
The Impact of Kidney Biopsy for Fabry Nephropathy Evaluation on Patients’ Management and Long-Term Outcomes: Experience of a Single Center

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Clinical Assessments and Study Endpoints
- Pathogenic variants: variants that are causative of a disease;
- Likely pathogenic variants: variants where the data support a high likelihood that it is pathogenic;
- Variants of uncertain significance (VUS): a genetic variant with unknown or questionable impact on clinical phenotype;
- Likely benign: variant where the data support a high likelihood that it is benign;
- Benign: a variant that is not considered to be the cause of the disease [18].
2.3. Statistical Analysis
3. Results
3.1. Patients
3.1.1. General Clinical and Biological Parameters
- Classic: c.797A > C in 5 patients, c.485G > A in 3 patients, c.779G > A, c.295C > T, c.836A > G each in the case of 2 patients, and c.863C > A, c.671delA, c.1224del66, c.1228A > G, c.141G > A, c.1121_1123delAAG each in the case of 1 patient;
- Late-onset: c.644A > G (N215S) in 1 patient.
3.1.2. Fabry Features
- Classic: c.797A > C in 5 patients, c.485G > A in 3 patients, c.779G > A, c.295C > T, c.836A > G each in the case of 2 patients, and c.863C > A, c.671delA, c.1224del66, c.1228A > G, c.141G > A, c.1121_1123delAAG each in the case of 1 patient;
- Late-onset: c.644A > G (N215S) in 1 patient.
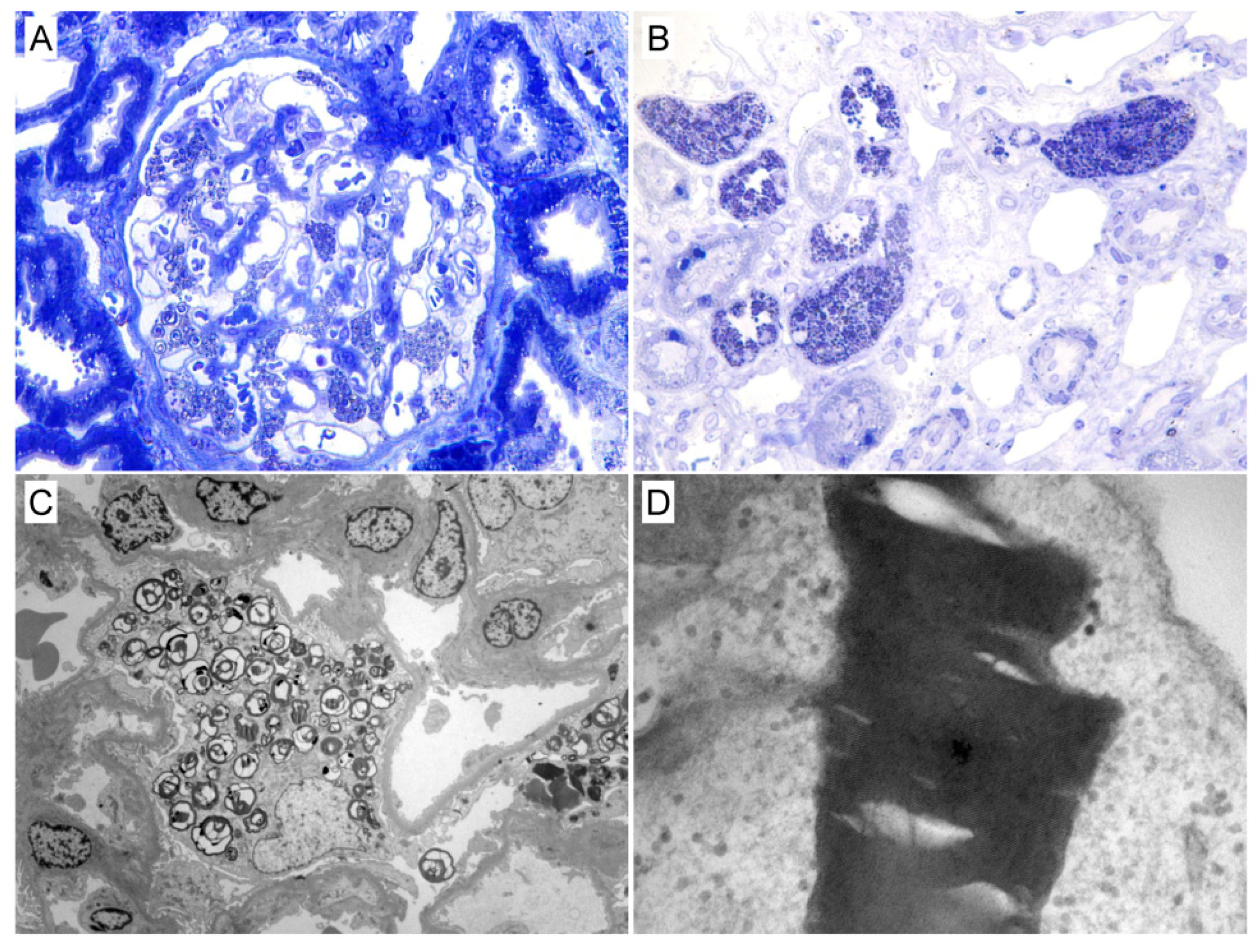
3.2. Kidney Biopsy Findings
3.3. Treatment
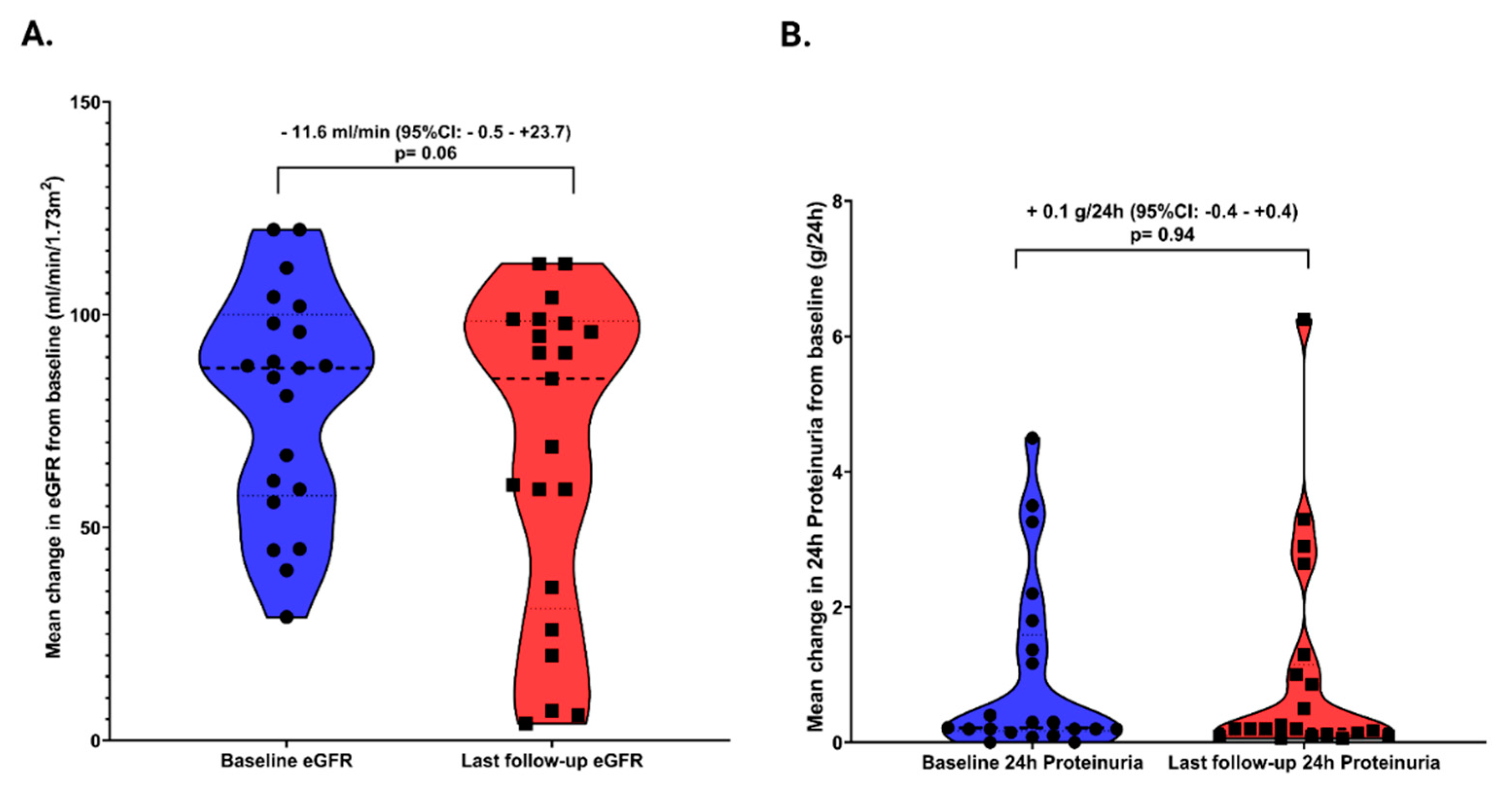
3.4. Outcomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Mauer, M.; Glynn, E.; Svarstad, E.; Tøndel, C.; Gubler, M.C.; West, M.; Sokolovskiy, A.; Whitley, C.; Najafian, B. Mosaicism of podocyte involvement is related to podocyte injury in females with Fabry disease. PLoS ONE 2014, 9, e112188. [Google Scholar] [CrossRef]
- Desnick, R.J.; Wasserstein, M.P.; Banikazemi, M. Fabry disease (alpha-galactosidase A deficiency): Renal involvement and enzyme replacement therapy. Contrib. Nephrol. 2001, 136, 174–192. [Google Scholar] [CrossRef]
- Gubler, M.C.; Lenoir, G.; Grunfeld, J.P.; Ulmann, A.; Droz, D.; Habib, R. Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. Kidney Int. 1978, 13, 223–235. [Google Scholar] [CrossRef]
- Tondel, C.; Bostad, L.; Hirth, A.; Svarstad, E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am. J. Kidney Dis. 2008, 51, 767–776. [Google Scholar] [CrossRef]
- Najafian, B.; Svarstad, E.; Bostad, L.; Gubler, M.C.; Tøndel, C.; Whitley, C.; Mauer, M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011, 79, 663–670. [Google Scholar] [CrossRef]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 2002, 13 (Suppl. 2), S134–S138. [Google Scholar] [CrossRef]
- Biegstraaten, M.; Arngrímsson, R.; Barbey, F.; Boks, L.; Cecchi, F.; Deegan, P.B.; Feldt-Rasmussen, U.; Geberhiwot, T.; Germain, D.P.; Hendriksz, C.; et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The European Fabry Working Group consensus document. Orphanet J. Rare Dis. 2015, 10, 36. [Google Scholar] [CrossRef]
- Van der Tol, L.; Svarstad, E.; Ortiz, A.; Tøndel, C.; Oliveira, J.P.; Vogt, L.; Waldek, S.; Hughes, D.A.; Lachmann, R.H.; Terryn, W.; et al. Chronic kidney disease and an uncertain diagnosis of Fabry disease: Approach to a correct diagnosis. Mol. Genet. Metab. 2015, 114, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Waldek, S.; Feriozzi, S. Fabry nephropathy: A review-how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.A.B.; Moura-Neto, J.A.; Dos Reis, M.A.; Vieira Neto, O.M.; Barreto, F.C. Renal Manifestations of Fabry Disease: A Narrative Review. Can. J. Kidney Health Dis. 2021, 8, 2054358120985627. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Brand, E. Precision medicine in Fabry disease. Nephrol. Dial. Transpl. 2021, 36, 14–23. [Google Scholar] [CrossRef]
- Hughes, D.A.; Aguiar, P.; Deegan, P.B.; Ezgu, F.; Frustaci, A.; Lidove, O.; Linhart, A.; Lubanda, J.C.; Moon, J.C.; Nicholls, K.; et al. Early indicators of disease progression in Fabry disease that may indicate the need for disease-specific treatment initiation: Findings from the opinion-based PREDICT-FD modified Delphi consensus initiative. BMJ Open 2020, 10, e035182. [Google Scholar] [CrossRef]
- Di Nora, C.; Livi, U. Heart transplantation in cardiac storage diseases: Data on Fabry disease and cardiac amyloidosis. Curr. Opin. Organ Transpl. 2020, 25, 211–217. [Google Scholar] [CrossRef]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Rombach, S.M.; Baas, M.C.; ten Berge, I.J.M.; Krediet, R.T.; Bemelman, F.J.; Hollak, C.E.M. The value of estimated GFR in comparison to measured GFR for the assessment of renal function in adult patients with Fabry disease. Nephrol. Dial. Transpl. 2010, 25, 2549–2556. [Google Scholar] [CrossRef][Green Version]
- Schwartz, G.J.; Work, D.F. Measurement and estimation of GFR in children and adolescents. Clin. J. Am. Soc. Nephrol. 2009, 4, 1832–1843. [Google Scholar] [CrossRef]
- KDIGO CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 136–150. [Google Scholar]
- Beck, M. The Mainz Severity Score Index (MSSI): Development and validation of a system for scoring the signs and symptoms of Fabry disease. Acta Paediatr. 2006, 95, 43–46. [Google Scholar] [CrossRef]
- Order of Romanian Ministry of Health and Romania National Insurance House (No. 536 M.O.525 BIS/21.08.2013) Regarding Therapeutic Protocol for Medical Practice on the Prescription, Monitoring and Reimbursement of Enzyme Replacement Therapy for Fabry Disease, Appendix 2, Page 9–15. Available online: http://cas.cnas.ro/castr/media/pageFiles/Ordinul%20536_2013_protocoale.pdf (accessed on 2 May 2022).
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef]
- Fogo, A.B.; Bostad, L.; Svarstad, E.; Cook, W.J.; Moll, S.; Barbey, F.; Geldenhuys, L.; West, M.; Ferluga, D.; Vujkovac, B.; et al. Scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol. Dial. Transpl. 2010, 25, 2168–2177. [Google Scholar] [CrossRef]
- Kim, I.Y.; Lee, H.J.; Cheon, C.K. Fabry nephropathy before and after enzyme replacement therapy: Important role of renal biopsy in patients with Fabry disease. Kidney Res. Clin. Pract. 2021, 40, 611–619. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, J.; Liang, S.; Wang, J.; Liu, Z. Clinic-Pathologic Features and Renal Outcome of Fabry Disease: Data from a Chinese Cohort. Am. J. Nephrol. 2018, 48, 137–146. [Google Scholar] [CrossRef]
- Germain, D.P.; Waldek, S.; Banikazemi, M.; Bushinsky, D.A.; Charrow, J.; Desnick, R.J.; Lee, P.; Loew, T.; Vedder, A.C.; Abichandani, R.; et al. Sustained, Long-Term Renal Stabilization After 54 Months of Agalsidase β Therapy in Patients with Fabry Disease. J. Am. Soc. Nephrol. 2007, 18, 1547–1557. [Google Scholar] [CrossRef]
- Valbuena, C.; Carvalho, E.; Bustorff, M.; Ganhão, M.; Relvas, S.; Nogueira, R.; Carneiro, F.; Oliveira, J.P. Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Arch. 2008, 453, 329–338. [Google Scholar] [CrossRef]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.H.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R.; et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef]
- Warnock, D.G.; Ortiz, A.; Mauer, M.; Linthorst, G.E.; Oliveira, J.P.; Serra, A.L.; Maródi, L.; Mignani, R.; Vujkovac, B.; Beitner-Johnson, D.; et al. Renal outcomes of agalsidase beta treatment for Fabry disease: Role of proteinuria and timing of treatment initiation. Nephrol. Dial. Transpl. 2012, 27, 1042–1049. [Google Scholar] [CrossRef]
- Arends, M.; Wijburg, F.A.; Wanner, C.; Vaz, F.M.; Van Kuilenburg, A.B.P.; Hughes, D.A.; Biegstraaten, M.; Mehta, A.; Hollak, C.E.M.; Langeveld, M. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol. Genet. Metab. 2017, 121, 157–161. [Google Scholar] [CrossRef]
- Lenders, M.; Schmitz, B.; Stypmann, J.; Duning, T.; Brand, S.M.; Kurschat, C.; Brand, E. Renal function predicts long-term outcome on enzyme replacement therapy in patients with Fabry disease. Nephrol. Dial. Transpl. 2017, 32, 2090–2097. [Google Scholar] [CrossRef]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Ronald, E.; Gordon, A.; Collins, B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef]
- Tøndel, C.; Bostad, L.; Larsen, K.K.; Hirth, A.; Vikse, B.E.; Houge, G.; Svarstad, E. Agalsidase benefits renal histology in young patients with Fabry disease. J. Am. Soc. Nephrol. 2013, 24, 137–148. [Google Scholar] [CrossRef]
- Najafian, B.; Tøndel, C.; Svarstad, E.; Sokolovkiy, A.; Smith, K.; Mauer, M. One Year of Enzyme Replacement Therapy Reduces Globotriaosylceramide Inclusions in Podocytes in Male Adult Patients with Fabry Disease. PLoS ONE 2016, 11, e0152812. [Google Scholar] [CrossRef]
- Ramaswami, U.; Beck, M.; Hughes, D.; Kampmann, C.; Botha, J.; Pintos-Morell, G.; West, M.L.; Niu, D.M.; Nicholls, K.; Giugliani, R.; et al. Cardio-Renal Outcomes With Long-Term Agalsidase Alfa Enzyme Replacement Therapy: A 10-Year Fabry Outcome Survey (FOS) Analysis. Drug Des. Dev. Ther. 2019, 13, 3705–3715. [Google Scholar] [CrossRef]
- Schiffmann, R.; Hughes, D.A.; Linthorst, G.E.; Ortiz, A.; Svarstad, E.; Warnock, D.G.; West, M.L.; Wanner, C.; Conference Participants. Screening, diagnosis, and management of patients with Fabry disease: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 284–293. [Google Scholar] [CrossRef]
- Hughes, D.A.; Aguiar, P.; Lidove, O.; Nicholls, K.; Nowak, A.; Thomas, M.; Torra, R.; Vujkovac, B.; West, M.L.; Feriozzi, S. Do clinical guidelines facilitate or impede drivers of treatment in Fabry disease? Orphanet. J. Rare Dis. 2022, 17, 42. [Google Scholar] [CrossRef]
- Svarstad, E.; Marti, H.P. The Changing Landscape of Fabry Disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 569–576. [Google Scholar] [CrossRef]
- Wanner, C.; Germain, D.P.; Hilz, M.J.; Spada, M.; Falissard, B.; Elliott, P.M. Therapeutic goals in Fabry disease: Recommendations of a European expert panel, based on current clinical evidence with enzyme replacement therapy. Mol. Genet. Metab. 2019, 126, 210–211. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Sex/Age (yr) | HTN/DM | Mutation | eGFR (mL/min/1.73 m2) | CKD Stage | UACR (mg/g) | Proteinuria (g/24 h) | RAAS Blocker | ERT Duration (mo) | Cardiac Involvement | Neurologic Manifestation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/17 | +/− | c.779G > A | 44.7 | 3 | 500 | 3.26 | + | 0 | − | + |
| 2 | M/10 | −/+ | c.797A > C | 87.5 | 2 | 10 | 0.08 | − | 0 | + | − |
| 3 | M/41 | −/− | c.863C > A | 45 | 3 | 100 | 1.37 | + | 0 | + | + |
| 4 | M/26 | −/− | c.836A > G | 89 | 2 | 345 | 0.3 | − | 0 | − | + |
| 5 | M/44 | −/− | c.644A > G | 29 | 4 | 600 | 3.5 | − | 0 | − | − |
| 6 | M/39 | −/− | c.671delA | 67 | 2 | 80 | 0.15 | + | 0 | + | + |
| 7 | M/29 | −/− | c.797A > C | 120 | 1 | 10 | 0.2 | − | 0 | − | + |
| 8 | F/50 | +/− | c.797A > C | 56 | 3 | 100 | 0.4 | + | 0 | + | + |
| 9 | F/55 | +/− | c.1224del66 | 61 | 2 | 819 | 1.8 | + | 0 | + | + |
| 10 | F/49 | −/− | c.779G > A | 104.2 | 1 | 100 | 0.2 | − | 0 | − | + |
| 11 | F/46 | −/− | c.797A > C | 96 | 1 | 10 | 0.1 | − | 0 | − | + |
| 12 | F/30 | −/− | c.797A > C | 88 | 2 | 10 | 0.2 | − | 0 | − | − |
| 13 | F/35 | −/− | c.295C > T | 81 | 2 | 20 | 0.3 | − | 0 | − | − |
| 14 | F/63 | +/− | c.1228A > G | 59 | 3 | 30 | 0.22 | + | 0 | + | + |
| 15 | F/35 | +/− | c.485G > A | 40 | 3 | 900 | 4.5 | − | 0 | − | + |
| 16 | F/61 | +/+ | c.141G > A | 85.3 | 2 | 20 | ND | + | 0 | + | + |
| 17 | M/43 | −/− | c.836A > G | 111 | 1 | 300 | 1.17 | + | 12 | − | + |
| 18 | M/32 | −/− | c.1121_1123delAAG | 120 | 1 | 20 | ND | − | 144 | + | + |
| 19 | M/58 | −/− | c.295C > T | 102 | 1 | 10 | 0.2 | − | 72 | − | + |
| 20 | M/37 | +/− | c.485G > A | 98 | 1 | 500 | 2.2 | + | 120 | + | + |
| 21 | F/57 | +/− | c.485G > A | 88 | 2 | 150 | 0.2 | + | 27 | − | + |
| Variables | Overall (n = 21) | (−) Combined Endpoint (n = 15) | (+) Combined Endpoint (n = 6) | p Value | |
|---|---|---|---|---|---|
| General features | Age at baseline, years, mean ± SD | 43.7 ± 14.2 | 45.7 ± 14.9 | 36.7 ± 9.5 | 0.27 |
| Age at diagnosis, years, mean ± SD | 36.1 ± 14.6 | 39.5 ± 15.1 | 27.7 ±10.1 | 0.10 | |
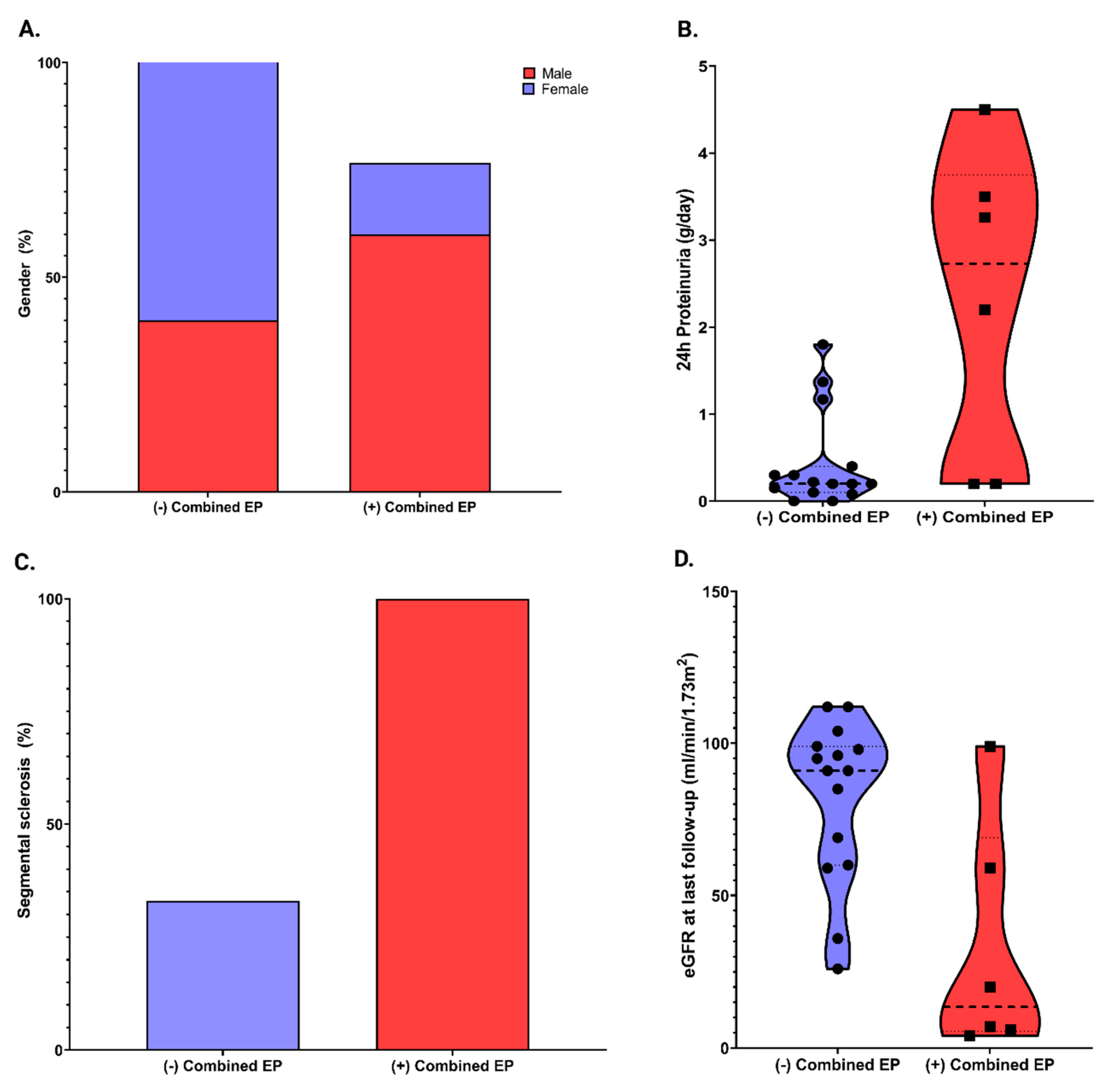
| Gender, n (%) | 0.06 | ||||
| Male | 11 (52.4%) | 6 (40%) | 5 (83.3%) | ||
| Female | 10(47.6%) | 9 (60%) | 1 (16.7%) | ||
| Hypertension, n (%) | 8 (31.8%) | 5 (33.3%) | 3 (50%) | 0.63 | |
| Diabetes, n (%) | 2 (9.5%) | 2 (13.3%) | 0 (0%) | 0.9 | |
| Obesity n (%) | 3 (14.3%) | 2 (13.3%) | 1 (16.7%) | 1 | |
| Dyslipidemia, n (%) | 11 (52.4%) | 8 (53.3%) | 3 (50%) | 1 | |
| Stroke, n (%) | 4 (19%) | 4 (26.7%) | 0 (0%) | 0.28 | |
| Heart failure, n (%) | 9 (42.9%) | 7 (46.7%) | 2 (33.3%) | 0.44 | |
| eGFR at baseline, ml/min/1.73 m2, mean ± SD | 79.6 ± 26.7 | 82.5 ± 21.3 | 72.2 ± 38.7 | 0.44 | |
| eGFR at last follow-up, ml/min/1.73 m2, mean ± SD | 68.0 ± 37.3 | 82.2 ± 26.4 | 32.5 ± 38.5 | 0.003 | |
| UACR at baseline, mg/g, median (IQR) | 100 (15–422.6) | 80 (20–300) | 500 (10–675) | 0.26 | |
| 24 h proteinuria at baseline, g/24 h, median (IQR) | 0.2 (0.1–1.5) | 0.4 (0.1–0.4) | 2.7 (0.2–3.7) | 0.02 | |
| Follow-up period, months, mean ± SD | 47.7 ± 19.1 | 49.2 ± 19.7 | 44.0 ± 18.4 | 0.58 | |
| Fabry features | Total MSSI at baseline, points, median (IQR) | 22 (10.5–25) | 22 (10–25) | 22 (12.7–24.5) | 0.91 |
| Neurologic manifestation at baseline, n (%) | 18 (85.7%) | 13 (86.6%) | 5 (83.3%) | 0.5 | |
| Hypertrophic cardiomyopathy at baseline, n (%) | 9 (42.8%) | 7 (46.6%) | 2 (33.3%) | 0.47 | |
| Arrhythmias at baseline (%) | 8 (38.1%) | 7 (46.7%) | 1 (16.7%) | 0.33 | |
| Pacemaker at baseline (%) | 1 (4.8%) | 1(6.7%) | 0 (0%) | 1 | |
| αGAL A α-GLA activity, nmol/h/mg, median (IQR) | 0.4 (0.1–1.2) | 0.6 (0.2–1.5) | 0.3 (0–0.4) | 0.13 | |
| Plasma Llyso-GL3 at baseline, ng/mL, median (IQR) | 7.6 (5.5–34.6) | 6.8 (4.9–22.5) | 27 (9.2–105.5) | 0.22 | |
| Kidney biopsy | Glomeruli number, median (IQR) | 8 (5–10) | 8 (5–12) | 6 (4–8) | 0.15 |
| Segmental sclerosis, n (%) | 11 (52.4%) | 5 (33%) | 6 (100%) | 0.009 | |
| Global sclerosis, n (%) | 6 (28.6%) | 3 (20%) | 3 (50%) | 0.29 | |
| Interstitial fibrosis, n (%) <25% 25–50% | 11 (52.4%) 10 (47.6%) | 9 (60%) 6 (40%) | 2 (33.3%) 4 (66.7%) | 0.36 | |
| Tubular atrophy, n (%) | 9 (42.9%) | 5 (33.3%) | 4 (66.7%) | 0.33 | |
| Arteriopathy, n (%) | 8 (38.1%) | 5 (33%) | 3 (50%) | 0.63 | |
| Podocyte GL3 deposits, n (%) | 21 (100%) | 15 (100%) | 6 (100%) | 1 | |
| Tubular GL3 deposits, n (%) | 21 (100%) | 21 (100%) | 21 (100%) | 1 | |
| Glomerular endothelial cell GL3 deposits, n (%) | 20 (95.2%) | 14 (93.3%) | 6 (100%) | 1 | |
| Treatment | FD-specific therapy at the moment of KB, n (%) | 5 (23.8%) | 3 (20%) | 2 (33%) | 0.6 |
| FD-specific therapy after KB, n (%) | 20 (95.2%) | 14 (93.3%) | 6 (100%) | 1 | |
| FD-specific therapy duration, months, median (IQR) | 36 (13–52.5) | 37 (12.2–49.5) | 30 (12.7–60) | 0.98 | |
| RAAS inhibitors, n (%) | 10 (47.6%) | 7 (46.7%) | 3 (50%) | 1 | |
| Outcomes | 50% decrease in eGFR, n (%) | 5 (23.8%) | 0 (0%) | 5 (83.3%) | <0.001 |
| ESKD KF, n (%) | 3 (14.3%) | 0 (%) | 3 (50%) | 0.01 | |
| Mortality, n (%) | 2 (9.5%) | 0 (0%) | 2 (33.3%) | 0.07 | |
| Combined endpoint, n (%) | 6 (28.6) | − | − | − | |
| Patient No. | Sex/Age (yr) | Mutation | Variant According to ACMG | Type of Mutation | Phenotype | α-GLA Activity (Reference Range > 2.8 µmol/L/h) | Lyso-GL3 (Reference Range 0–3.5 ng/mL) | MSSI Score |
|---|---|---|---|---|---|---|---|---|
| 1 | M/17 | c.779G > A | Pathogenic | Missense | Classic | 0.5 | NA | 26 |
| 2 | M/10 | c.797A > C | Pathogenic | Missense | Classic | 0 | 101.1 | 9 |
| 3 | M/41 | c.863C > A | Pathogenic | Missense | Classic | 0.3 | 98 | 24 |
| 4 | M/26 | c.836A > G | Pathogenic | Missense | Classic | 0.13 | 22.5 | 25 |
| 5 | M/44 | c.644A > G | Pathogenic | Missense | Late-onset | 0.4 | 34.6 | 20 |
| 6 | M/39 | c.671delA | Pathogenic | Deletion | Classic | 2 | 5.5 | 32 |
| 7 | M/29 | c.797A > C | Pathogenic | Missense | Classic | 0 | 129.2 | 9 |
| 8 | F/50 | c.797A > C | Pathogenic | Missense | Classic | 1.2 | 6.7 | 33 |
| 9 | F/55 | c.1224del66 | Pathogenic | Deletion | Classic | 1 | 7.6 | 22 |
| 10 | F/49 | c.779G > A | Pathogenic | Missense | Classic | NA | 6.8 | 3 |
| 11 | F/46 | c.797A > C | Pathogenic | Missense | Classic | 0.4 | 10.7 | 14 |
| 12 | F/30 | c.797A > C | Pathogenic | Missense | Classic | 0.7 | 3.9 | 10 |
| 13 | F/35 | c.295C > T | Pathogenic | Nonsense | Classic | 1.5 | 4.4 | 7 |
| 14 | F/63 | c.1228A > G | Pathogenic | Missense | Classic | 1.8 | 4.9 | 42 |
| 15 | F/35 | c.485G > A | Pathogenic | Nonsense | Classic | 0.32 | 5.8 | 14 |
| 16 | F/61 | c.141G > A | Pathogenic | Nonsense | Classic | 3.3 | 3.3 | 13 |
| 17 | M/43 | c.836A > G | Pathogenic | Missense | Classic | 0.1 | 42.3 | 25 |
| 18 | M/32 | c.1121_1123delAAG | Pathogenic | Deletion | Classic | 0.5 | 16.8 | 22 |
| 19 | M/58 | c.295C > T | Pathogenic | Nonsense | Classic | 0.1 | NA | 24 |
| 20 | M/37 | c.485G > A | Pathogenic | Nonsense | Classic | 0.1 | 19.4 | 24 |
| 21 | F/57 | c.485G > A | Pathogenic | Nonsense | Classic | 0.3 | 6.3 | 11 |
| Patient No. | Sex/Age (yr) | Global Sclerosis | Segmental Sclerosis | Glomerular Hyaline | Interstitial Fibrosis | Tubular Atrophy | Arterio- Pathy | Podocyte GL3 Deposits | Tubular GL3 Deposits | Glomerular Endothelial Cell GL3 Deposits |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/17 | + | + | − | + | NA | + | + | + | + |
| 2 | M/10 | − | − | − | − | − | − | + | + | + |
| 3 | M/41 | − | − | − | + | NA | + | + | + | + |
| 4 | M/26 | − | − | − | − | − | − | + | + | + |
| 5 | M/44 | − | + | + | + | + | + | + | + | + |
| 6 | M/39 | + | + | + | + | + | − | + | + | + |
| 7 | M/29 | − | + | − | − | − | − | + | + | + |
| 8 | F/50 | + | + | − | + | + | + | + | + | + |
| 9 | F/55 | + | + | + | + | + | + | + | + | + |
| 10 | F/49 | − | − | − | − | + | − | + | + | + |
| 11 | F/46 | − | − | − | − | − | − | + | + | + |
| 12 | F/30 | − | − | − | − | − | − | + | + | + |
| 13 | F/35 | − | − | − | − | − | − | + | + | + |
| 14 | F/63 | − | − | − | − | NA | − | + | + | − |
| 15 | F/35 | + | + | − | − | − | + | + | + | + |
| 16 | F/61 | − | − | − | + | NA | + | + | + | + |
| 17 * | M/43 | − | + | − | + | + | + | + | + | + |
| 18 * | M/32 | − | − | − | − | − | − | + | + | + |
| 19 * | M/58 | − | + | − | + | + | − | + | + | + |
| 20 * | M/37 | + | + | − | + | + | − | + | + | + |
| 21 * | F/57 | − | + | + | − | − | − | + | + | + |
| Variables | Treatment before KB (n = 5) | Treatment after (KB) (n = 16) | p Value |
|---|---|---|---|
| Combined EP, n (%) | 2 (40%) | 4 (25%) | 0.58 |
| Segmental sclerosis, n (%) | 4 (80%) | 7 (44%) | 0.30 |
| Global sclerosis, n (%) | 1 (20%) | 5 (31.3%) | 1 |
| Interstitial fibrosis, n (%) <25% 25–50% | 2 (40%) 3 (60%) | 9 (56.3%) 7 (43.8%) | 0.63 |
| Tubular atrophy, n (%) | 3 (60%) | 6 (37.5%) | 0.61 |
| Arteriopathy, n (%) | 1 (20%) | 7 (43.8%) | 0.60 |
| Podocyte GL3 deposits, n (%) | 5 (100%) | 16 (100%) | 1 |
| Tubular GL3 deposits, n (%) | 5 (100%) | 16 (100%) | 1 |
| Glomerular endothelial cell GL3 deposits, n (%) | 5 (100%) | 16 (100%) | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusu, E.-E.; Zilisteanu, D.-S.; Ciobotaru, L.-M.; Gherghiceanu, M.; Procop, A.; Jurcut, R.-O.; Dulamea, A.O.; Sorohan, B.M. The Impact of Kidney Biopsy for Fabry Nephropathy Evaluation on Patients’ Management and Long-Term Outcomes: Experience of a Single Center. Biomedicines 2022, 10, 1520. https://doi.org/10.3390/biomedicines10071520
Rusu E-E, Zilisteanu D-S, Ciobotaru L-M, Gherghiceanu M, Procop A, Jurcut R-O, Dulamea AO, Sorohan BM. The Impact of Kidney Biopsy for Fabry Nephropathy Evaluation on Patients’ Management and Long-Term Outcomes: Experience of a Single Center. Biomedicines. 2022; 10(7):1520. https://doi.org/10.3390/biomedicines10071520
Chicago/Turabian StyleRusu, Elena-Emanuela, Diana-Silvia Zilisteanu, Lucia-Mihaela Ciobotaru, Mihaela Gherghiceanu, Alexandru Procop, Ruxandra-Oana Jurcut, Adriana Octaviana Dulamea, and Bogdan Marian Sorohan. 2022. "The Impact of Kidney Biopsy for Fabry Nephropathy Evaluation on Patients’ Management and Long-Term Outcomes: Experience of a Single Center" Biomedicines 10, no. 7: 1520. https://doi.org/10.3390/biomedicines10071520
APA StyleRusu, E.-E., Zilisteanu, D.-S., Ciobotaru, L.-M., Gherghiceanu, M., Procop, A., Jurcut, R.-O., Dulamea, A. O., & Sorohan, B. M. (2022). The Impact of Kidney Biopsy for Fabry Nephropathy Evaluation on Patients’ Management and Long-Term Outcomes: Experience of a Single Center. Biomedicines, 10(7), 1520. https://doi.org/10.3390/biomedicines10071520