
Antibacterial and Anti-Inflammatory Effects of Apolipoprotein E

 , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Endotoxins
2.3. Proteins and Peptides
2.4. Cells
2.5. Animals
2.6. Viable Count Assay (VCA)
2.7. NF-κB Activity Assay
2.8. MTT Viability Assay
2.9. Mouse Model of Subcutaneous Inflammation and In Vivo Imaging
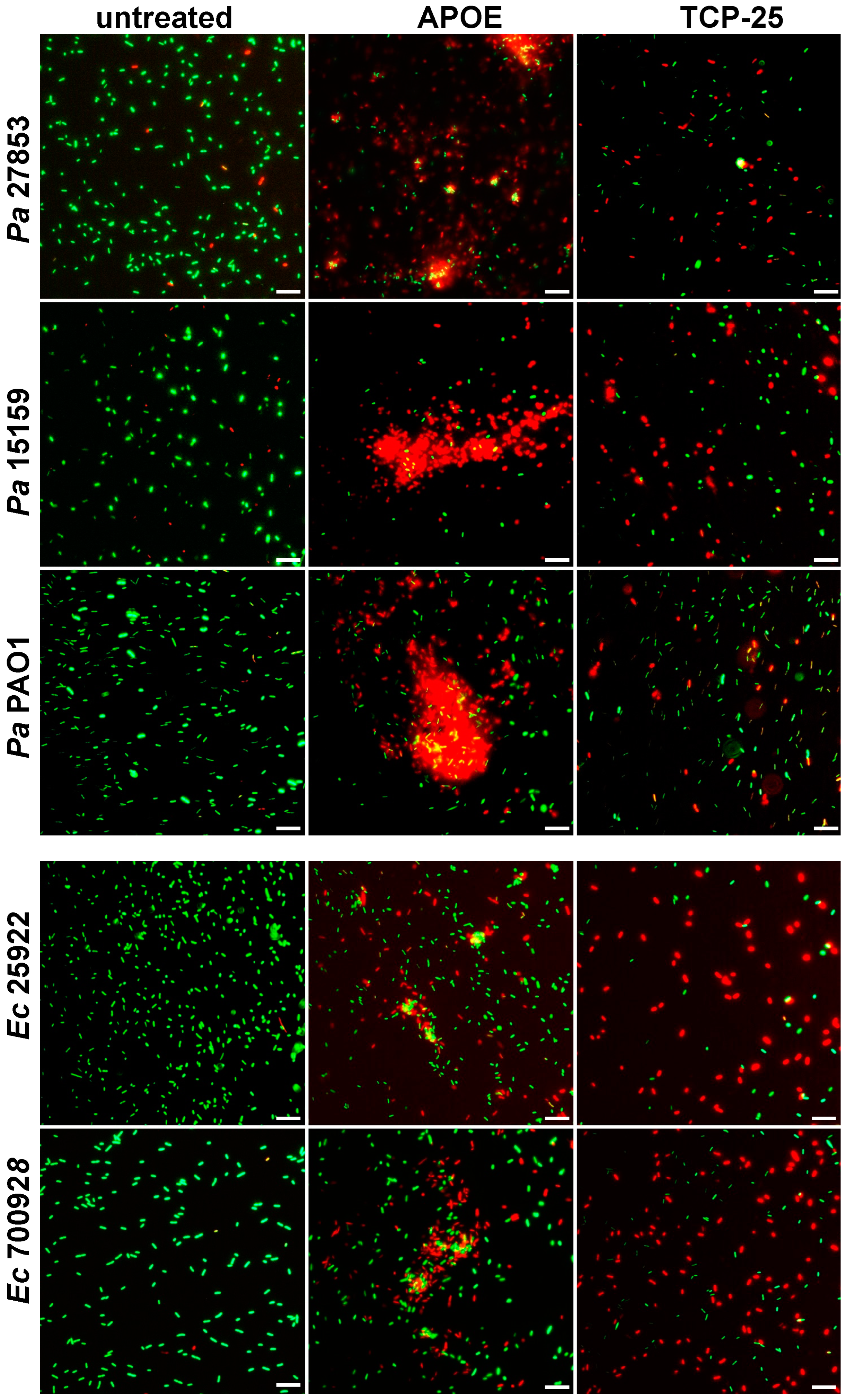
2.10. Fluorescence Microscopy
Live/Dead Bacteria
2.11. Thioflavin T Assays (ThT)
2.12. Transmission Electron Microscopy (TEM)
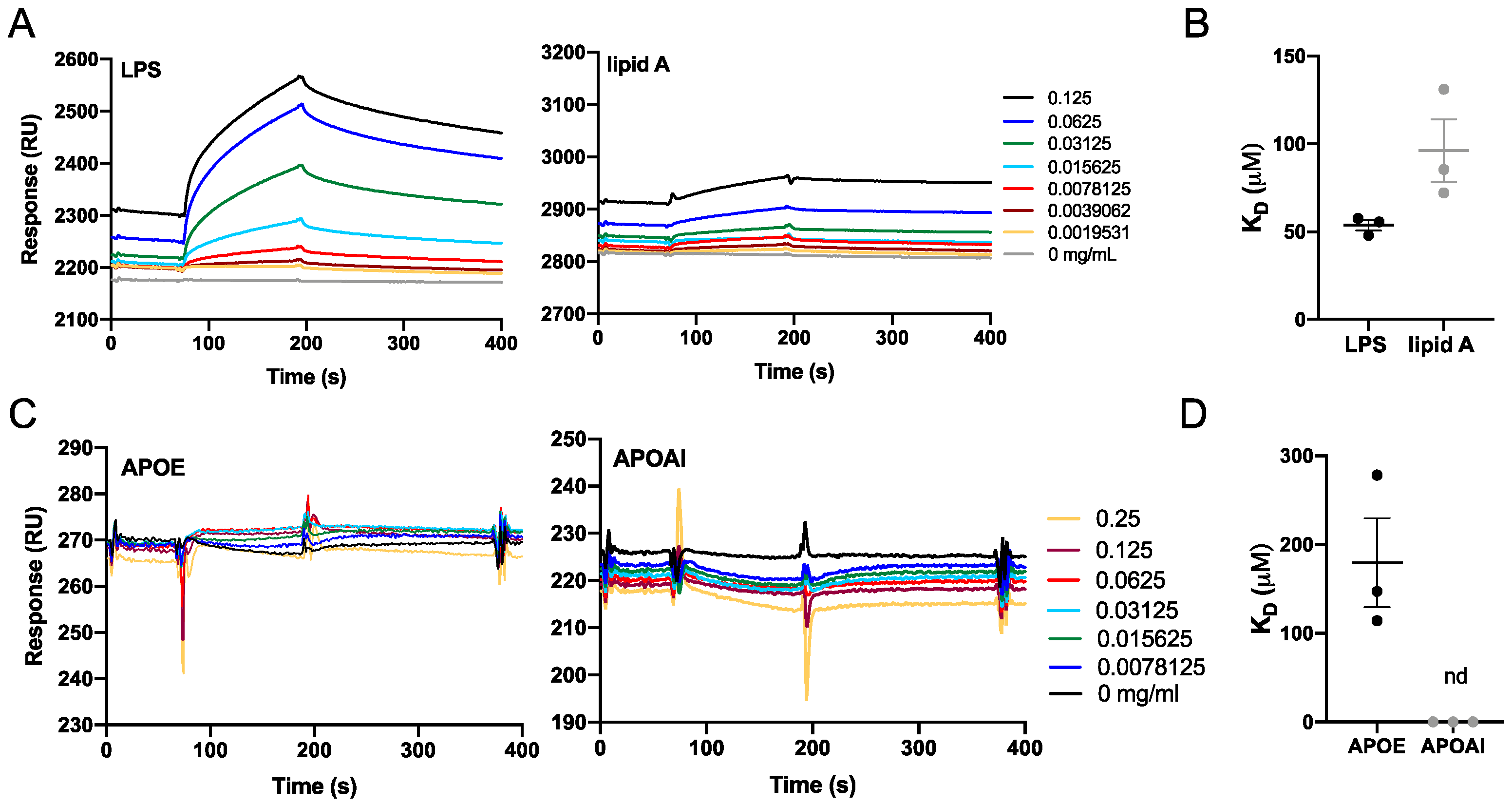
2.13. Biacore Analysis
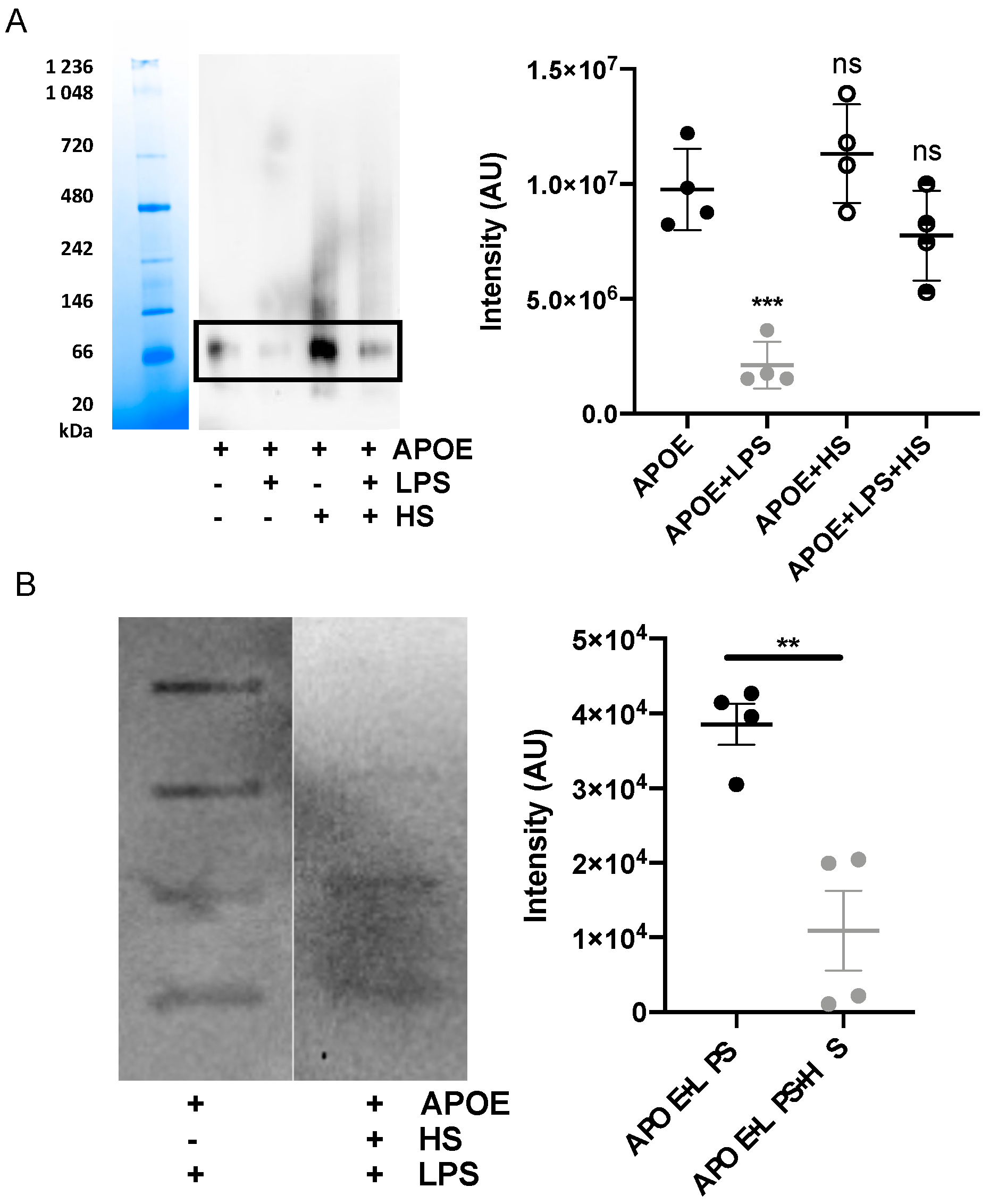
2.14. Blue Native-Polyacrylamide Gel Electrophoresis and Western Blot
2.15. Slot-Blot Assay
2.16. Blood Stimulation Assay and ELISA
2.17. Molecular Dynamics Simulations
2.18. Statistical Analysis
3. Results
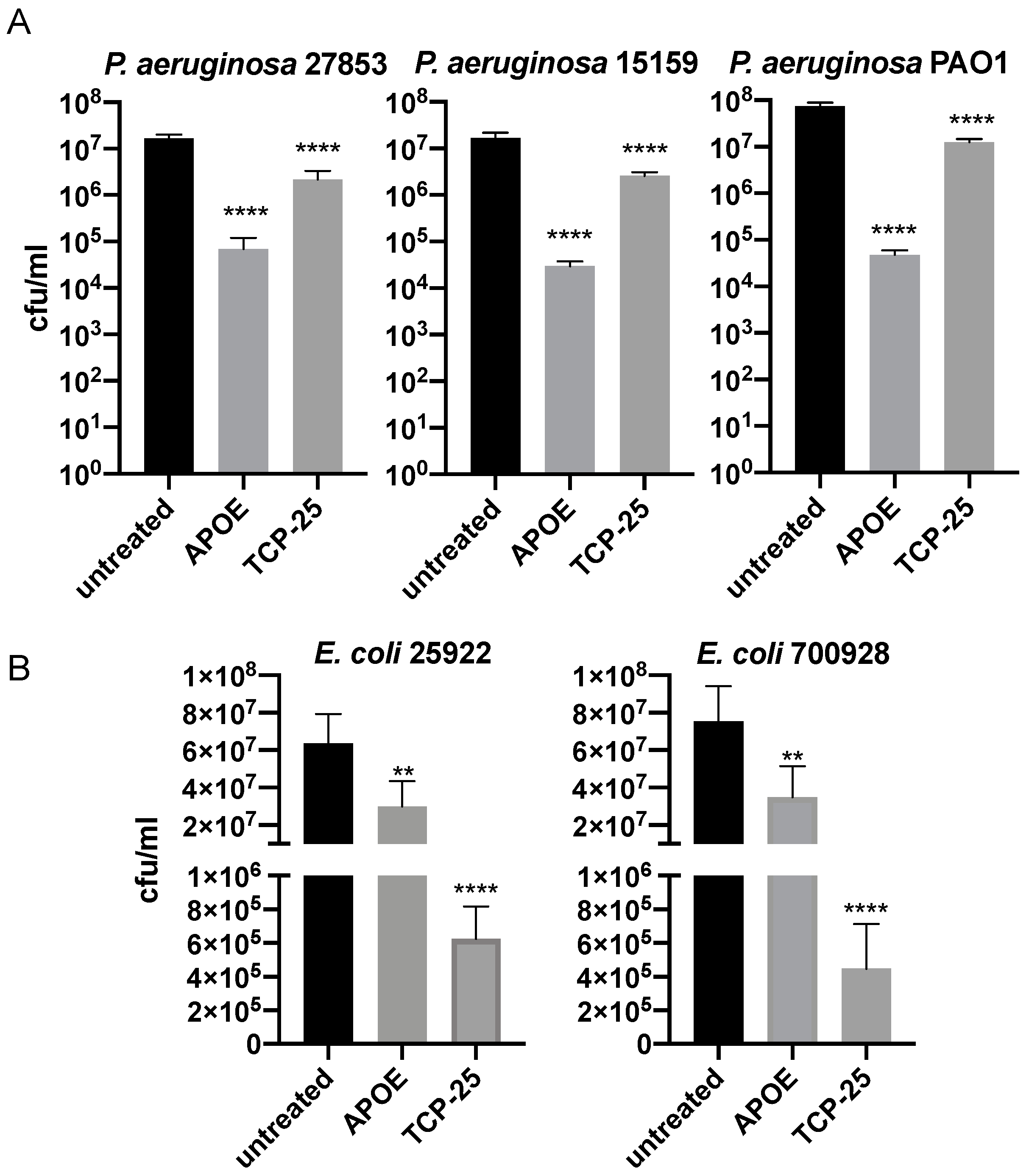
3.1. Antibacterial Activity of APOE In Vitro
3.2. Interaction of APOE with Bacterial Products
3.3. Bacterial Products Induce the Formation of APOE Aggregates
3.4. Heparin Blocks Interaction between APOE and LPS
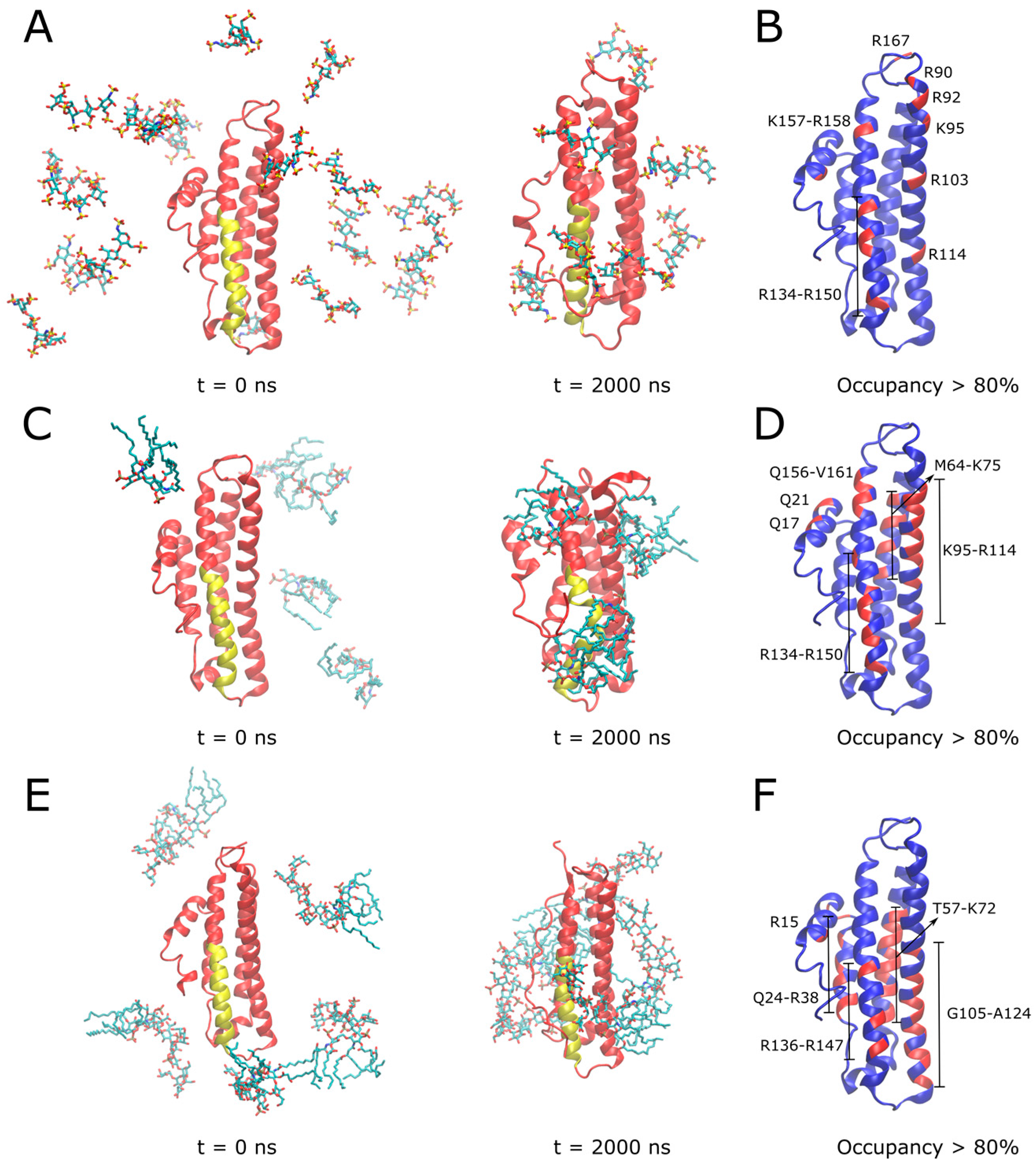
3.5. Molecular Dynamics Simulations of APOE and Its Interactions with Heparin, Lipid A and LPS
3.6. Immunomodulatory Activity of APOE In Vitro
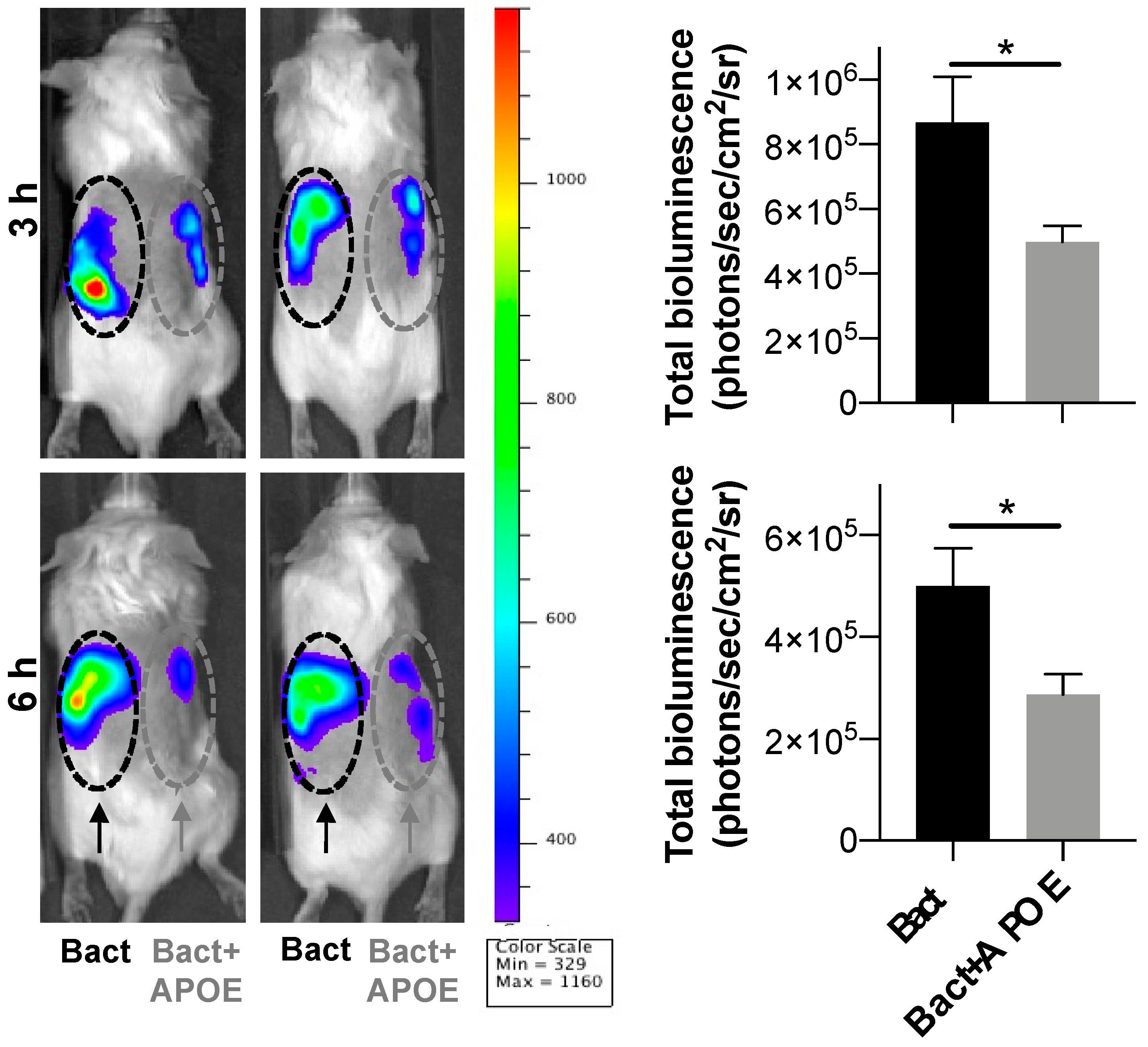
3.7. Immunomodulatory Activity of APOE In Vivo
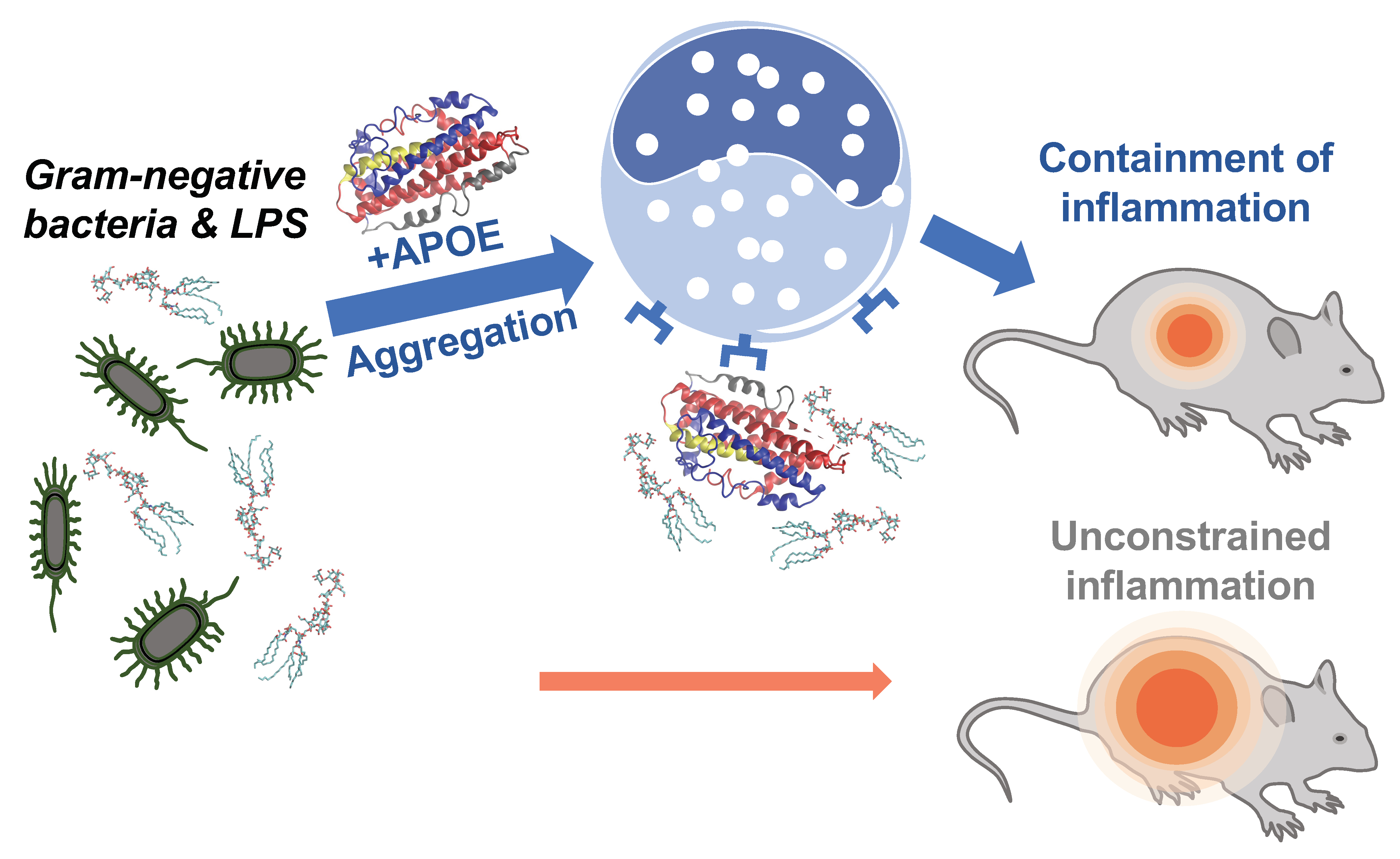
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- White, C.R.; Garber, D.W.; Anantharamaiah, G.M. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: A review. J. Lipid Res. 2014, 55, 2007–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrlova, J.; Hong, H.S.; Bricarello, D.A.; Harishchandra, G.; Lorigan, G.A.; Jin, L.W.; Voss, J.C. A differential association of Apolipoprotein E isoforms with the amyloid-beta oligomer in solution. Proteins 2011, 79, 402–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.C. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Laskowitz, D.T.; Thekdi, A.D.; Thekdi, S.D.; Han, S.K.; Myers, J.K.; Pizzo, S.V.; Bennett, E.R. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 2001, 167, 74–85. [Google Scholar] [CrossRef]
- Saito, H.; Dhanasekaran, P.; Nguyen, D.; Baldwin, F.; Weisgraber, K.H.; Wehrli, S.; Phillips, M.C.; Lund-Katz, S. Characterization of the heparin binding sites in human apolipoprotein E. J. Biol. Chem. 2003, 278, 14782–14787. [Google Scholar] [CrossRef] [Green Version]
- Azuma, M.; Kojimab, T.; Yokoyama, I.; Tajiri, H.; Yoshikawa, K.; Saga, S.; Del Carpio, C.A. A synthetic peptide of human apoprotein E with antibacterial activity. Peptides 2000, 21, 327–330. [Google Scholar] [CrossRef]
- Croy, J.E.; Brandon, T.; Komives, E.A. Two apolipoprotein E mimetic peptides, ApoE(130–149) and ApoE(141–155)2, bind to LRP1. Biochemistry 2004, 43, 7328–7335. [Google Scholar] [CrossRef]
- Laskowitz, D.T.; Fillit, H.; Yeung, N.; Toku, K.; Vitek, M.P. Apolipoprotein E-derived peptides reduce CNS inflammation: Implications for therapy of neurological disease. Acta Neurol. Scand. Suppl. 2006, 185, 15–20. [Google Scholar] [CrossRef]
- Pane, K.; Sgambati, V.; Zanfardino, A.; Smaldone, G.; Cafaro, V.; Angrisano, T.; Pedone, E.; Di Gaetano, S.; Capasso, D.; Haney, E.F.; et al. A new cryptic cationic antimicrobial peptide from human apolipoprotein E with antibacterial activity and immunomodulatory effects on human cells. FEBS J. 2016, 283, 2115–2131. [Google Scholar] [CrossRef] [Green Version]
- Petruk, G.; Elven, M.; Hartman, E.; Davoudi, M.; Schmidtchen, A.; Puthia, M.; Petrlova, J. The role of full-length apoE in clearance of Gram-negative bacteria and their endotoxins. J. Lipid Res. 2021, 62, 100086. [Google Scholar] [CrossRef]
- Puthia, M.; Butrym, M.; Petrlova, J.; Stromdahl, A.C.; Andersson, M.A.; Kjellstrom, S.; Schmidtchen, A. A dual-action peptide-containing hydrogel targets wound infection and inflammation. Sci. Transl. Med. 2020, 12, eaax6601. [Google Scholar] [CrossRef] [PubMed]
- Petrlova, J.; Petruk, G.; Huber, R.G.; McBurnie, E.W.; van der Plas, M.J.A.; Bond, P.J.; Puthia, M.; Schmidtchen, A. Thrombin-derived C-terminal fragments aggregate and scavenge bacteria and their proinflammatory products. J. Biol. Chem. 2020, 295, 3417–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, S.; Khakzad, H.; Bergdahl, G.E.; Lood, R.; Ekstrom, S.; Linke, D.; Malmstrom, L.; Happonen, L.; Malmstrom, J. Streptococcus pyogenes Forms Serotype- and Local Environment-Dependent Interspecies Protein Complexes. mSystems 2021, 6, e0027121. [Google Scholar] [CrossRef] [PubMed]
- Khakzad, H.; Happonen, L.; Karami, Y.; Chowdhury, S.; Bergdahl, G.E.; Nilges, M.; Tran Van Nhieu, G.; Malmstrom, J.; Malmstrom, L. Structural determination of Streptococcus pyogenes M1 protein interactions with human immunoglobulin G using integrative structural biology. PLoS Comput. Biol. 2021, 17, e1008169. [Google Scholar] [CrossRef] [PubMed]
- Petrlova, J.; Hansen, F.C.; van der Plas, M.J.A.; Huber, R.G.; Morgelin, M.; Malmsten, M.; Bond, P.J.; Schmidtchen, A. Aggregation of thrombin-derived C-terminal fragments as a previously undisclosed host defense mechanism. Proc. Natl. Acad. Sci. USA 2017, 114, E4213–E4222. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Q.; Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA 2011, 108, 14813–14818. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nivedha, A.K.; Thieker, D.F.; Makeneni, S.; Hu, H.; Woods, R.J. Vina-Carb: Improving Glycosidic Angles during Carbohydrate Docking. J. Chem. Theory Comput. 2016, 12, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquino, R.S.; Pereira, M.S.; Vairo, B.C.; Cinelli, L.P.; Santos, G.R.; Fonseca, R.J.; Mourao, P.A. Heparins from porcine and bovine intestinal mucosa: Are they similar drugs? Thromb. Haemost. 2010, 103, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Simmonett, A.C.; Brooks, B.R. A compression strategy for particle mesh Ewald theory. J. Chem. Phys. 2021, 154, 054112. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Papareddy, P.; Rydengard, V.; Pasupuleti, M.; Walse, B.; Morgelin, M.; Chalupka, A.; Malmsten, M.; Schmidtchen, A. Proteolysis of human thrombin generates novel host defense peptides. PLoS Pathog. 2010, 6, e1000857. [Google Scholar] [CrossRef] [Green Version]
- Kaneva, A.M.; Bojko, E.R.; Potolitsyna, N.N.; Odland, J.O. Plasma levels of apolipoprotein-E in residents of the European North of Russia. Lipids Health Dis. 2013, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Barra, R.M.; Fenjves, E.S.; Taichman, L.B. Secretion of apolipoprotein E by basal cells in cultures of epidermal keratinocytes. J. Investig. Dermatol. 1994, 102, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Kodvawala, A.; Hui, D.Y. Apolipoprotein E inhibits toll-like receptor (TLR)-3- and TLR-4-mediated macrophage activation through distinct mechanisms. Biochem. J. 2010, 428, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.A.; Fenjves, E.S.; Williams, D.L.; Taichman, L.B. Synthesis and secretion of apolipoprotein E by cultured human keratinocytes. J. Investig. Dermatol. 1989, 92, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouchet, E.; Lefevre, M.; Verrier, E.R.; Oudot, M.A.; Baumert, T.F.; Schuster, C. Extracellular lipid-free apolipoprotein E inhibits HCV replication and induces ABCG1-dependent cholesterol efflux. Gut 2017, 66, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanfardino, A.; Bosso, A.; Gallo, G.; Pistorio, V.; Di Napoli, M.; Gaglione, R.; Dell’Olmo, E.; Varcamonti, M.; Notomista, E.; Arciello, A.; et al. Human apolipoprotein E as a reservoir of cryptic bioactive peptides: The case of ApoE 133–167. J. Pept. Sci. 2018, 24, e3095. [Google Scholar] [CrossRef]
- Dobson, C.B.; Sales, S.D.; Hoggard, P.; Wozniak, M.A.; Crutcher, K.A. The receptor-binding region of human apolipoprotein E has direct anti-infective activity. J. Infect. Dis. 2006, 193, 442–450. [Google Scholar] [CrossRef]
- Kunjithapatham, R.; Oliva, F.Y.; Doshi, U.; Pérez, M.; Avila, J.; Muñoz, V. Role for the alpha-helix in aberrant protein aggregation. Biochemistry 2005, 44, 149–156. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Ma, J.; Liao, X.L.; Lou, B.; Wu, M.P. Role of apolipoprotein A-I in protecting against endotoxin toxicity. Acta Biochim. Biophys. Sin. 2004, 36, 419–424. [Google Scholar] [CrossRef]
- Laupland, K.B.; Church, D.L. Population-based epidemiology and microbiology of community-onset bloodstream infections. Clin. Microbiol. Rev. 2014, 27, 647–664. [Google Scholar] [CrossRef] [Green Version]
- Marston, H.D.; Dixon, D.M.; Knisely, J.M.; Palmore, T.N.; Fauci, A.S. Antimicrobial Resistance. JAMA 2016, 316, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Llewelyn, M.J.; Fitzpatrick, J.M.; Darwin, E.; SarahTonkin, C.; Gorton, C.; Paul, J.; Peto, T.E.A.; Yardley, L.; Hopkins, S.; Walker, A.S. The antibiotic course has had its day. BMJ 2017, 358, j3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonk, A.G.; De Bont, N.; Netea, M.G.; Demacker, P.N.; van der Meer, J.W.; Stalenhoef, A.F.; Kullberg, B.J. Apolipoprotein-E-deficient mice exhibit an increased susceptibility to disseminated candidiasis. Med Mycol. 2004, 42, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puthia, M.; Marzinek, J.K.; Petruk, G.; Ertürk Bergdahl, G.; Bond, P.J.; Petrlova, J. Antibacterial and Anti-Inflammatory Effects of Apolipoprotein E. Biomedicines 2022, 10, 1430. https://doi.org/10.3390/biomedicines10061430
Puthia M, Marzinek JK, Petruk G, Ertürk Bergdahl G, Bond PJ, Petrlova J. Antibacterial and Anti-Inflammatory Effects of Apolipoprotein E. Biomedicines. 2022; 10(6):1430. https://doi.org/10.3390/biomedicines10061430
Chicago/Turabian StylePuthia, Manoj, Jan K. Marzinek, Ganna Petruk, Gizem Ertürk Bergdahl, Peter J. Bond, and Jitka Petrlova. 2022. "Antibacterial and Anti-Inflammatory Effects of Apolipoprotein E" Biomedicines 10, no. 6: 1430. https://doi.org/10.3390/biomedicines10061430
APA StylePuthia, M., Marzinek, J. K., Petruk, G., Ertürk Bergdahl, G., Bond, P. J., & Petrlova, J. (2022). Antibacterial and Anti-Inflammatory Effects of Apolipoprotein E. Biomedicines, 10(6), 1430. https://doi.org/10.3390/biomedicines10061430