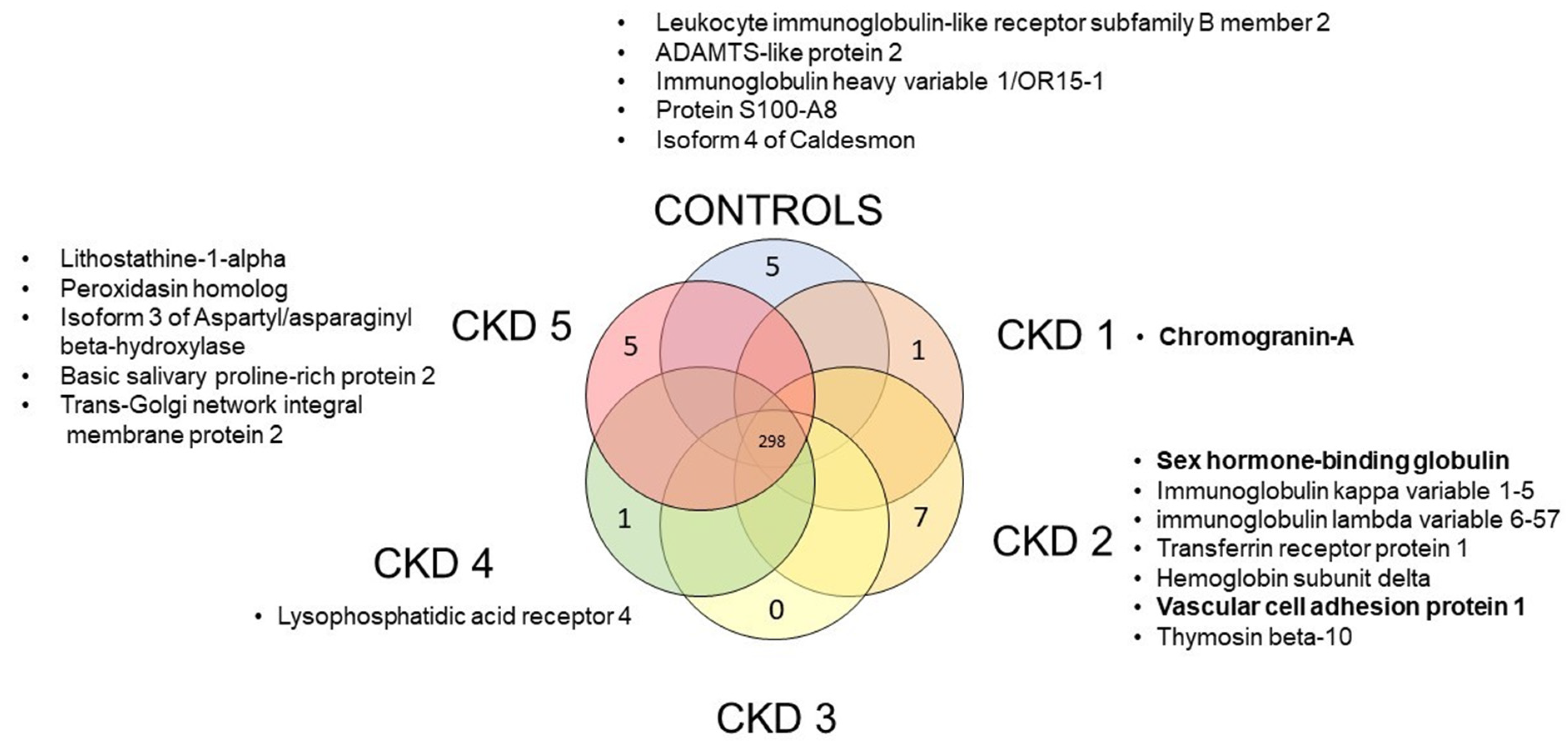
3.1. Protein Identification
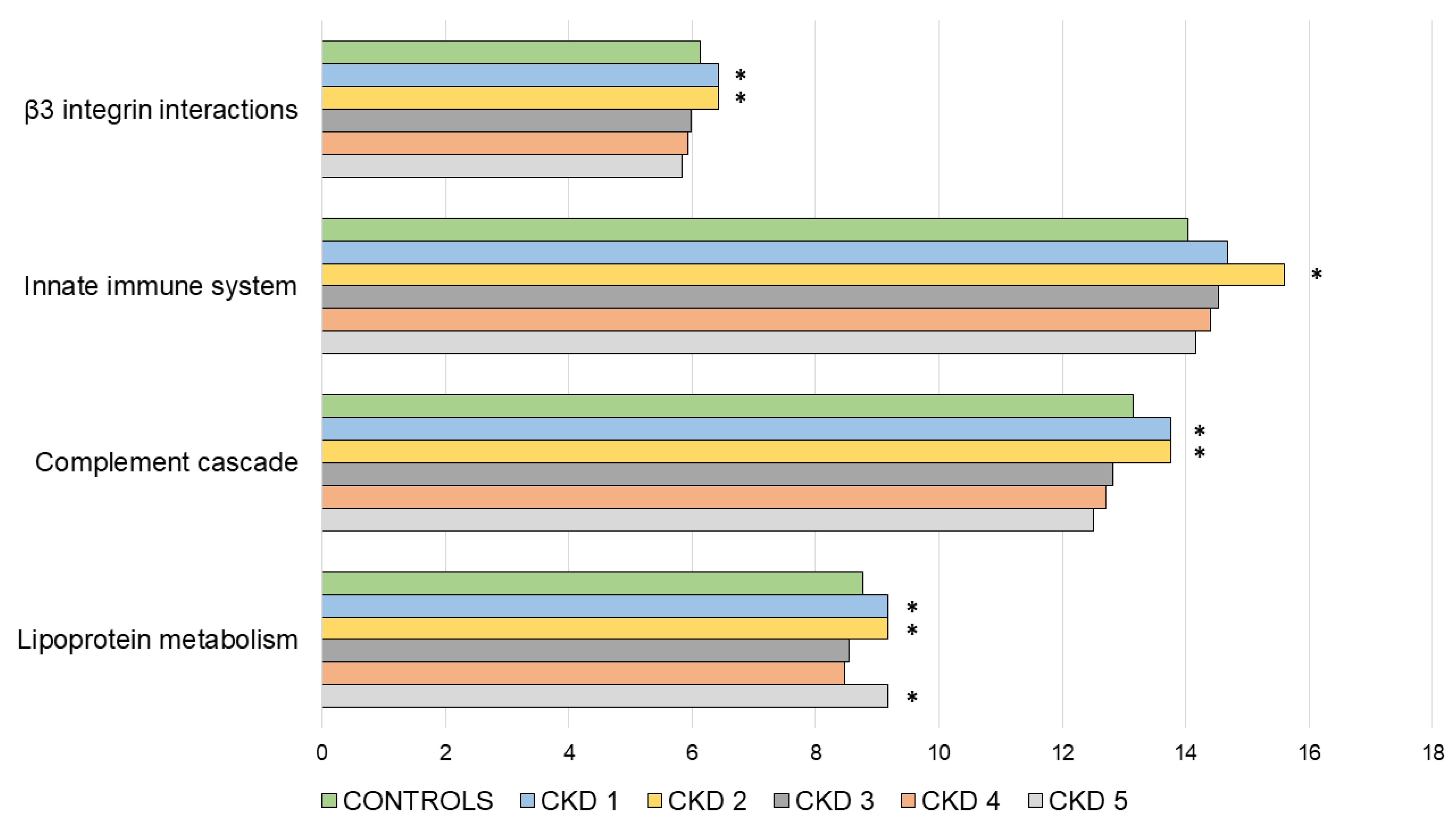
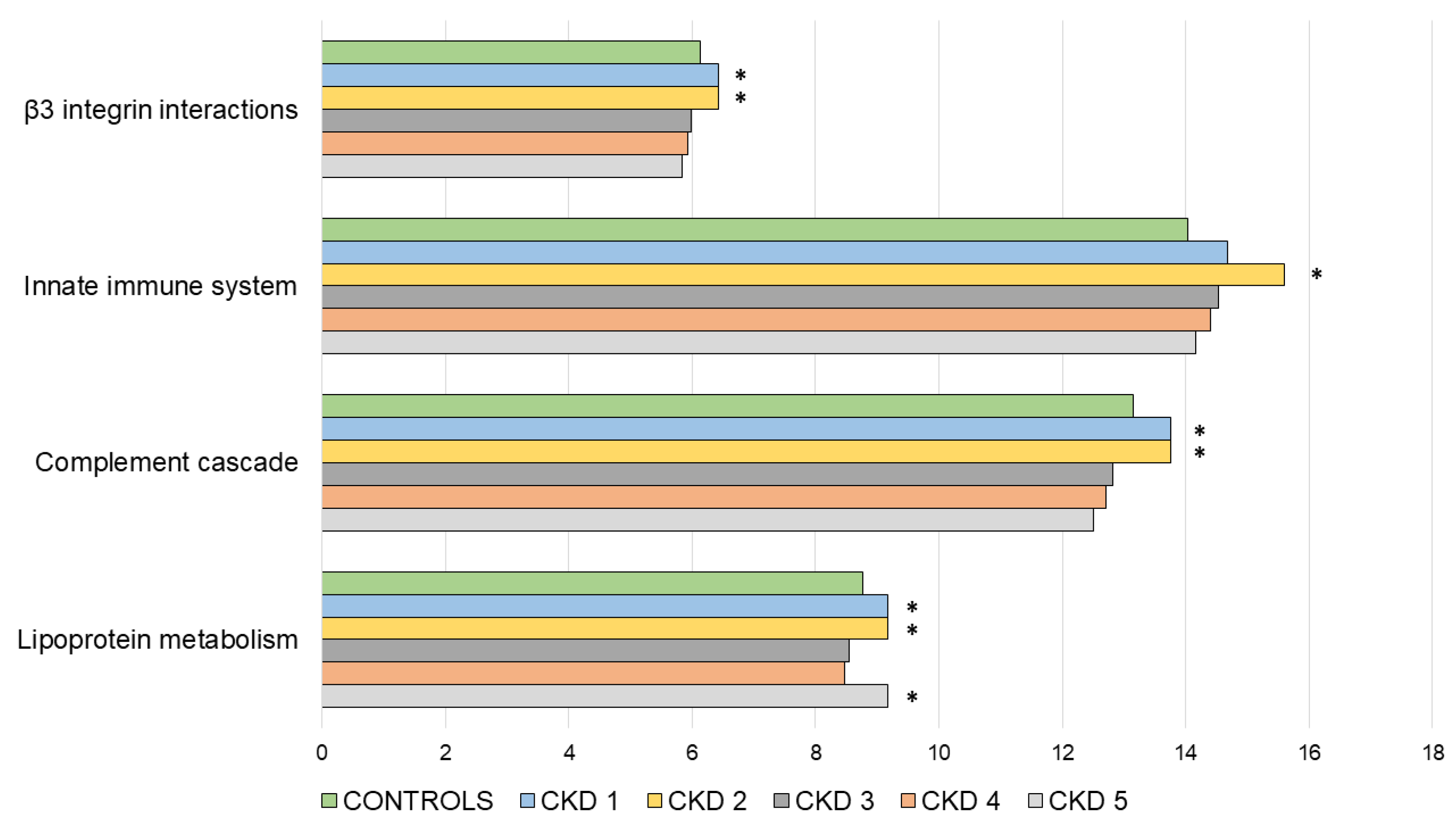
3.2. Functional Enrichment Analysis
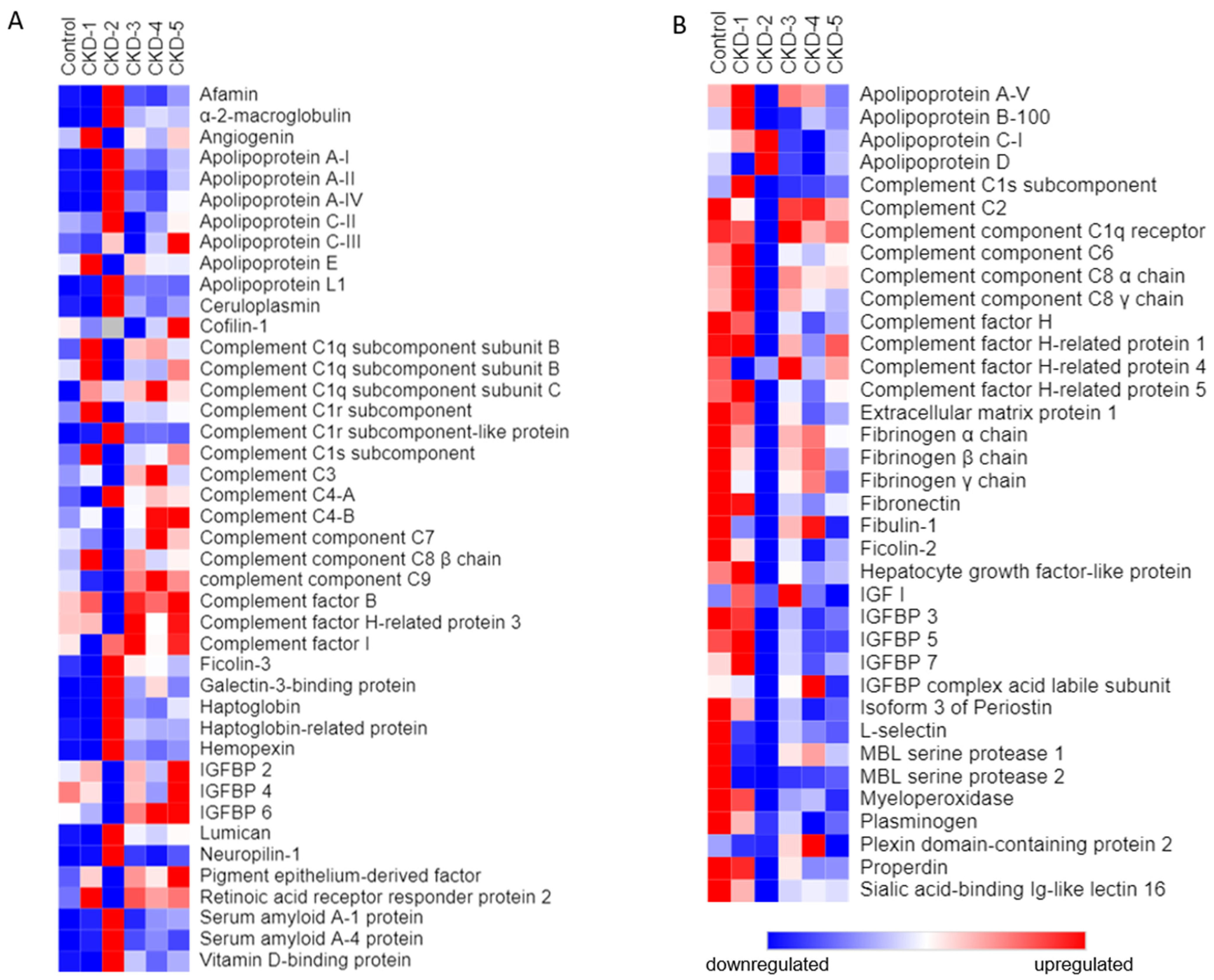
3.3. Outlier Analysis
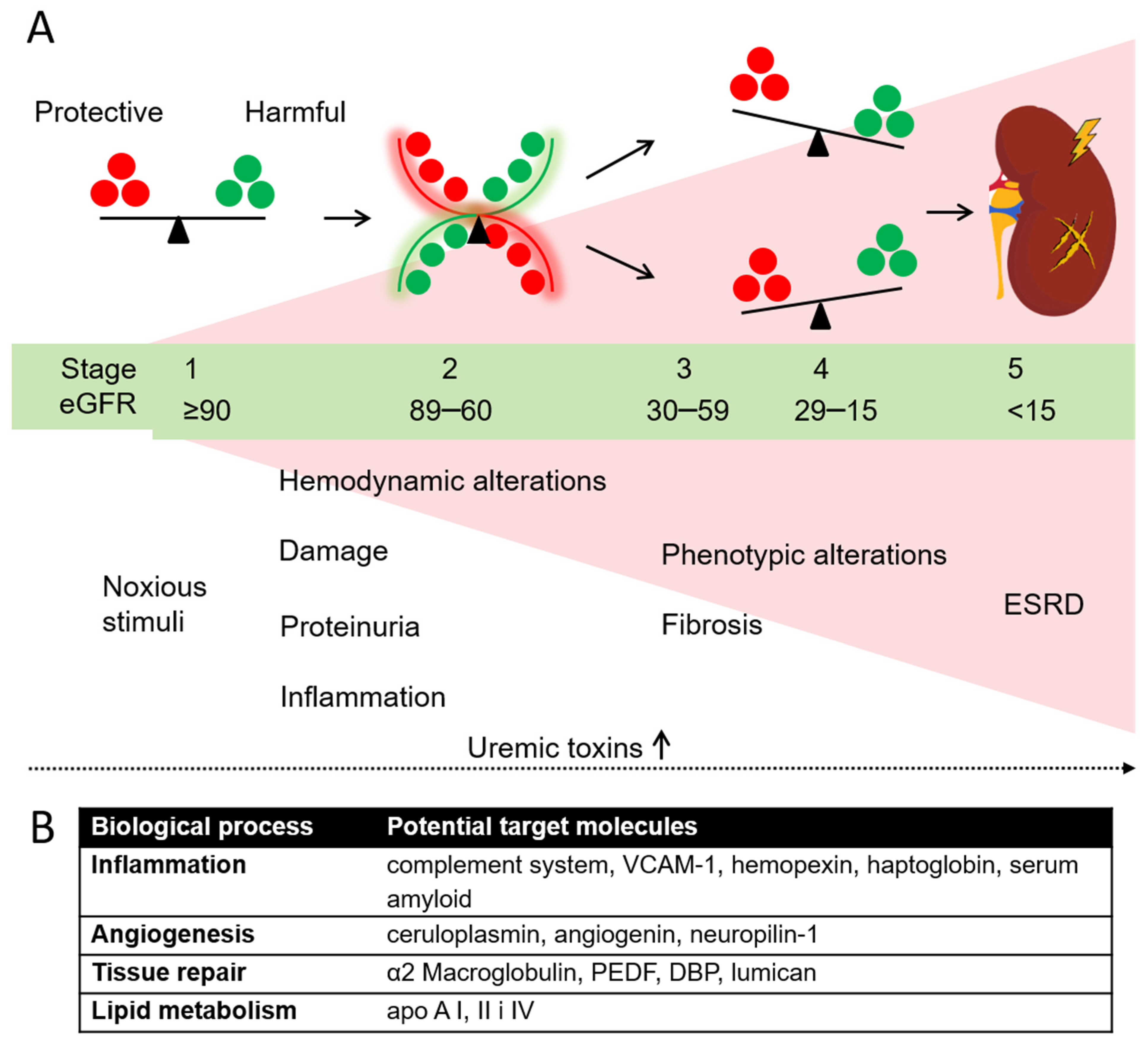
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manns, B.; Hemmelgarn, B.; Tonelli, M.; Au, F.; So, H.; Weaver, R.; Quinn, A.E.; Klarenbach, S. The Cost of Care for People with Chronic Kidney Disease. Can. J. kidney Health Dis. 2019, 6, 2054358119835521. [Google Scholar] [CrossRef] [Green Version]
- Vojtusek, I.K.; Laganovic, M.; Burek Kamenaric, M.; Bulimbasic, S.; Hrkac, S.; Salai, G.; Ivkovic, V.; Coric, M.; Novak, R.; Grgurevic, L. First Characterization of ADAMTS-4 in Kidney Tissue and Plasma of Patients with Chronic Kidney Disease—A Potential Novel Diagnostic Indicator. Diagnostics 2022, 12, 648. [Google Scholar] [CrossRef] [PubMed]
- KDIGO. 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Off. J. Int. Soc. Nephrol. 2013, 3, 5–14. [Google Scholar]
- Cao, Q.; Chen, X.-M.; Huang, C.; Pollock, C.A. MicroRNA as novel biomarkers and therapeutic targets in diabetic kidney disease: An update. FASEB Bioadvances 2019, 1, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Neovius, M.; Jacobson, S.H.; Eriksson, J.K.; Elinder, C.-G.; Hylander, B. Mortality in chronic kidney disease and renal replacement therapy: A population-based cohort study. BMJ Open 2014, 4, e004251. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, G.; Mullen, W.; Duranton, F.; Filip, S.; Gayrard, N.; Husi, H.; Schepers, E.; Neirynck, N.; Schanstra, J.P.; Jankowski, J.; et al. New insights in molecular mechanisms involved in chronic kidney disease using high-resolution plasma proteome analysis. Nephrol. Dial. Transpl. 2015, 30, 1842–1852. [Google Scholar] [CrossRef] [Green Version]
- Romanova, Y.; Laikov, A.; Markelova, M.; Khadiullina, R.; Makseev, A.; Hasanova, M.; Rizvanov, A.; Khaiboullina, S.; Salafutdinov, I. Proteomic Analysis of Human Serum from Patients with Chronic Kidney Disease. Biomolecules 2020, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Higashisaka, K.; Takeya, S.; Kamada, H.; Obana, M.; Maeda, M.; Kabayama, M.; Yamamoto, K.; Ishida, N.; Isaka, R.; Tsujino, H.; et al. Identification of biomarkers of chronic kidney disease among kidney-derived proteins. Clin. Proteom. 2022, 19, 3. [Google Scholar] [CrossRef]
- Jalal, D.; Sanford, B.; Renner, B.; Ten Eyck, P.; Laskowski, J.; Cooper, J.; Sun, M.; Zakharia, Y.; Spitz, D.; Dokun, A.; et al. Detection of pro angiogenic and inflammatory biomarkers in patients with CKD. Sci. Rep. 2021, 11, 8786. [Google Scholar] [CrossRef]
- Luczak, M.; Formanowicz, D.; Marczak, Ł.; Pawliczak, E.; Wanic-Kossowska, M.; Figlerowicz, M.; Stobiecki, M. Deeper insight into chronic kidney disease-related atherosclerosis: Comparative proteomic studies of blood plasma using 2DE and mass spectrometry. J. Transl. Med. 2015, 13, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gansevoort, R.T.; de Jong, P.E. The Case for Using Albuminuria in Staging Chronic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 465–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using Stage Tips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Fonseka, P.; Pathan, M.; Chitti, S.V.; Kang, T.; Mathivanan, S. FunRich enables enrichment analysis of OMICs datasets. J. Mol. Biol. 2021, 433, 166747. [Google Scholar] [CrossRef] [PubMed]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus/ (accessed on 7 February 2022).
- Lee, P.-T.; Liao, I.-C.; Lee, C.-H.; Hsu, L.-W.; Liu, P.-Y. Expression of Vascular Cell Adhesion Molecule-1 in Peripheral Artery Disease is Enriched in Patients with Advanced Kidney Disease. Acta Cardiol. Sin. 2021, 37, 591–599. [Google Scholar]
- Gasparin, A.A.; de Andrade, N.P.B.; Hax, V.; Palominos, P.E.; Siebert, M.; Marx, R.; Schaefer, P.G.; Veronese, F.V.; Monticielo, O.A. Urinary soluble VCAM-1 is a useful biomarker of disease activity and treatment response in lupus nephritis. BMC Rheumatol. 2020, 4, 67. [Google Scholar] [CrossRef]
- Jia, Y.; Xu, H.; Yu, Q.; Tan, L.; Xiong, Z. Identification and verification of vascular cell adhesion protein 1 as an immune-related hub gene associated with the tubulointerstitial injury in diabetic kidney disease. Bioengineered 2021, 12, 6655–6673. [Google Scholar] [CrossRef]
- Tramonti, G.; Ferdeghini, M.; Annichiarico, C.; Norpoth, M.; Donadio, C.; Bianchi, R.; Bianchi, C. Relationship between renal function and blood level of chromogranin A. Ren. Fail. 2001, 23, 449–457. [Google Scholar] [CrossRef]
- Bech, P.R.; Ramachandran, R.; Dhillo, W.S.; Martin, N.M.; Bloom, S.R. Quantifying the Effects of Renal Impairment on Plasma Concentrations of the Neuroendocrine Neoplasia Biomarkers Chromogranin A, Chromogranin B, and Cocaine- and Amphetamine-Regulated Transcript. Clin. Chem. 2012, 58, 941–943. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wang, H.; Su, X.; Cao, A.; Yao, X.; Wang, Y.; Zhu, B.; Wang, H.; Fang, J. Serum chromogranin A correlated with albuminuria in diabetic patients and is associated with early diabetic nephropathy. BMC Nephrol. 2022, 23, 41. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, C.; Zhang, X.; Wang, Y.; Wan, H.; Chen, Y.; Zhang, W.; Xia, F.; Lu, Y.; Wang, N. Association between sex hormone-binding globulin and kidney function in men: Results from the SPECT-China study. Chin. Med. J. 2022. Available online: https://pubmed.ncbi.nlm.nih.gov/35170516/ (accessed on 3 March 2022).
- Farrar, C.A.; Zhou, W.; Sacks, S.H. Role of the lectin complement pathway in kidney transplantation. Immunobiology 2016, 221, 1068–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, S.H.; Zhou, W.; Pani, A.; Campbell, R.D.; Martin, J. Complement C3 gene expression and regulation in human glomerular epithelial cells. Immunology 1993, 79, 348–354. [Google Scholar] [PubMed]
- Michels, M.A.H.M.; Volokhina, E.B.; van de Kar, N.C.A.J.; van den Heuvel, L.P.W.J. The role of properdin in complement-mediated renal diseases: A new player in complement-inhibiting therapy? Pediatr. Nephrol. 2019, 34, 1349–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaarkeuken, H.; Siezenga, M.A.; Zuidwijk, K.; van Kooten, C.; Rabelink, T.J.; Daha, M.R.; Berger, S.P. Complement activation by tubular cells is mediated by properdin binding. Am. J. Physiol. Physiol. 2008, 295, F1397–F1403. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yang, B. Erythropoietin Receptor/β Common Receptor: A Shining Light on Acute Kidney Injury Induced by Ischemia-Reperfusion. Front. Immunol. 2021, 12, 697796. [Google Scholar] [CrossRef]
- Zhang, Y.; Meyer, N.C.; Fervenza, F.C.; Lau, W.; Keenan, A.; Cara-Fuentes, G.; Shao, D.; Akber, A.; Fremeaux-Bacchi, V.; Sethi, S.; et al. C4 Nephritic Factors in C3 Glomerulopathy: A Case Series. Am. J. Kidney Dis. 2017, 70, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Corvillo, F.; Bravo García-Morato, M.; Nozal, P.; Garrido, S.; Tortajada, A.; Rodríguez de Córdoba, S.; López-Trascasa, M. Serum properdin consumption as a biomarker of C5 convertase dysregulation in C3 glomerulopathy. Clin. Exp. Immunol. 2016, 184, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Thiel, S.; Jensen, L.; Degn, S.E.; Nielsen, H.J.; Gál, P.; Dobó, J.; Jensenius, J.C. Mannan-binding lectin (MBL)-associated serine protease-1 (MASP-1), a serine protease associated with humoral pattern-recognition molecules: Normal and acute-phase levels in serum and stoichiometry of lectin pathway components. Clin. Exp. Immunol. 2012, 169, 38–48. [Google Scholar] [CrossRef]
- Man-Kupisinska, A.; Michalski, M.; Maciejewska, A.; Swierzko, A.S.; Cedzynski, M.; Lugowski, C.; Lukasiewicz, J. A New Ligand-Based Method for Purifying Active Human Plasma-Derived Ficolin-3 Complexes Supports the Phenomenon of Crosstalk between Pattern-Recognition Molecules and Immunoglobulins. PLoS ONE 2016, 11, e0156691. [Google Scholar] [CrossRef] [Green Version]
- Smedbråten, Y.V.; Sagedal, S.; Mjøen, G.; Hartmann, A.; Fagerland, M.W.; Rollag, H.; Mollnes, T.E.; Thiel, S. High Ficolin-3 Level at the Time of Transplantation Is an Independent Risk Factor for Graft Loss in Kidney Transplant Recipients. Transplantation 2015, 99, 791–796. [Google Scholar] [CrossRef]
- Graw, J.A.; Hildebrandt, P.; Krannich, A.; Balzer, F.; Spies, C.; Francis, R.C.; Kuebler, W.M.; Weber-Carstens, S.; Menk, M.; Hunsicker, O. The role of cell-free hemoglobin and haptoglobin in acute kidney injury in critically ill adults with ARDS and therapy with VV ECMO. Crit. Care 2022, 26, 50. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, R.J.; Dieter, B.P.; Meek, R.L.; Tuttle, K.R. An Inflammatory Nexus: Serum Amyloid A and inflammation in Diabetic Kidney Disease. Inflamm. Cell Signal. 2015, 2, 1–5. [Google Scholar]
- Anderberg, R.J.; Meek, R.L.; Hudkins, K.L.; Cooney, S.K.; Alpers, C.E.; Leboeuf, R.C.; Tuttle, K.R. Erratum: Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab Investig. 2015, 95, 697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudhana Rao, A.; Anand, U.; Anand, C.V. Myeloperoxidase in Chronic Kidney Disease. Indian J. Clin. Biochem. 2011, 26, 28–31. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-M.; Kwon, Y.-E.; Kim, S.; Oh, D.-J. Changes in FGF-23, Neutrophil/Platelet Activation Markers, and Angiogenin in Advanced Chronic Kidney Disease and Their Effect on Arterial Stiffness. Kidney Blood Press Res. 2019, 44, 1166–1178. [Google Scholar] [CrossRef]
- Mami, I.; Bouvier, N.; El Karoui, K.; Gallazzini, M.; Rabant, M.; Laurent-Puig, P.; Li, S.; Tharaux, P.L.; Beaune, P.; Thervet, E.; et al. Angiogenin Mediates Cell-Autonomous Translational Control under Endoplasmic Reticulum Stress and Attenuates Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Gacche, R.N. Compensatory angiogenesis and tumor refractoriness. Oncogenesis 2015, 4, e153. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Bandyopadhyay, C.; Bottero, V.; Veettil, M.V.; Wilson, L.; Pins, M.R.; Johnson, K.E.; Warshall, C.; Chandran, B. Angiogenin interacts with the plasminogen activation system at the cell surface of breast cancer cells to regulate plasmin formation and cell migration. Mol. Oncol. 2014, 8, 483–507. [Google Scholar] [CrossRef] [Green Version]
- Mulay, S.R.; Eberhard, J.N.; Pfann, V.; Marschner, J.A.; Darisipudi, M.N.; Daniel, C.; Romoli, S.; Desai, J.; Grigorescu, M.; Kumar, S.V.; et al. Oxalate-induced chronic kidney disease with its uremic and cardiovascular complications in C57BL/6 mice. Am. J. Physiol. Physiol. 2016, 310, F785–F795. [Google Scholar] [CrossRef] [Green Version]
- Kennedy-Lydon, T.M.; Crawford, C.; Wildman, S.S.P.; Peppiatt-Wildman, C.M. Renal pericytes: Regulators of medullary blood flow. Acta Physiol. 2013, 207, 212–225. [Google Scholar] [CrossRef]
- Gagnon, M.L.; Bielenberg, D.R.; Gechtman, Z.; Miao, H.-Q.; Takashima, S.; Soker, S.; Klagsbrun, M. Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: In vivo expression and antitumor activity. Proc. Natl. Acad Sci. USA 2000, 97, 2573–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadasz, Z.; Ben-Izhak, O.; Bejar, J.; Sabo, E.; Kessel, A.; Storch, S.; Toubi, E. The involvement of immune semaphorins and neuropilin-1 in lupus nephritis. Lupus 2011, 20, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Fan, Y.; Wu, Y.; Pepoy, M.; Hazen, S.L.; Tang, W.H.W. Plasma Ceruloplasmin, a Regulator of Nitric Oxide Activity, and Incident Cardiovascular Risk in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 462–467. [Google Scholar] [CrossRef] [Green Version]
- Lazzaro, M.; Bettegazzi, B.; Barbariga, M.; Codazzi, F.; Zacchetti, D.; Alessio, M. Ceruloplasmin potentiates nitric oxide synthase activity and cytokine secretion in activated microglia. J. Neuroinflammation 2014, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Gao, H.; Liang, Y.; Zhang, J.; Wang, J.; Qiu, J. Cofilin1 is involved in hypertension-induced renal damage via the regulation of NF-κB in renal tubular epithelial cells. J. Transl. Med. 2015, 13, 323. [Google Scholar] [CrossRef] [Green Version]
- Coşkun, A.; Ucal, Y.; Berber, I.; Çakır, Ü.; Serteser, M.; Moldur, D.E.; Derelioğlu, E.İ.; Yozgatlı, T.K.; Özpınar, A.; Ünsal, İ. Cofilin-1 as a potential biomarker to evaluate acute kidney injury. Turk. J. Biochem. 2019, 44, 9–15. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, K.; Xu, Y.; Zhou, H.; He, W.; Zhang, W.; Cai, L.; Lin, X.; Fang, Z.; Luo, Z.; et al. Characterization of acute renal allograft rejection by human serum proteomic analysis. J. Huazhong Univ. Sci. Technol. (Med. Sci.) 2009, 29, 585–591. [Google Scholar] [CrossRef]
- Speeckaert, M.; Huang, G.; Delanghe, J.R.; Taes, Y.E.C. Biological and clinical aspects of the vitamin D binding protein (Gc-globulin) and its polymorphism. Clin. Chim. Acta. 2006, 372, 33–42. [Google Scholar] [CrossRef]
- Kalousova, M.; Dusilova-Sulkova, S.; Zakiyanov, O.; Kostirova, M.; Safranek, R.; Tesar, V.; Zima, T. Vitamin D Binding Protein Is Not Involved in Vitamin D Deficiency in Patients with Chronic Kidney Disease. Biomed Res. Int. 2015, 2015, 492365. [Google Scholar] [CrossRef]
- Wang, J.J.; Zhang, S.X.; Mott, R.; Chen, Y.; Knapp, R.R.; Cao, W.; Ma, J.X. Anti-inflammatory effects of pigment epithelium-derived factor in diabetic nephropathy. Am. J. Physiol. Physiol. 2008, 294, F1166–F1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motomiya, Y.; Yamagishi, S.; Adachi, H.; Abe, A. Increased Serum Concentrations of Pigment Epithelium-Derived Factor in Patients with End-Stage Renal Disease. Clin. Chem. 2006, 52, 1970–1971. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Pigment Epithelium-derived Factor (PEDF) and Cardiometabolic Disorders. Curr. Pharm. Des. 2014, 20, 2377–2386. [Google Scholar] [CrossRef]
- Feige, J.-J.; Negoescu, A.; Keramidas, M.; Souchelnitskiy, S.; Chambaz, E.M. Alpha-2-Macroglobulin: A Binding Protein for Transforming Growth Factor-β and Various Cytokines. Horm. Res. Paediatr. 1996, 45, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Lagrange, J.; Lecompte, T.; Knopp, T.; Lacolley, P.; Regnault, V. Alpha-2-macroglobulin in hemostasis and thrombosis: An underestimated old double-edged sword. J. Thromb. Haemost. 2022, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.H.; Chen, J.Y. Glomerular deposition of alpha 2-macroglobulin in glomerular diseases. Nephrol. Dial. Transplant. 1997, 12, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- lu, X.-M.; Ma, L.; Jin, Y.-N.; Yu, Y.-Q. Lumican overexpression exacerbates lipopolysaccharide-induced renal injury in mice. Mol. Med. Rep. 2015, 12, 4089–4094. [Google Scholar] [CrossRef] [Green Version]
- Vij, N.; Roberts, L.; Joyce, S.; Chakravarti, S. Lumican Regulates Corneal Inflammatory Responses by Modulating Fas-Fas Ligand Signaling. Investig. Opthalmology Vis. Sci. 2005, 46, 88. [Google Scholar] [CrossRef]
- Krishnan, A.; Li, X.; Kao, W.-Y.; Viker, K.; Butters, K.; Masuoka, H.; Knudsen, B.; Gores, G.; Charlton, M. Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab Investig. 2012, 92, 1712–1725. [Google Scholar] [CrossRef]
- Neiman, M.; Hedberg, J.J.; Dönnes, P.R.; Schuppe-Koistinen, I.; Hanschke, S.; Schindler, R.; Uhlén, M.; Schwenk, J.M.; Nilsson, P. Plasma Profiling Reveals Human Fibulin-1 as Candidate Marker for Renal Impairment. J. Proteome. Res. 2011, 10, 4925–4934. [Google Scholar] [CrossRef]
- Barbagallo, C.M.; Cefalù, A.B.; Giammanco, A.; Noto, D.; Caldarella, R.; Ciaccio, M.; Averna, M.R.; Nardi, E. Lipoprotein Abnormalities in Chronic Kidney Disease and Renal Transplantation. Life 2021, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Kim, D.K.; Oh, K.-H.; Joo, K.W.; Lim, C.S.; Kim, Y.S.; Han, S.S. Apolipoprotein B is a risk factor for end-stage renal disease. Clin. Kidney J. 2021, 14, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boes, E.; Fliser, D.; Ritz, E.; König, P.; Lhotta, K.; Mann, J.F.E.; Müller, G.A.; Neyer, U.; Riegel, W.; Riegler, P.; et al. Apolipoprotein A-IV Predicts Progression of Chronic Kidney Disease: The Mild to Moderate Kidney Disease Study. J. Am. Soc. Nephrol. 2006, 17, 528–536. [Google Scholar] [CrossRef]
- Diz, A.P.; Truebano, M.; Skibinski, D.O.F. The consequences of sample pooling in proteomics: An empirical study. Electrophoresis 2009, 30, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, E.M.; Linhardt, R.J. Heparin-Binding Domains in Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1549–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, N.S.; Mancera, R.L. Prediction of heparin binding sites in bone morphogenetic proteins (BMPs). Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 1374–1381. [Google Scholar] [CrossRef]
- Ori, A.; Free, P.; Courty, J.; Wilkinson, M.C.; Fernig, D.G. Identification of Heparin-binding Sites in Proteins by Selective Labeling. Mol. Cell Proteom. 2009, 8, 2256–2265. [Google Scholar] [CrossRef] [Green Version]
- Casu, B.; Naggi, A.; Torri, G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Aviezer, D.; Levy, E.; Safran, M.; Svahn, C.; Buddecke, E.; Schmidt, A.; David, G.; Vlodavsky, I.; Yayon, A. Differential structural requirements of heparin and heparan sulfate proteoglycans that promote binding of basic fibroblast growth factor to its receptor. J. Biol. Chem. 1994, 269, 114–121. [Google Scholar] [CrossRef]
- Alberdi, E.; Hyde, C.C.; Becerra, S.P. Pigment Epithelium-Derived Factor (PEDF) Binds to Glycosaminoglycans: Analysis of the Binding Site. Biochemistry 1998, 37, 10643–10652. [Google Scholar] [CrossRef]
- Palhares, L.C.G.F.; London, J.A.; Kozlowski, A.M.; Esposito, E.; Chavante, S.F.; Ni, M.; Yates, E.A. Chemical Modification of Glycosaminoglycan Polysaccharides. Molecules 2021, 26, 5211. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chi, K.; Wu, D.; Hong, Q. Insulin-Like Growth Factor Binding Proteins in Kidney Disease. Front. Pharmacol. 2021, 12, 807119. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y. The insulin-like growth factor system in chronic kidney disease: Pathophysiology and therapeutic opportunities. Kidney Res. Clin. Pract. 2012, 31, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Healthy | CKD Stage 1 | CKD Stage 2 | CKD Stage 3 | CKD Stage 4 | CKD Stage 5 |
|---|---|---|---|---|---|---|
| N | 15 | 15 | 15 | 15 | 15 | 15 |
| Age (years) | 42.9 ± 9.4 | 36.7 ± 15.2 | 54.5 ± 18.2 | 56.5 ± 14.5 | 60.5 ± 16.1 | 60.7 ± 15.4 |
| Gender: N (%) female | 7 (46%) | 8 (53%) | 7 (46%) | 4 (26%) | 9 (60%) | 6 (40%) |
| BMI (kg/m2) | 24.3 ± 2.7 | 25.1 ± 4.7 | 28.7 ± 6.43 | 25.7 ± 5.22 | 26.3 ± 4.06 | 28.3 ± 5.23 |
| Serum creatinine (μmol/L) | 72.7 ± 10.9 | 74.1 ± 14.3 | 88.7 ± 15.2 | 158 ± 23.7 | 237 ± 52.2 | 476 ± 136 |
| eGFR (mL/min/1.73 m2) | 104 ± 10.1 | 102 ± 10.2 | 74.7 ± 8.8 | 39.3 ± 7.2 | 21.7 ± 4.7 | 10.3 ± 2.3 |
| Proteinuria (g/24 h) | 0.2 (0–0.5) | 0.36 (0.2–1.5) | 0.7 (0.3–1.3) | 0.49 (0.4–1.2) | 0.43 (0.4–3.1) | 1.88 (0.7–3.2) |
| BUN (mmol/L) | 5 ± 1 | 5 ± 1.3 | 6.2 ± 1.6 | 10.5 ± 3.3 | 16.6 ± 4.9 | 24.3 ± 4.9 |
| Serum cholesterol (mmol/L) | 4.4 ± 0.6 | 5.4 ± 2.5 | 4.2 ± 0.8 | 4.5 ± 0.8 | 5 ± 1.4 | 5 ± 1.7 |
| Serum triglycerides (mmol/L) | 1.1 (0.7–1.4) | 1 (0.8–1.2) | 1.4 (1–1.8) | 1.7 (1.2–2) | 2 (1.6–3.4) | 1.6 (1.4–2.8) |
| Serum HDL (mmol/L) | 1.6 ± 0.3 | 1.6 ± 0.4 | 1.3 ± 0.4 | 1.3 ± 0.4 | 1.1 ± 06 | 1.1 ± 0.3 |
| Serum LDL (mmol/L) | 2.4 ± 0.5 | 3.3 ± 2.4 | 2.2 ± 0.7 | 2.1 ± 0.8 | 2.8 ± 1.4 | 2.7 ± 1.3 |
| Serum albumin (g/L) | 44 ± 3 | 39 ± 6 | 38 ± 3 | 40 ± 3 | 37 ± 6.6 | 40 ± 4 |
| Comorbidities | ||||||
| Hypertension | 0 | 7 (47%) | 14 (93%) | 13.(86%) | 15 (100%) | 15 (100%) |
| Diabetes | 0 | 0 | 3 (20%) | 4 (26%) | 5 (33%) | 2 (13%) |
| Smoker | 2 (13%) | 2 (13%) | 1 (7%) | 6 (40%) | 3 (20%) | 8 (53%) |
| Hyperlipidemia | 0 | 5 (33%) | 12 (80%) | 10 (67%) | 12 (80%) | 9 (60%) |
| Atherosclerosis | 0 | 0 | 3 (20%) | 4 (26%) | 5 (33%) | 8 (53%) |
| Underlying Disease | ||||||
| Primary glomerular KD | 0 | 13 (86%) | 15 (100%) | 9 (60%) | 5 (33%) | 6 (40%) |
| Hypertensive/atherosclerotic KD | 0 | 0 | 0 | 2 (13%) | 5 (33%) | 2 (13%) |
| Autosomal dominant polycystic KD | 0 | 1 (7%) | 0 | 0 | 0 | 1 (7%) |
| Diabetic KD | 0 | 0 | 0 | 0 | 1 (7%) | 1 (7%) |
| Other specific cause | 0 | 1 (7%) | 0 | 2 (13%) | 2 (13%) | 2 (13%) |
| Unknown | 0 | 0 | 0 | 1 (7%) | 2 (13%) | 3 (20%) |
| Reference | Outlier Protein Name | N | Hospital Admission Disease | Study Groups | Tested Sample | Main Points and Conclusions |
|---|---|---|---|---|---|---|
| Po-Tseng Lee et al., 2021 [16] | VCAM-1 | 51 | Peripheral arterial disease (PAD) | 3 groups:A/normal kidney function; B/CKD; C/HD | serum/arterial tissue | - Link with underlying kidney disease - The HD group had a higher Concentration of VCAM-1 than the other two groups - Serum level and the tissue expression of VCAM-1 were significantly higher in PAD patients with advanced kidney disease |
| Gasparin et al., 2020 [17] | VCAM-1 | 62 | Systemic lupus erythematosus (SLE) | 2 groups:A/without active lupus nephritis (LN); B/with active LN | urine | VCAM-1 level was elevated in patients with active compared to inactive LN |
| Y Jia et al., 2021 [18] | VCAM-1 | 22 | Diabetes mellitus | 2 groups:A/Control; B/Diabetic kidney disease (DKD) | renal tubular cells, infiltrated immune cells | VCAM1 expression was upregulated in renal tubular cells, which might interact with infiltrated immune cells, thus promoting fibrosis. |
| Tramonti G et al., 2009 [19] | CGA | 102 | Kidney related disease | Patients with different values of GFR | serum | CGA accumulates in the blood in renal failure |
| Bech PR et al., 2012 [20] | CGA | 147 | Kidney related disease | 2 groups: A/normal kidney function; B/CKD | plasma | CGA accumulates in the blood in renal failure |
| Yu et al., 2022 [21] | CGA | 219 | Type 2 diabetes mellitus (T2DM) | 3 groups of patients with DN based on their urine albumin to creatinine ratios | serum | Serum CgA increased gradually with the degree of DN |
| Zhang H 2022 [22] | SHBP | 5027 | Screening for methabolic disease and other risk factors | Measurement of SHBP and testing correlation with eGFR | Serum, plasma | Lower serum SHBG levels were significantly associated with lower eGFR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grgurevic, L.; Novak, R.; Salai, G.; Hrkac, S.; Mocibob, M.; Vojtusek, I.K.; Laganovic, M. Stage II of Chronic Kidney Disease—A Tipping Point in Disease Progression? Biomedicines 2022, 10, 1522. https://doi.org/10.3390/biomedicines10071522
Grgurevic L, Novak R, Salai G, Hrkac S, Mocibob M, Vojtusek IK, Laganovic M. Stage II of Chronic Kidney Disease—A Tipping Point in Disease Progression? Biomedicines. 2022; 10(7):1522. https://doi.org/10.3390/biomedicines10071522
Chicago/Turabian StyleGrgurevic, Lovorka, Rudjer Novak, Grgur Salai, Stela Hrkac, Marko Mocibob, Ivana Kovacevic Vojtusek, and Mario Laganovic. 2022. "Stage II of Chronic Kidney Disease—A Tipping Point in Disease Progression?" Biomedicines 10, no. 7: 1522. https://doi.org/10.3390/biomedicines10071522
APA StyleGrgurevic, L., Novak, R., Salai, G., Hrkac, S., Mocibob, M., Vojtusek, I. K., & Laganovic, M. (2022). Stage II of Chronic Kidney Disease—A Tipping Point in Disease Progression? Biomedicines, 10(7), 1522. https://doi.org/10.3390/biomedicines10071522