
KHK, PNPLA3 and PPAR as Novel Targets for the Anti-Steatotic Action of Bempedoic Acid

 , , ,
, , ,  ,
,  ,
,
Abstract
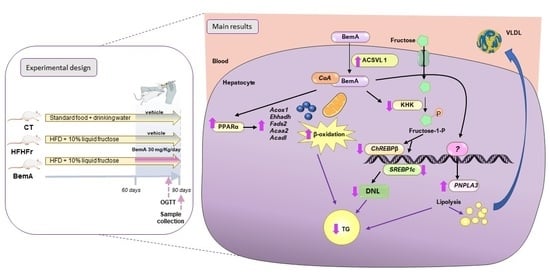
:
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Oral Glucose Tolerance Test
2.3. Sample Preparation
2.4. Serum Analytes
2.5. Histological Studies
2.6. RNA Extraction and Quantitative RT-PCR Analysis
2.7. Protein Extraction and Western Blot
2.8. Liver Lipidomic Analysis
2.9. PPAR Array and qPCR Validation
2.10. PPAR Luciferase Assay
2.11. β-Oxidation Activity
2.12. Statistical Analysis
3. Results
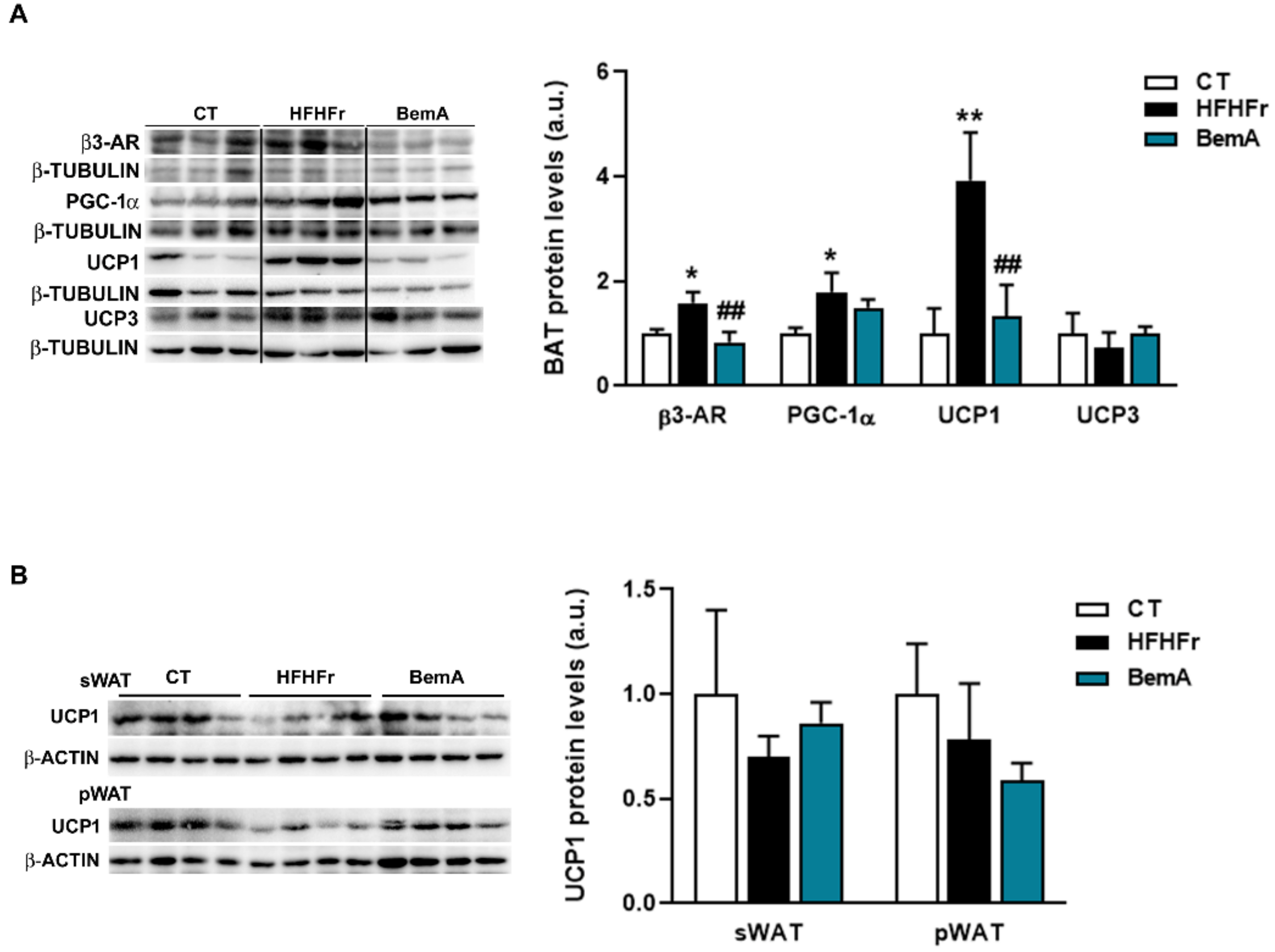
3.1. Treatment with Bempedoic Acid Reduces Body Weight and Adiposity, and Increases Liver Weight, without Affecting the Expression of Thermogenesis Markers in WAT
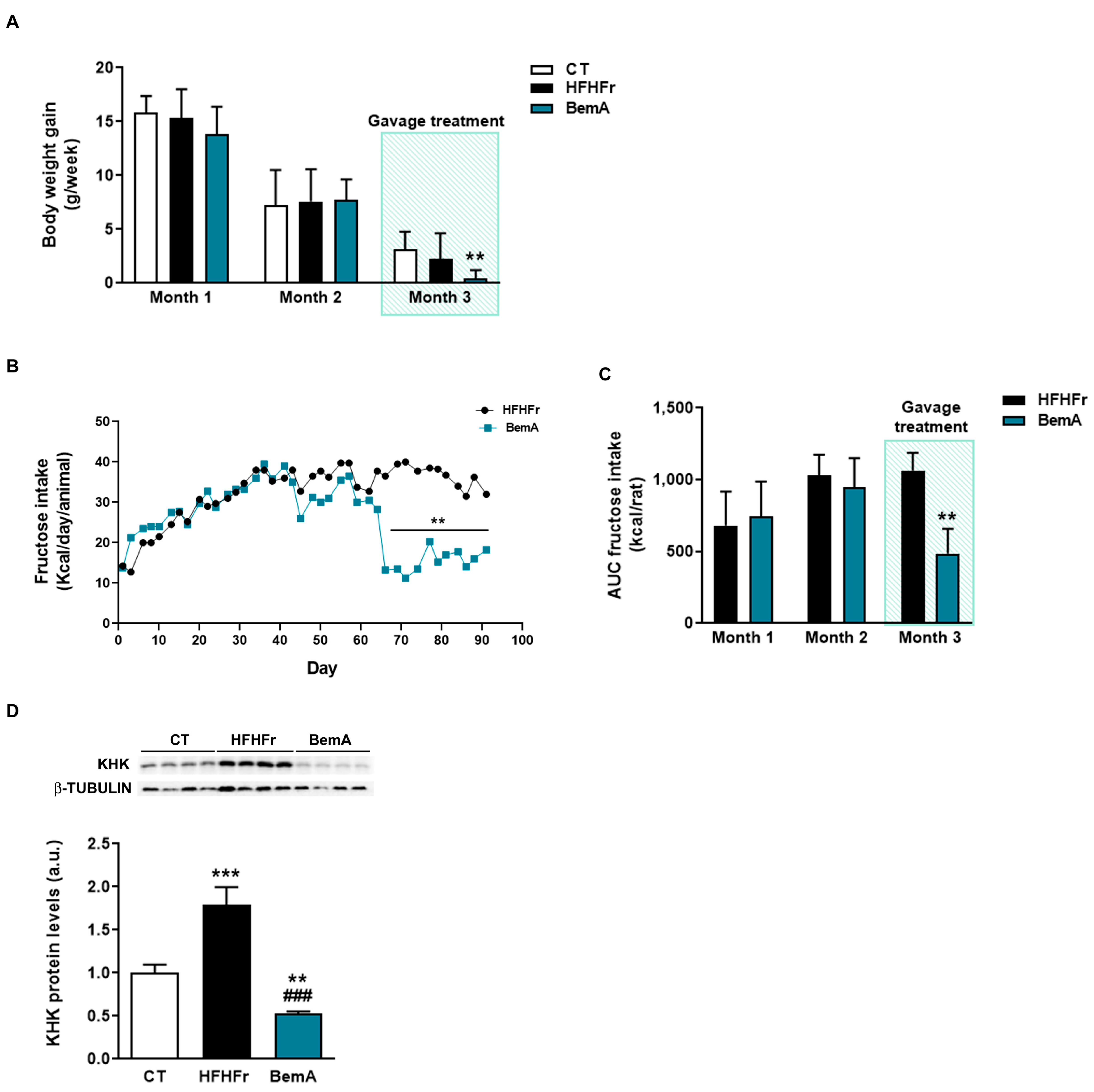
3.2. Rats Treated with Bempedoic Acid Reduce Their Fructose Ingestion and the Expression of Hepatic Ketohexokinase
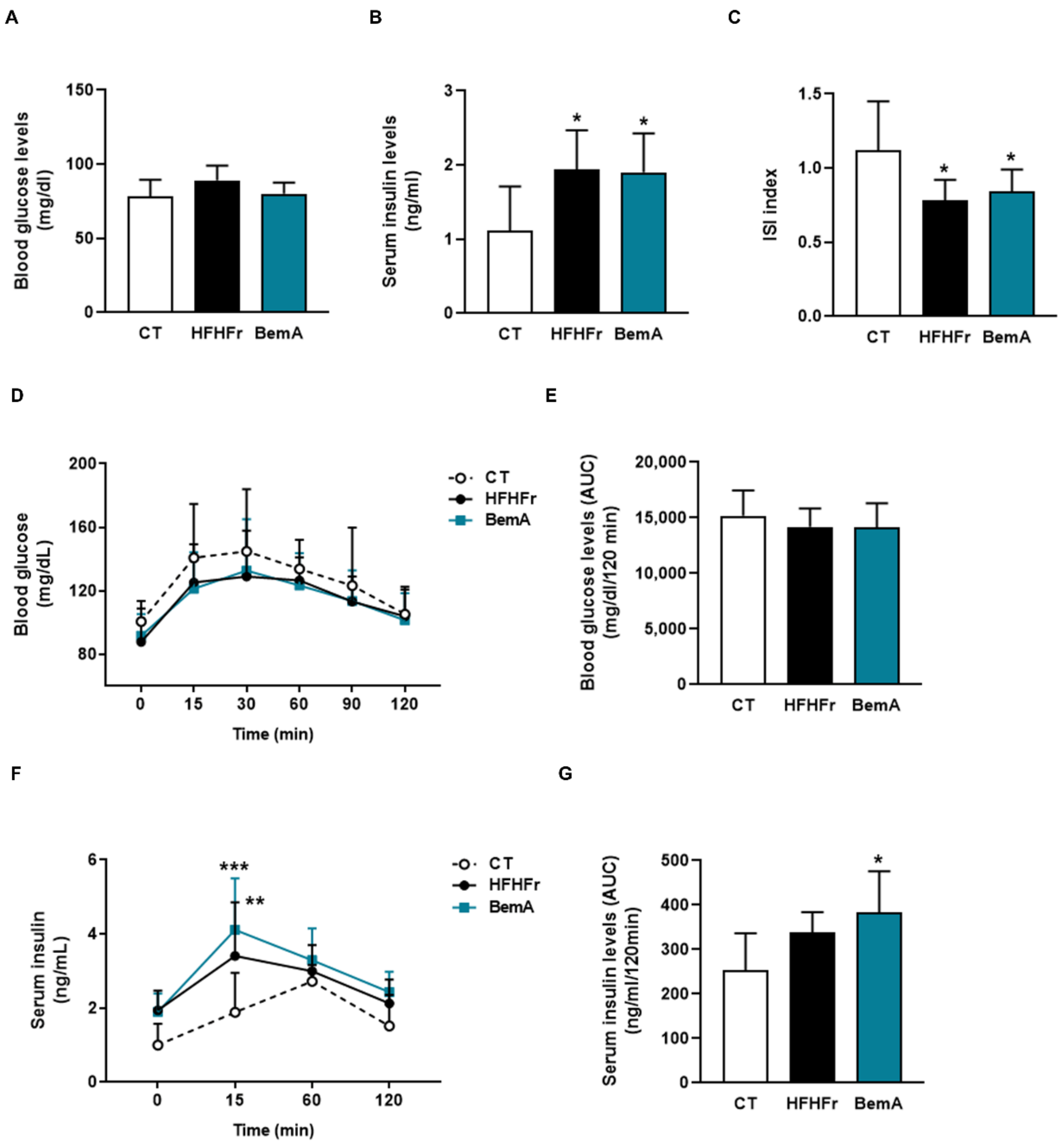
3.3. Bempedoic Acid Does Not Alter Insulin Sensitivity in High-Fat, High-Fructose Rats
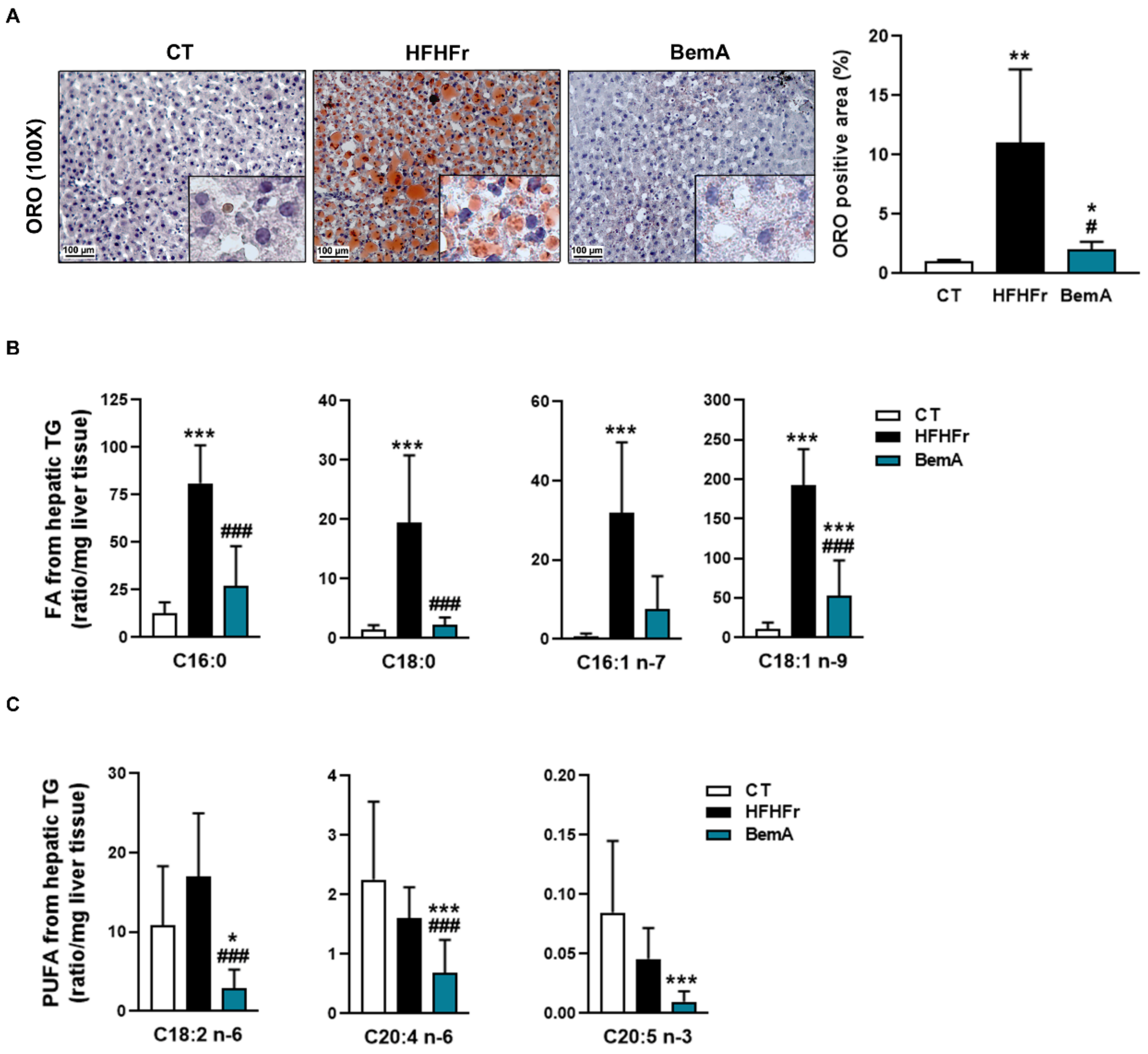
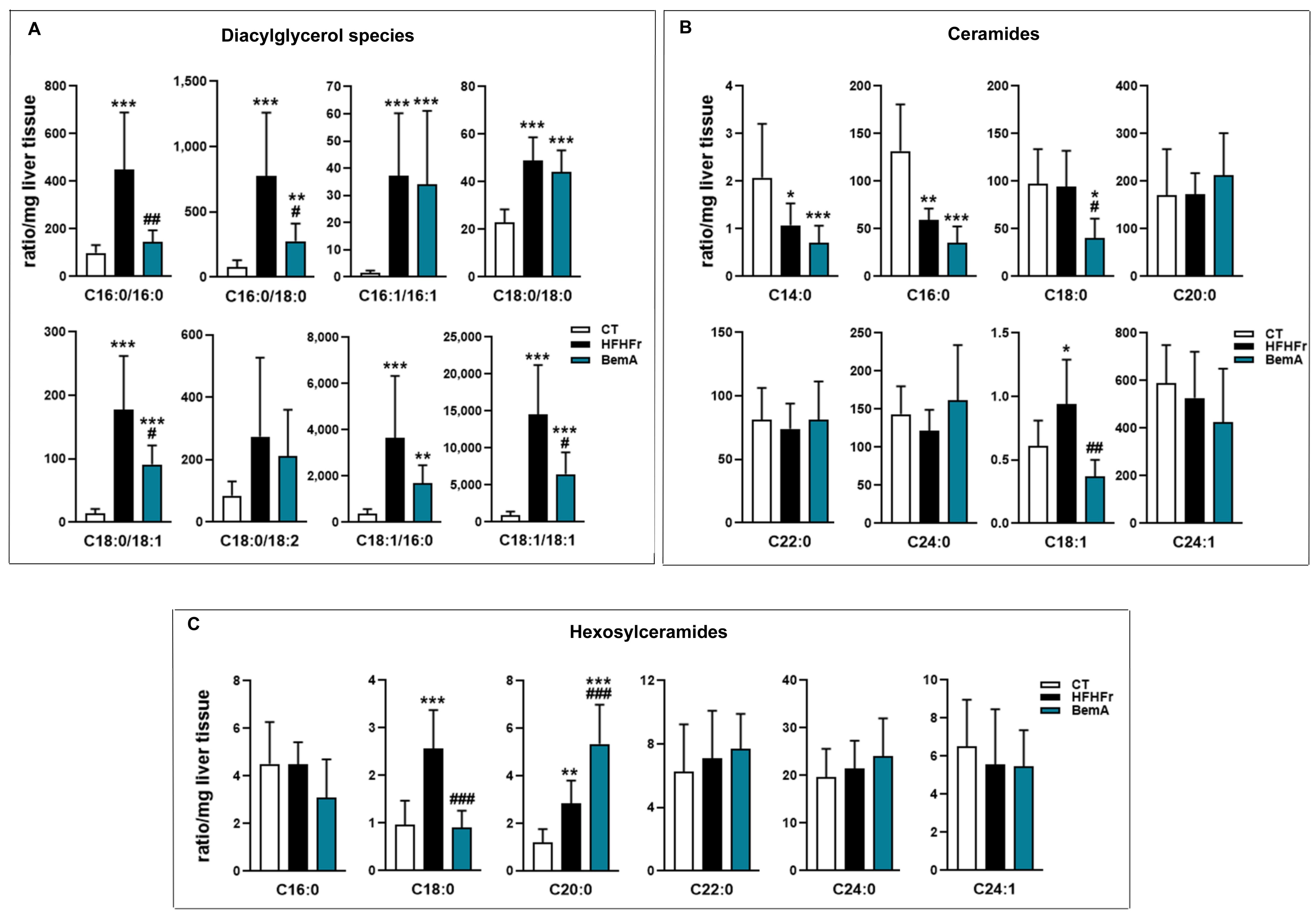
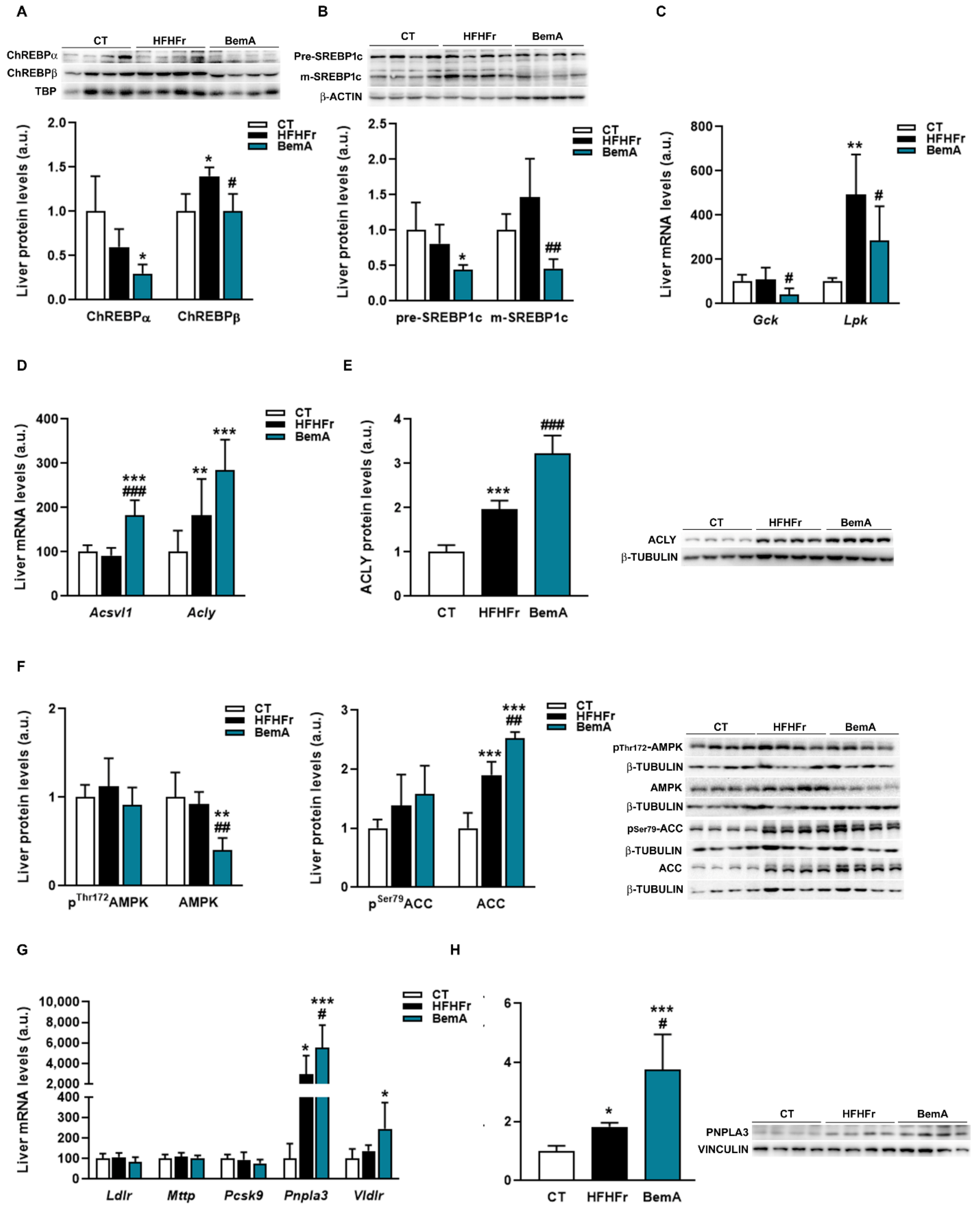
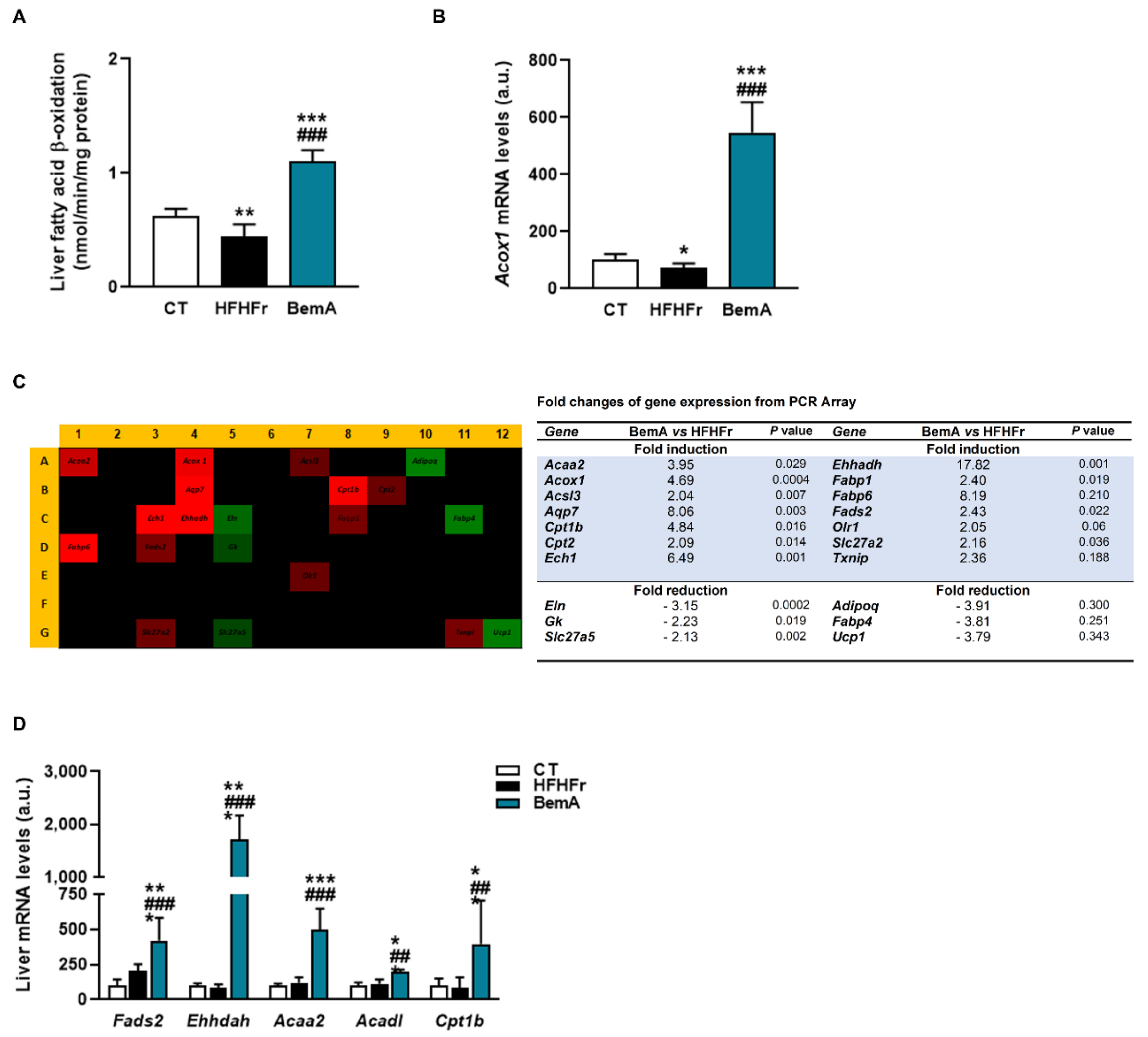
3.4. Bempedoic Acid Reduces Hepatic TG Accumulation Induced by the High-Fat, High-Fructose Diet and Significantly Alters Hepatic Lipid Metabolism
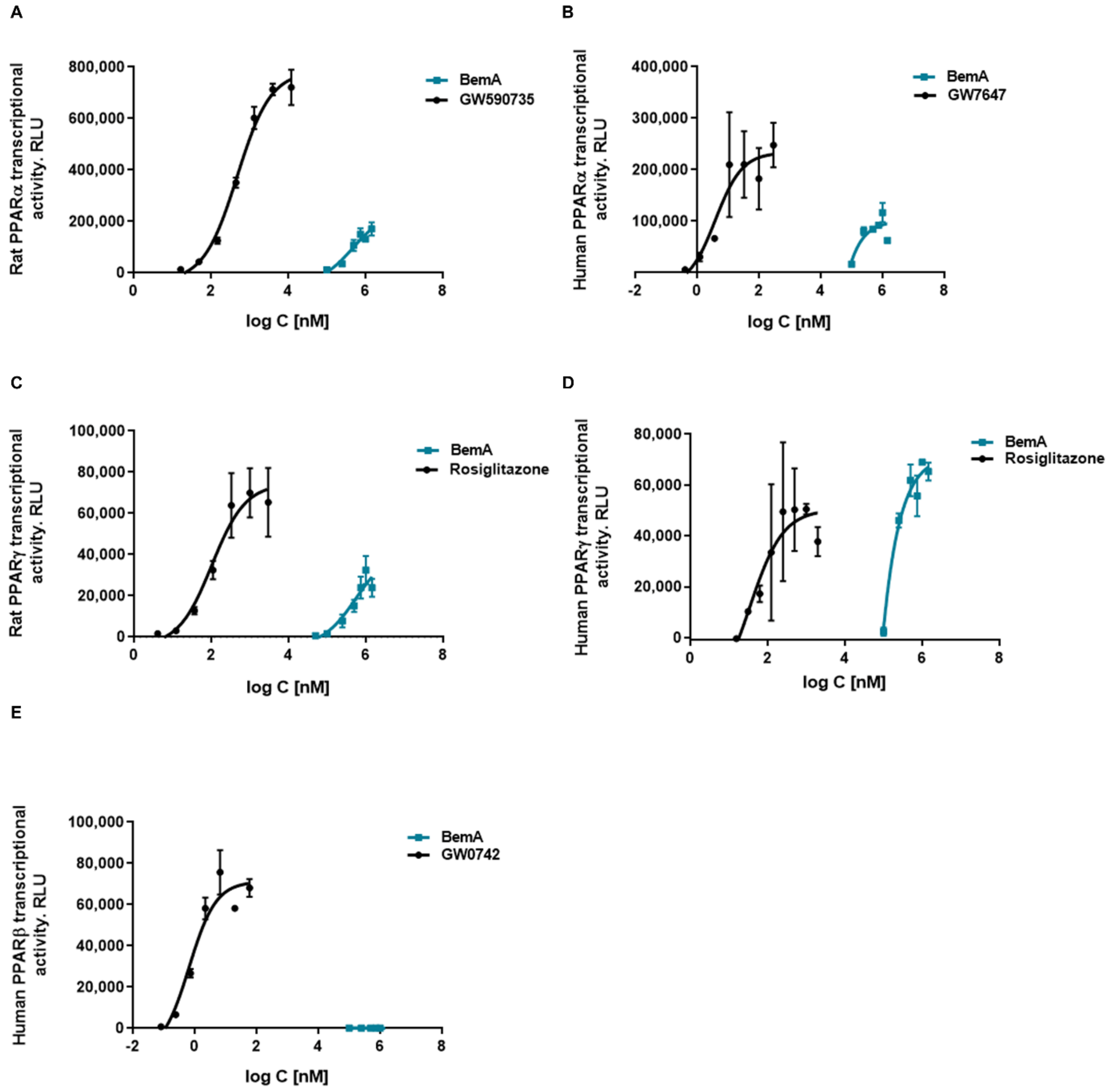
3.5. Bempedoic Acid Behaves as an Agonist of Human and Rat PPAR α/γ
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruscica, M.; Banach, M.; Sahebkar, A.; Corsini, A.; Sirtori, C.R. ETC-1002 (Bempedoic acid) for the management of hyperlipidemia: From preclinical studies to phase 3 trials. Expert Opin. Pharmacother. 2019, 20, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Zhang, L.; Xu, S.; Shen, A.-Z. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog. Lipid Res. 2019, 77, 101006. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 223–238. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- El-Agroudy, N.N.; Kurzbach, A.; Rodionov, R.N.; O’Sullivan, J.; Roden, M.; Birkenfeld, A.L.; Pesta, D.H. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends Endocrinol. Metab. 2019, 30, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, A.M.; Roglans, N.; Bentanachs, R.; Gené, M.; Sala-Vila, A.; Lázaro, I.; Rodríguez-Morató, J.; Sánchez, R.M.; Laguna, J.C.; Alegret, M. Effects of a Low Dose of Caffeine Alone or as Part of a Green Coffee Extract, in a Rat Dietary Model of Lean Non-Alcoholic Fatty Liver Disease without Inflammation. Nutrients 2020, 12, 3240. [Google Scholar] [CrossRef]
- Velázquez, A.M.; Bentanachs, R.; Sala-Vila, A.; Lázaro, I.; Rodríguez-Morató, J.; Sánchez, R.M.; Alegret, M.; Roglans, N.; Laguna, J.C. ChREBP-driven DNL and PNPLA3 Expression Induced by Liquid Fructose Are Essential in the Production of Fatty Liver and Hypertriglyceridemia in a High-fat Diet-fed Rat Model. Mol. Nutr. Food Res. 2022, 66, 2101115. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, L.; Wang, H.; Li, S.; Yu, Y.; You, J.; Zeng, R.; Gao, X.; Rui, L.; Li, W.; et al. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology 2009, 49, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, S.; Jiang, L.; Zhou, Y.; Li, Z.; Shao, M.; Li, W.; Liu, Y. Deficiency in hepatic ATP-citrate lyase affects VLDL-triglyceride mobilization and liver fatty acid composition in mice. J. Lipid Res. 2010, 51, 2516–2526. [Google Scholar] [CrossRef] [Green Version]
- Bentanachs, R.; Velázquez, A.M.; Sánchez, R.M.; Alegret, M.; Laguna, J.C.; Roglans, N. Bempedoic acid as a PPARα activator: New perspectives for hepatic steatosis treatment in a female rat experimental model. Clin. Investig. Arterioscler. 2022, 34, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Pinkosky, S.L.; Filippov, S.; Srivastava, R.; Hanselman, J.C.; Bradshaw, C.D.; Hurley, T.R.; Cramer, C.T.; Spahr, M.A.; Brant, A.F.; Houghton, J.L.; et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid Res. 2013, 54, 134–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lazarow, P.B. Assay of peroxisomal ß-oxidation of fatty acids. Methods Enzymol. 1981, 72, 315–319. [Google Scholar] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; MacLean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Mazzolini, G.; Sowa, J.-P.; Atorrasagasti, C.; Kücükoglu, Ö.; Syn, W.-K.; Canbay, A. Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence. Cells 2020, 9, 2458. [Google Scholar] [CrossRef]
- Miller, C.O.; Yang, X.; Lu, K.; Cao, J.; Herath, K.; Rosahl, T.W.; Askew, R.; Pavlovic, G.; Zhou, G.; Li, C.; et al. Ketohexokinase knockout mice, a model for essential fructosuria, exhibit altered fructose metabolism and are protected from diet-induced metabolic defects. Am. J. Physiol. Metab. 2018, 315, E386–E393. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubuquoy, C.; Robichon, C.; Lasnier, F.; Langlois, C.; Dugail, I.; Foufelle, F.; Girard, J.; Burnol, A.-F.; Postic, C.; Moldes, M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J. Hepatol. 2011, 55, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A Sequence Variation (I148M) in PNPLA3 Associated with Nonalcoholic Fatty Liver Disease Disrupts Triglyceride Hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- Qiao, A.; Liang, J.; Ke, Y.; Li, C.; Cui, Y.; Shen, L.; Zhang, H.; Cui, A.; Liu, X.; Liu, C.; et al. Mouse patatin-like phospholipase domain-containing 3 influences systemic lipid and glucose homeostasis. Hepatology 2011, 54, 509–521. [Google Scholar] [CrossRef]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Kotronen, A.; Johansson, L.E.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; Fisher, R.; et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Ka-man, V.L.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Mitsche, M.A.; Hobbs, H.H.; Cohen, J.C. Patatin-like phospholipase domain–containing protein 3 promotes transfers of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J. Biol. Chem. 2018, 293, 6958–6968. [Google Scholar] [CrossRef] [Green Version]
- Banini, B.A.; Kumar, D.P.; Cazanave, S.; Seneshaw, M.; Mirshahi, F.; Santhekadur, P.K.; Wang, L.; Guan, H.P.; Oseini, A.M.; Alonso, C.; et al. Identification of a Metabolic, Transcriptomic, and Molecular Signature of Patatin-Like Phospholipase Domain Containing 3–Mediated Acceleration of Steatohepatitis. Hepatology 2020, 73, 1290–1306. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Ståhlman, M.; Taskinen, M.-R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Maglio, C.; Pirazzi, C.; Burza, M.A.; Adiels, M.; Burch, L.; Donnelly, L.A.; Colhoun, H.; Doney, A.; Dillon, J.; et al. Paradoxical Lower Serum Triglyceride Levels and Higher Type 2 Diabetes Mellitus Susceptibility in Obese Individuals with the PNPLA3 148M Variant. PLoS ONE 2012, 7, e39362. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.; Gonzalez, F.J. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Liver Physiol. 2017, 312, G283–G299. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Am. J. Dig. Dis. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Hernandez, A.V.; Banach, M. Efficacy and safety of bempedoic acid for the treatment of hypercholesterolemia: A systematic review and meta-analysis. PLoS Med. 2020, 17, e1003121. [Google Scholar] [CrossRef]
- Holden, P.R.; Tugwood, J.D. Peroxisome proliferator-activated receptor alpha: Role in rodent liver cancer and species differences. J. Mol. Endocrinol. 1999, 22, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C.; Peters, J.M.; Klaunig, J.E. The PPARα-dependent rodent liver tumor response is not relevant to humans: Addressing misconceptions. Arch. Toxicol. 2017, 92, 83–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of Fenofibrate and Niacin on Intrahepatic Triglyceride Content, Very Low-Density Lipoprotein Kinetics, and Insulin Action in Obese Subjects with Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef]
- Oscarsson, J.; Önnerhag, K.; Risérus, U.; Sundén, M.; Johansson, L.; Jansson, P.-A.; Moris, L.; Nilsson, P.M.; Eriksson, J.W.; Lind, L. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: A double-blind, randomized, placebo-controlled study. J. Clin. Lipidol. 2018, 12, 1390–1403.e4. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CT | HFHFr | BemA | |
|---|---|---|---|
| Total calorie intake (kcal/90 days/rat) | 3648 ± 133 | 5914 ± 178 *** | 5219 ± 420 *** # |
| Body weight (g) | 262 ± 12 | 269 ± 14 | 244 ± 9 * ## |
| Liver weight (g) | 7.9 ± 0.6 | 9.5 ± 1.5 | 15.8 ± 1.4 *** # |
| Liver weight/BW (%) | 3.0 ± 0.3 | 3.6 ± 0.6 | 6.4 ± 0.5 *** # |
| pWAT/BW (%) | 0.9 ± 0.1 | 0.9 ± 0.2 | 0.6 ± 0.3 # |
| sWAT/BW (%) | 1.2 ± 0.3 | 1.5 ± 0.5 | 1.0 ± 0.1 # |
| Blood TG (mg/dL) | 118 ± 15 | 197 ± 65 * | 231 ± 71 ** |
| Blood Chol (mg/dL) | 160 ± 3 | 158 ± 5 | 181 ± 21 # |
| NEFA (mmol/L) | 0.2 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.1 |
| ALT (UI/L) | 26 ± 6 | 25 ± 6 | 32 ± 8 |
| AST (UI/L) | 43 ± 6 | 37 ± 13 | 40 ± 8 |
| Adiponectin (µg/mL) | 32 ± 5 | 36 ± 15 | 26 ± 7 |
| Leptin (ng/mL) | 3.3 ± 0.8 | 4.0 ± 1.2 | 1.3 ± 0.3 *** ### |
| PPAR Isoform | Compound | EC50 | Emax |
|---|---|---|---|
| Rat PPARα | BemA | 781 (0–1737) µM | 261675 (107,548–415,802) |
| GW590735 | 561 (392–730) nM | 761689 (704,593–818,785) | |
| Rat PPARγ | BemA | 876 (0–2803) µM | 181745 (0–381,356) |
| Rosiglitazone | 78 (31–125) nM | 177321 (153,254–201,388) | |
| Human PPARα | BemA | 168 (0–670) µM | 107342 (29,995–184,688) |
| GW7647 | 5.3 (0–11.1) nM | 236160 (181,649–290,670) | |
| Human PPARγ | BemA | 332 (0–962) µM | 87195 (32,506–141,883) |
| Rosiglitazone | 76 (0–176) nM | 51652 (35,820–67,485) | |
| Human PPARβ | BemA | - | - |
| GW0742 | 0.98 (0–2.10) nM | 72221 (54,980–89,461) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velázquez, A.M.; Bentanachs, R.; Sala-Vila, A.; Lázaro, I.; Rodríguez-Morató, J.; Sánchez, R.M.; Laguna, J.C.; Roglans, N.; Alegret, M. KHK, PNPLA3 and PPAR as Novel Targets for the Anti-Steatotic Action of Bempedoic Acid. Biomedicines 2022, 10, 1517. https://doi.org/10.3390/biomedicines10071517
Velázquez AM, Bentanachs R, Sala-Vila A, Lázaro I, Rodríguez-Morató J, Sánchez RM, Laguna JC, Roglans N, Alegret M. KHK, PNPLA3 and PPAR as Novel Targets for the Anti-Steatotic Action of Bempedoic Acid. Biomedicines. 2022; 10(7):1517. https://doi.org/10.3390/biomedicines10071517
Chicago/Turabian StyleVelázquez, Ana Magdalena, Roger Bentanachs, Aleix Sala-Vila, Iolanda Lázaro, Jose Rodríguez-Morató, Rosa María Sánchez, Juan Carlos Laguna, Núria Roglans, and Marta Alegret. 2022. "KHK, PNPLA3 and PPAR as Novel Targets for the Anti-Steatotic Action of Bempedoic Acid" Biomedicines 10, no. 7: 1517. https://doi.org/10.3390/biomedicines10071517
APA StyleVelázquez, A. M., Bentanachs, R., Sala-Vila, A., Lázaro, I., Rodríguez-Morató, J., Sánchez, R. M., Laguna, J. C., Roglans, N., & Alegret, M. (2022). KHK, PNPLA3 and PPAR as Novel Targets for the Anti-Steatotic Action of Bempedoic Acid. Biomedicines, 10(7), 1517. https://doi.org/10.3390/biomedicines10071517