
Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Chemicals and Small Molecule Inhibitor Erastin
2.3. Oncolytic Viruses vvDD and vvDD-IL15Rα
2.4. Cell Viability Assays
2.5. Mice and Murine Tumor Models
2.6. Analysis of Immune Cells in the Tumor Microenvironment
2.7. Flow Cytometry
2.8. RT-qPCR
2.9. IFN-γ ELISpot Assays
2.10. Statistical Analysis
3. Results
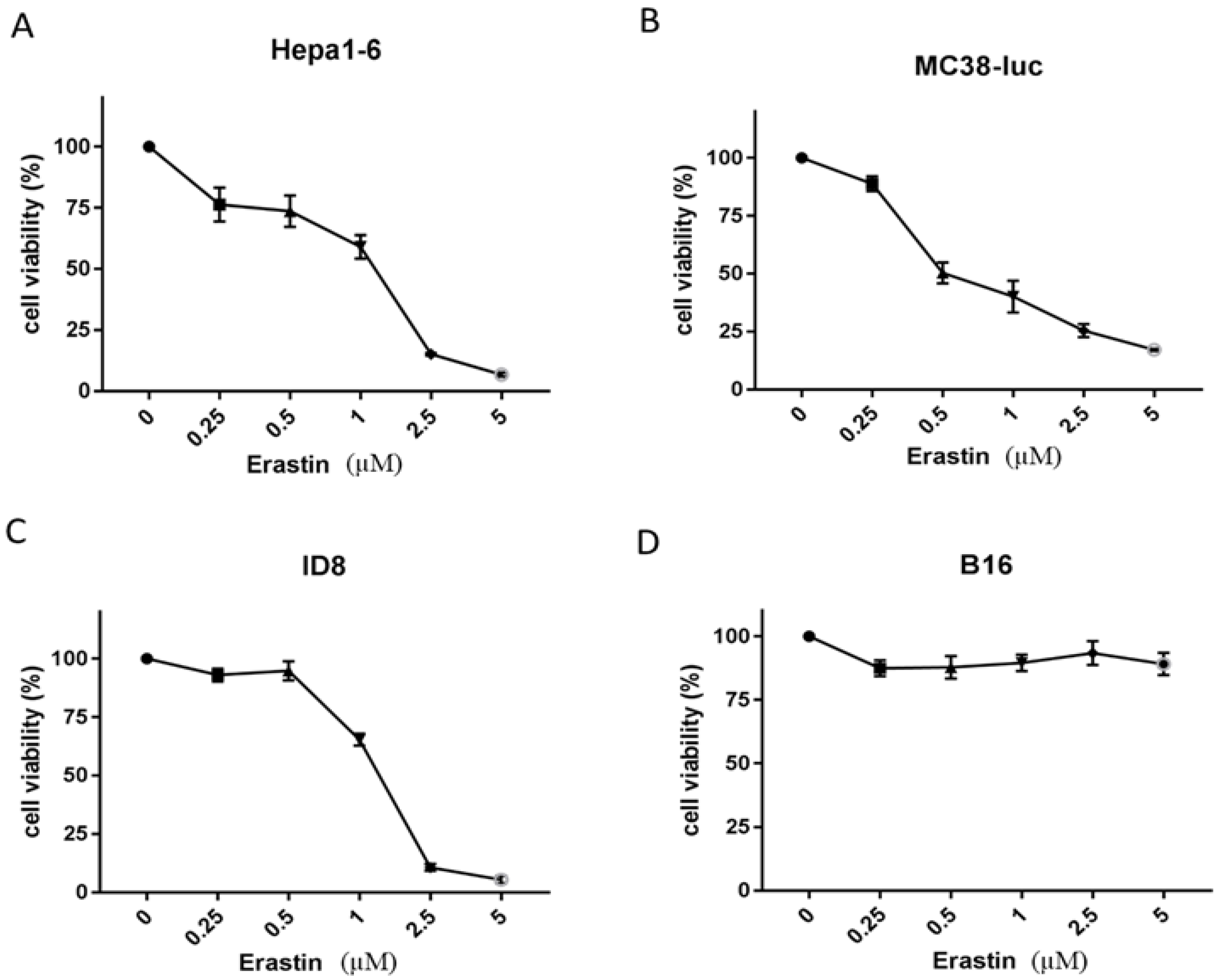
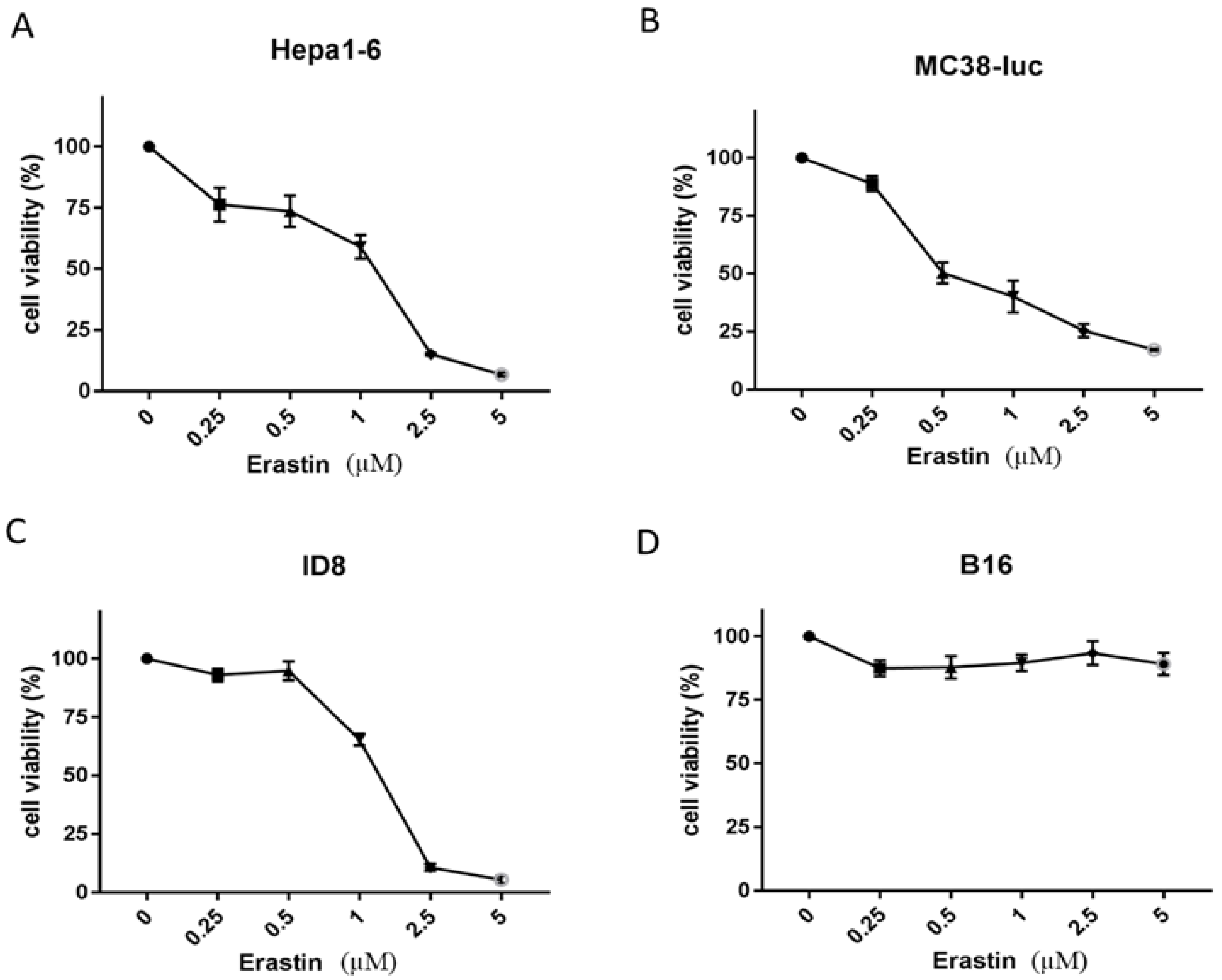
3.1. Erastin Exhibits Various Extents of Cytotoxicity to Different Cancer Cells
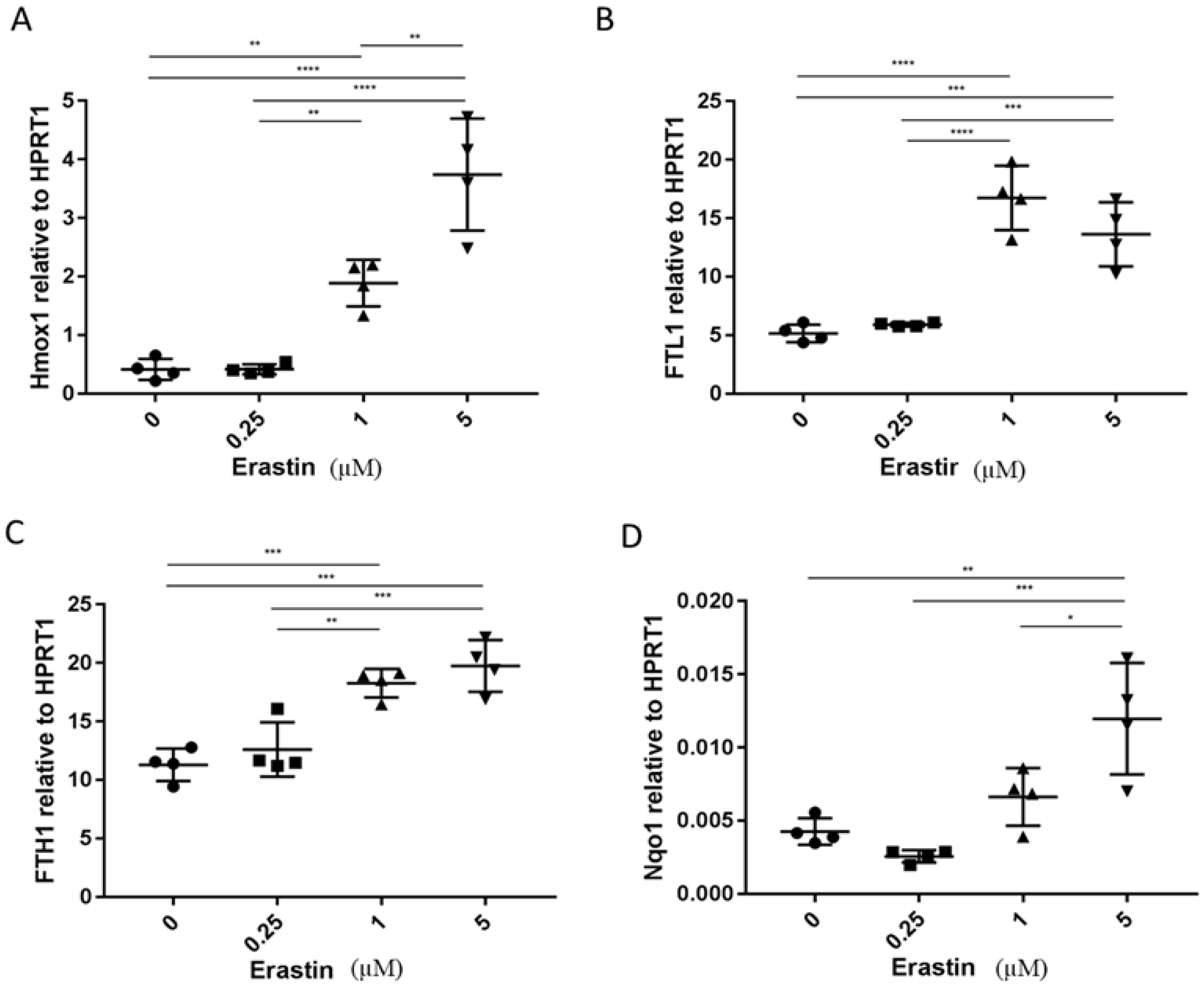
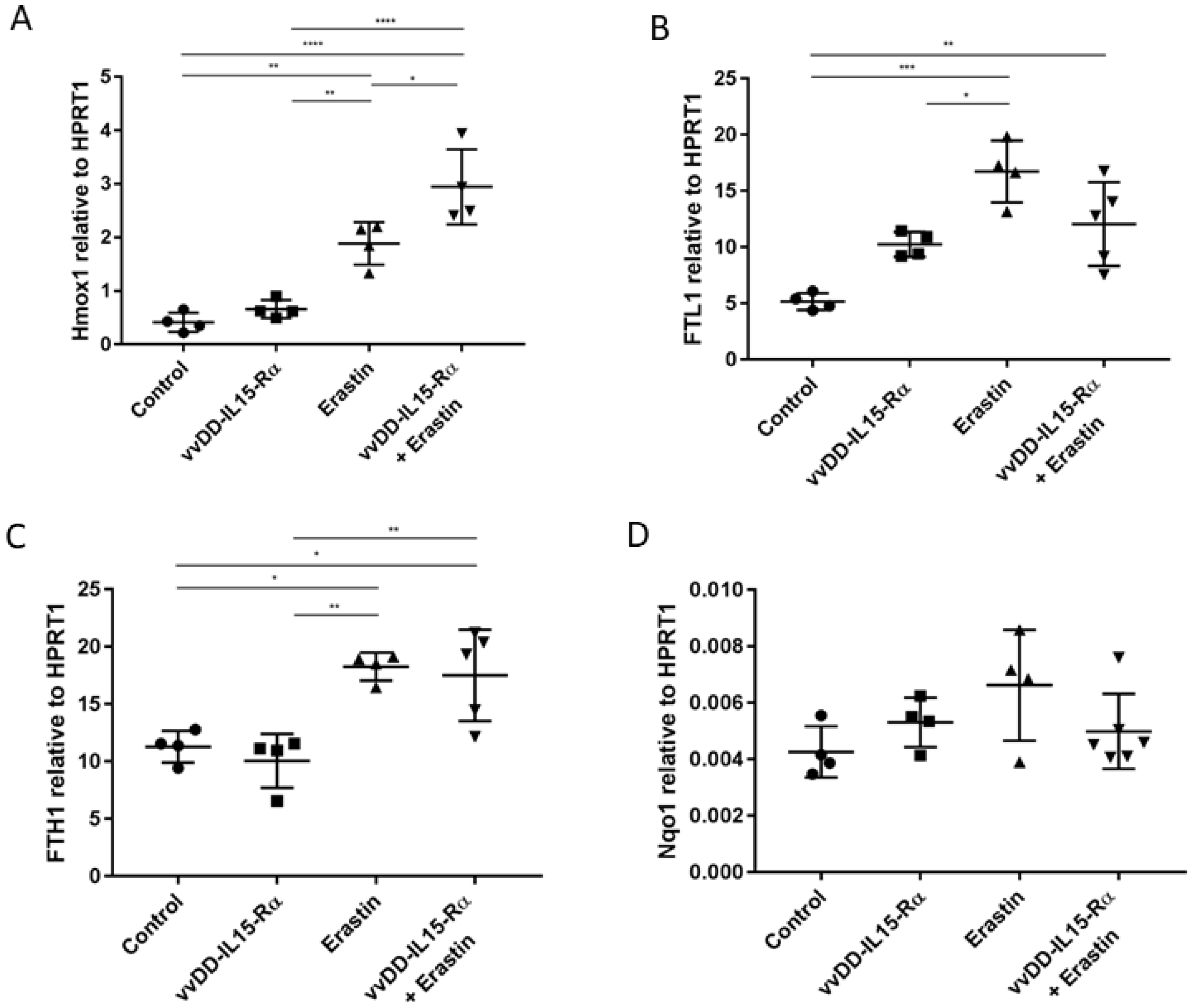
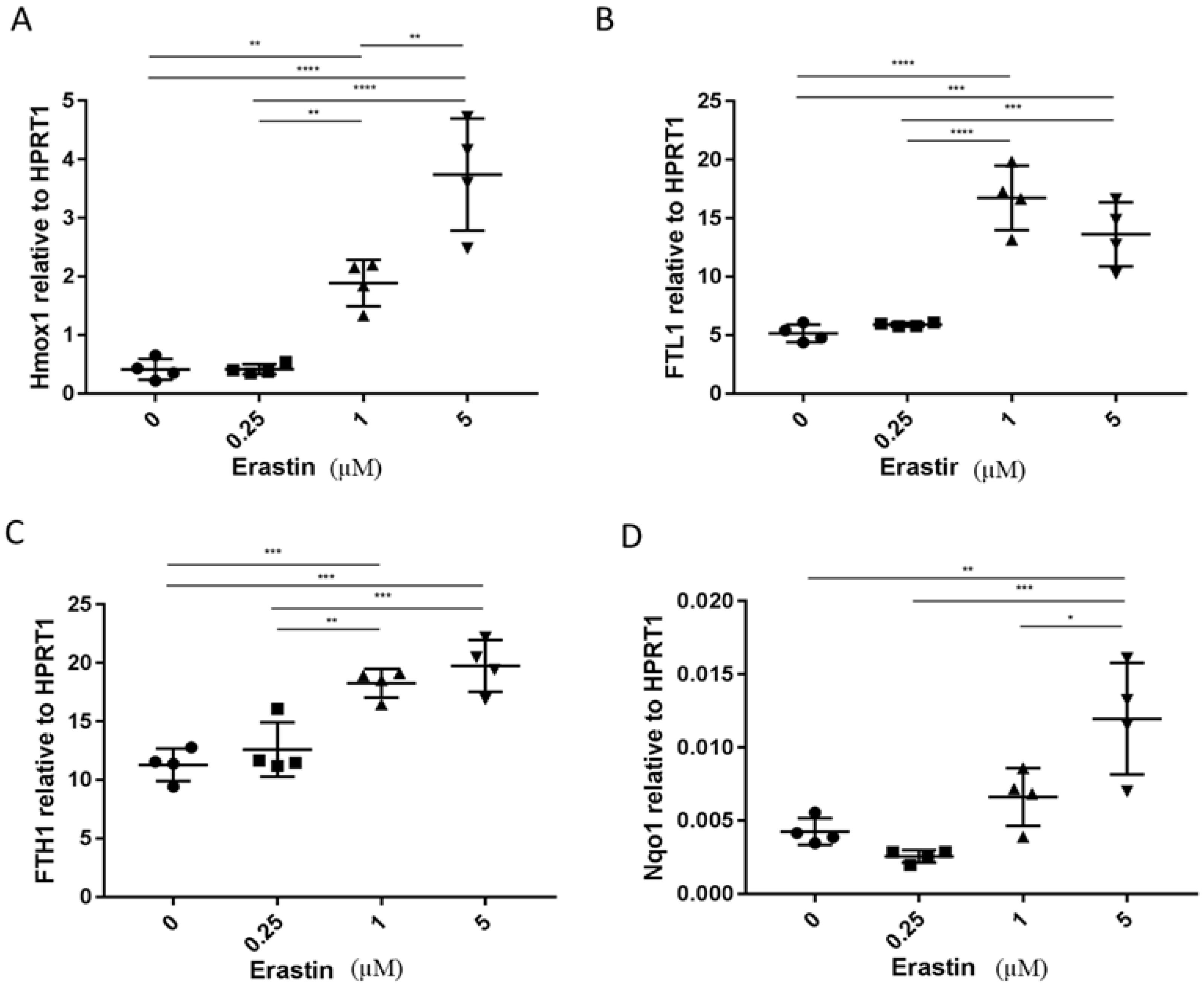
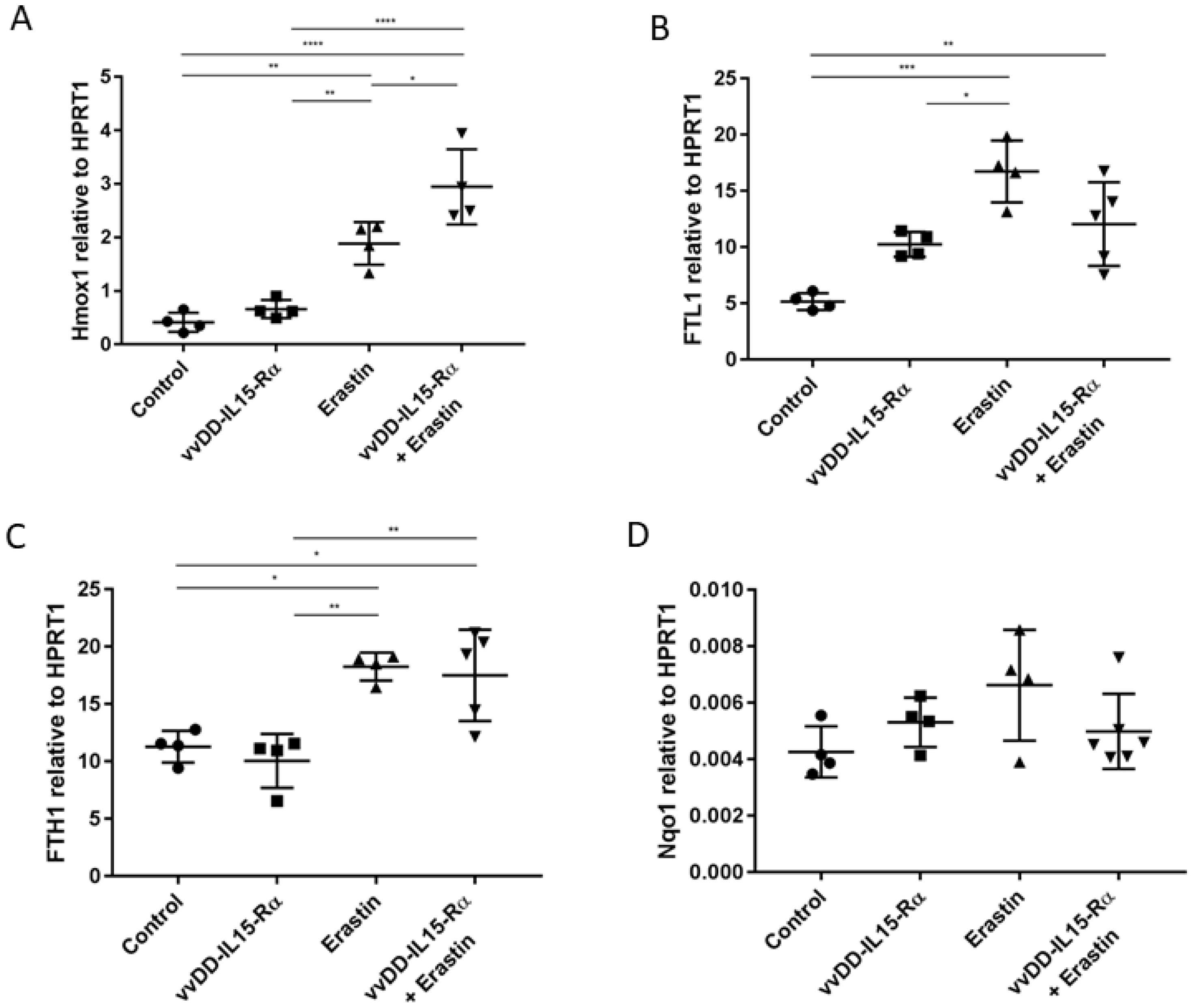
3.2. The Analysis of Expression of Marker Genes for Ferroptosis
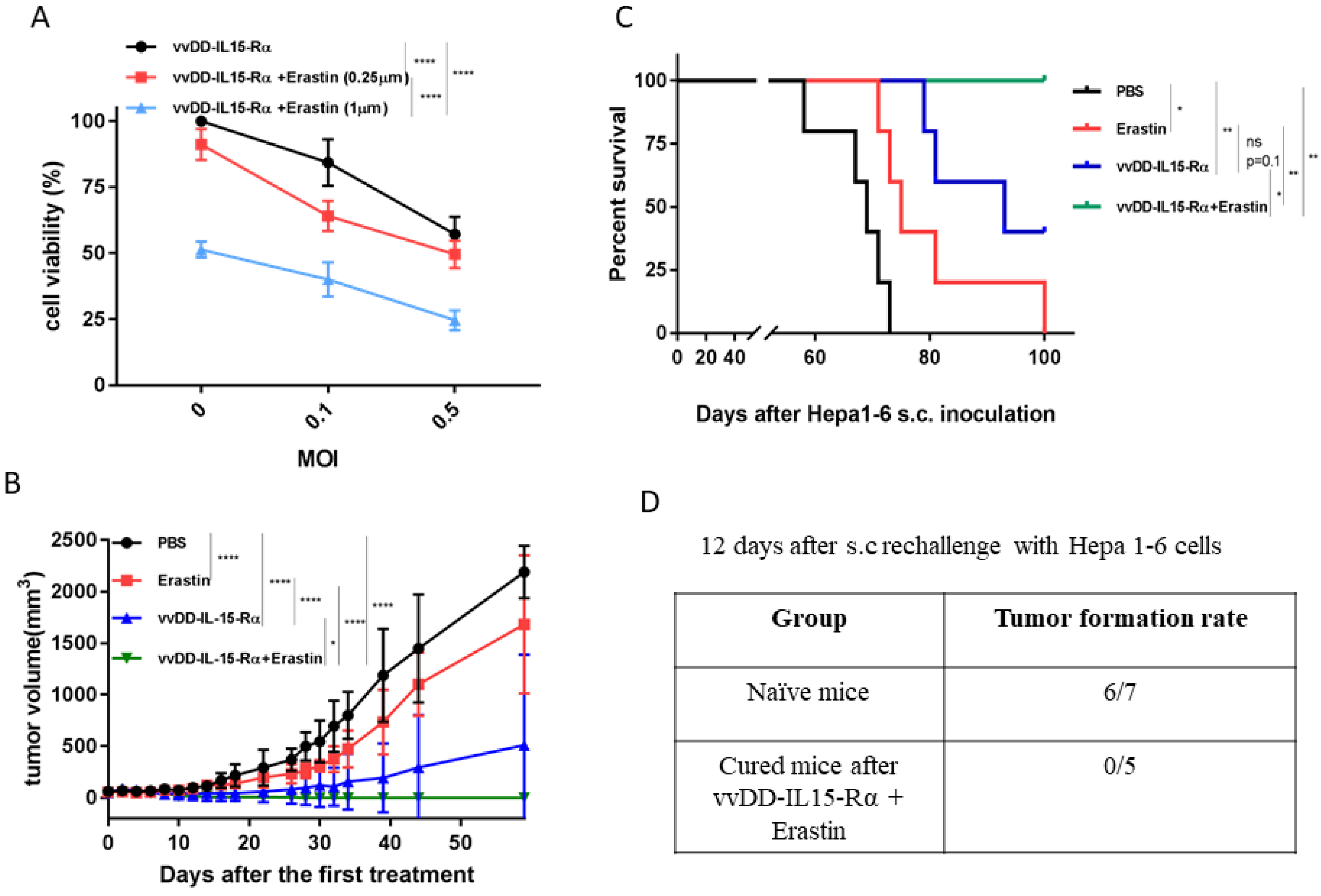
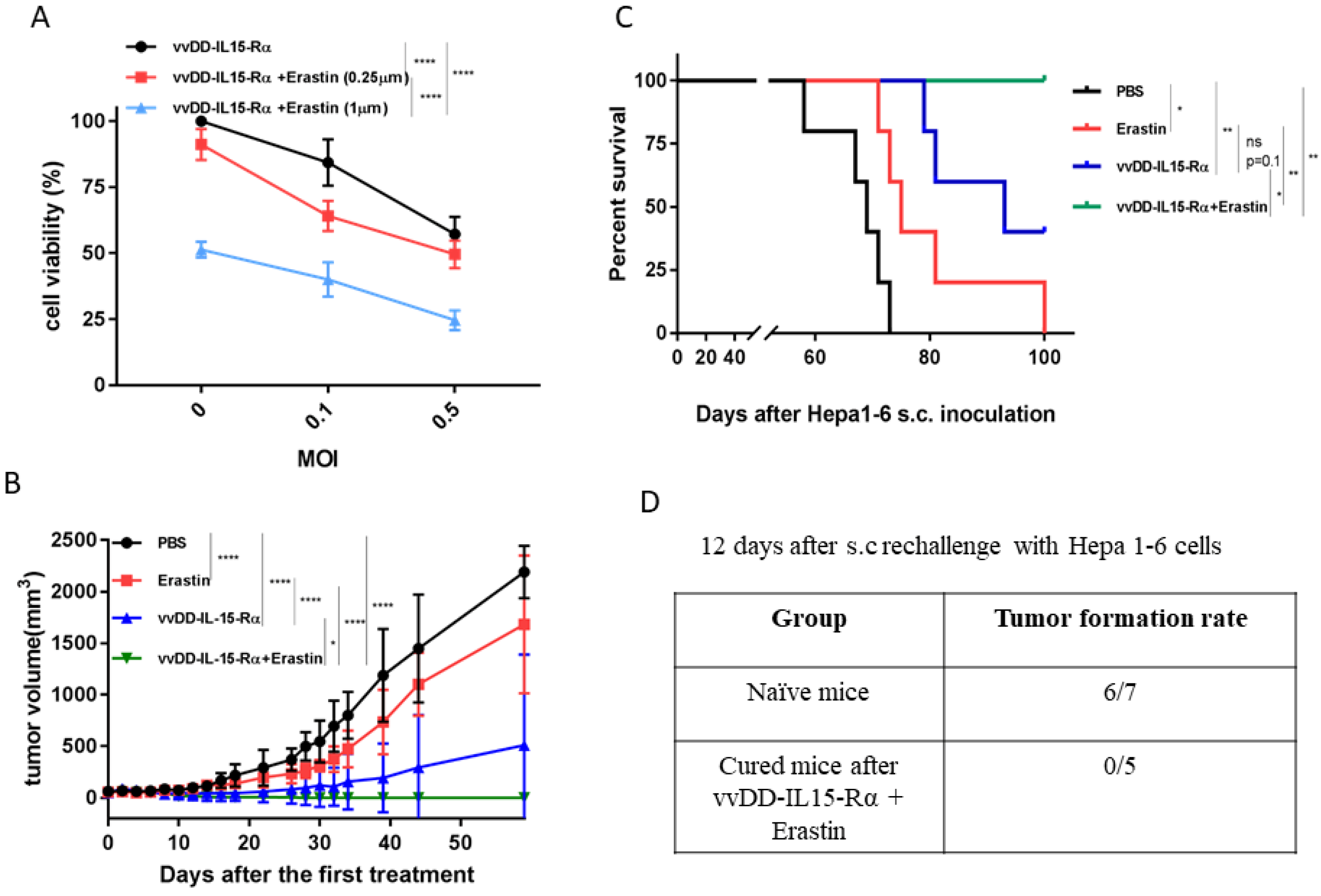
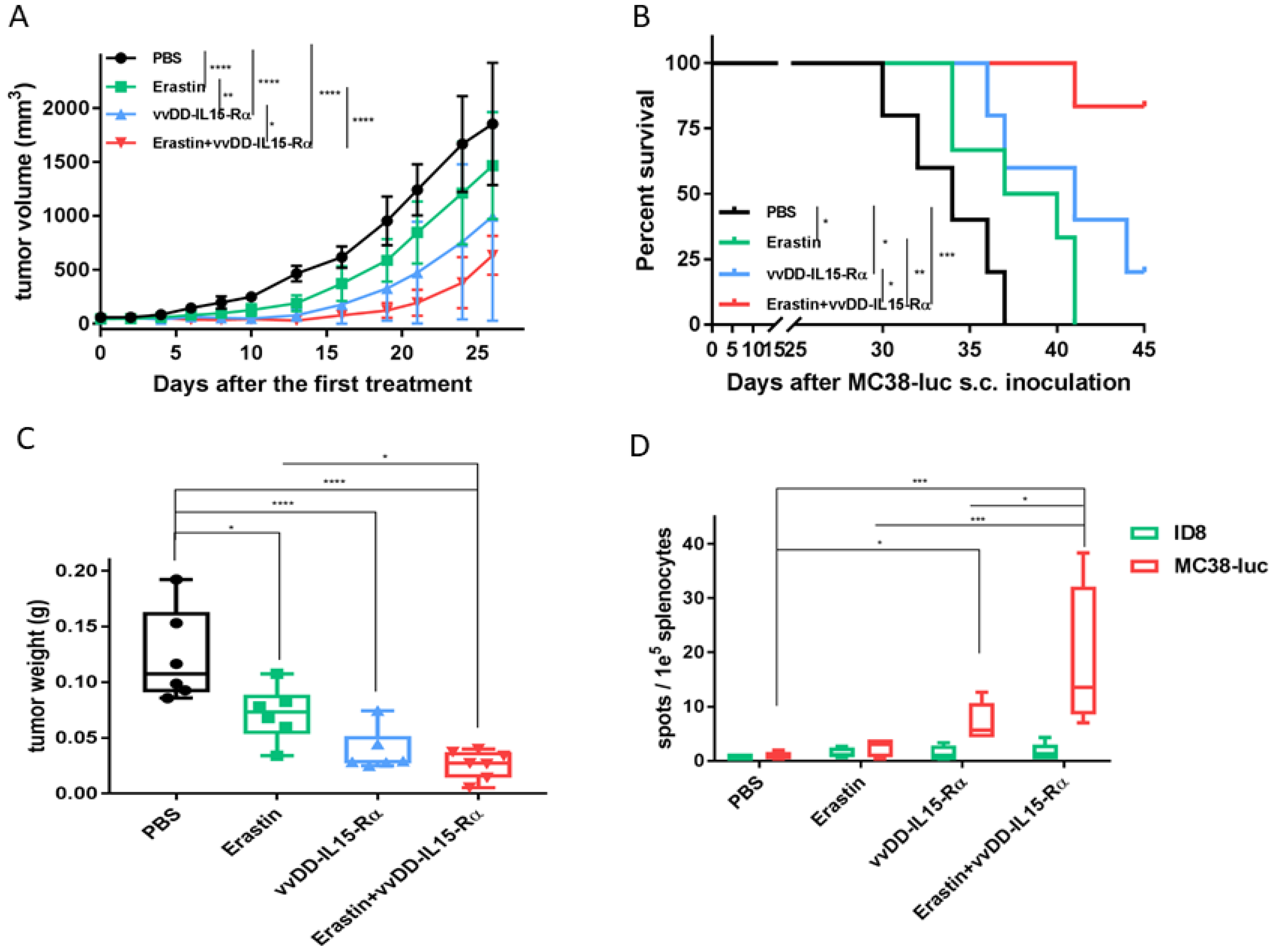
3.3. The Therapeutic Effects of Erastin, OV, or Their Combination on Hepatoma and Colon Cancer Models in Syngeneic Mice
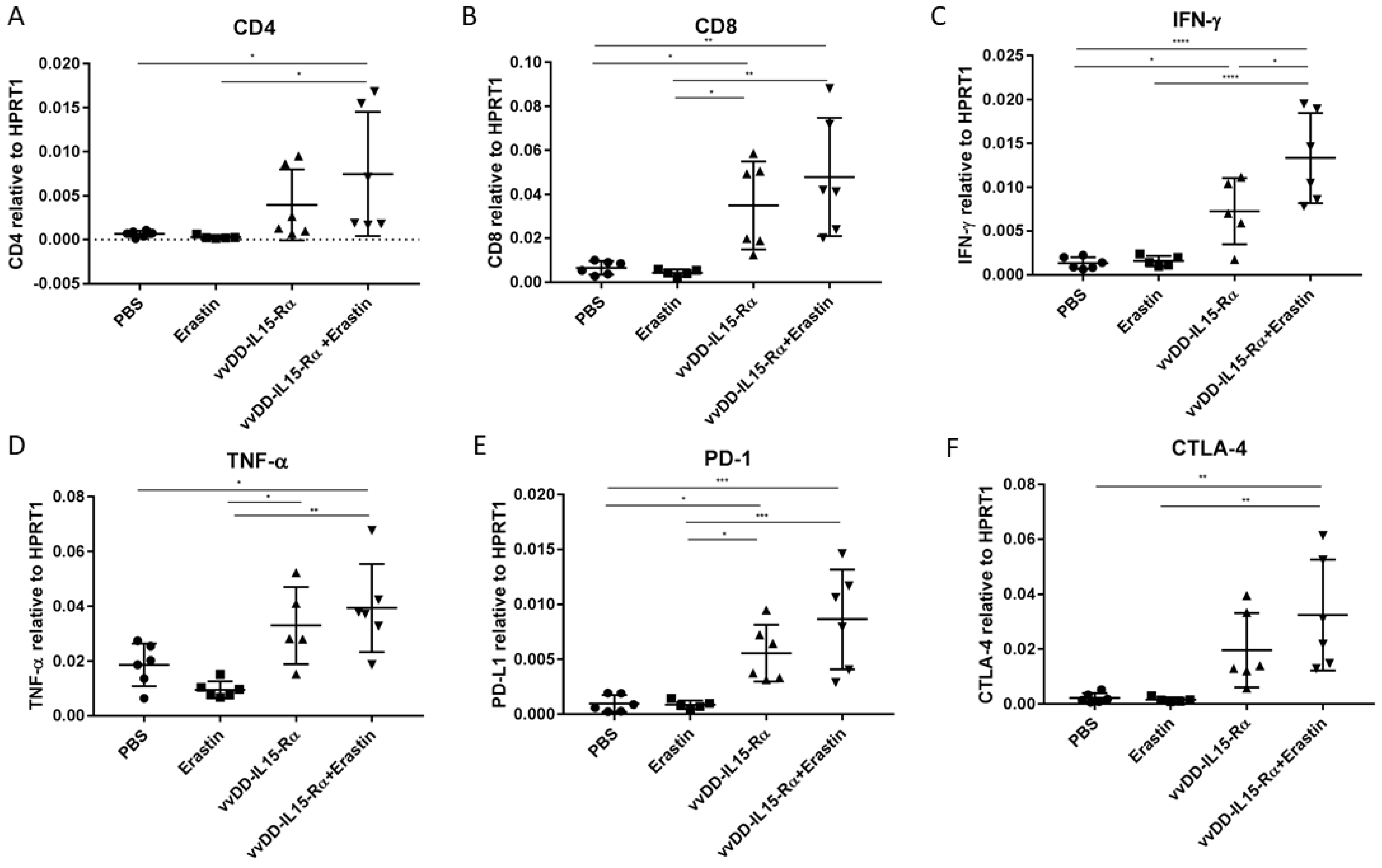
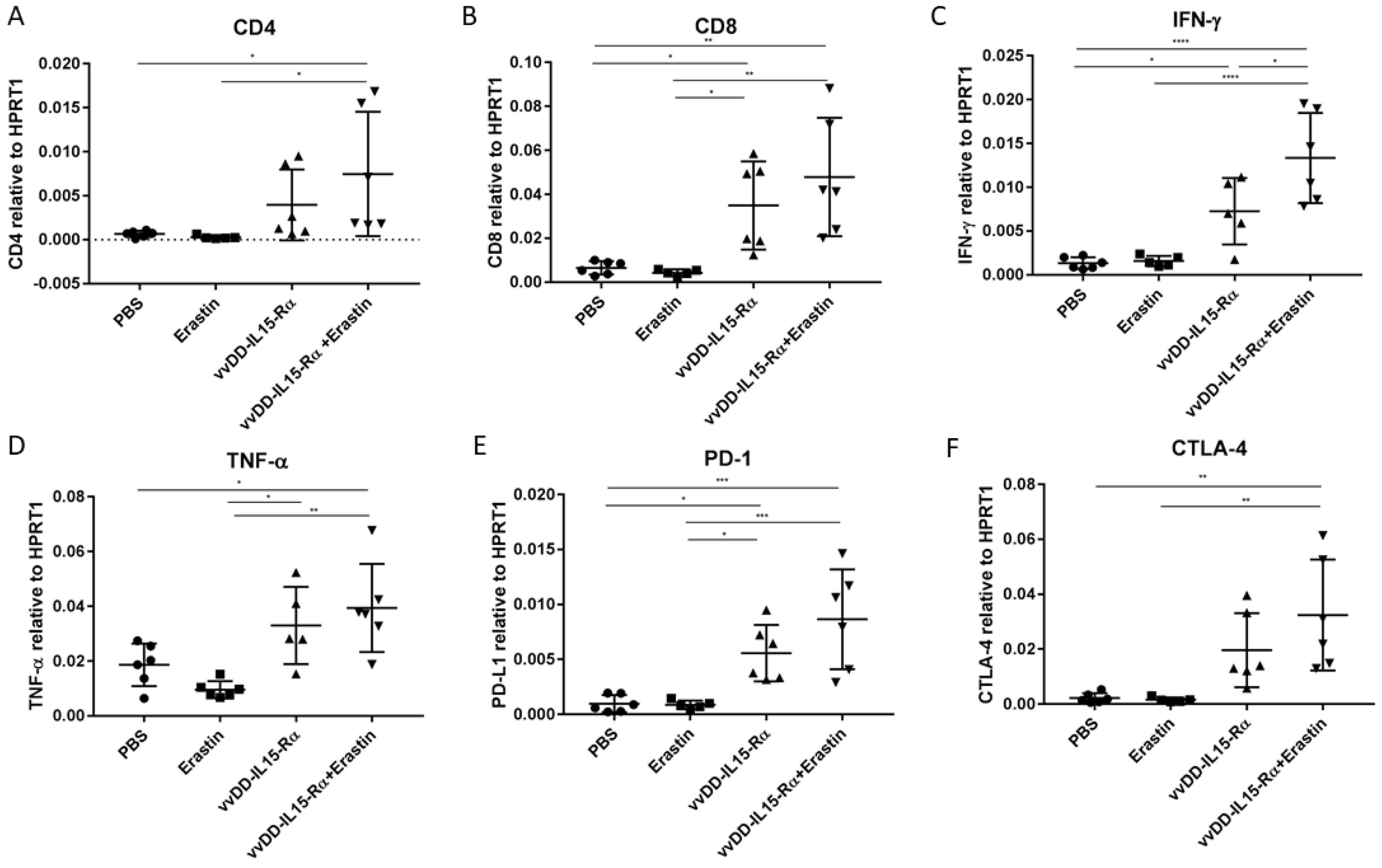
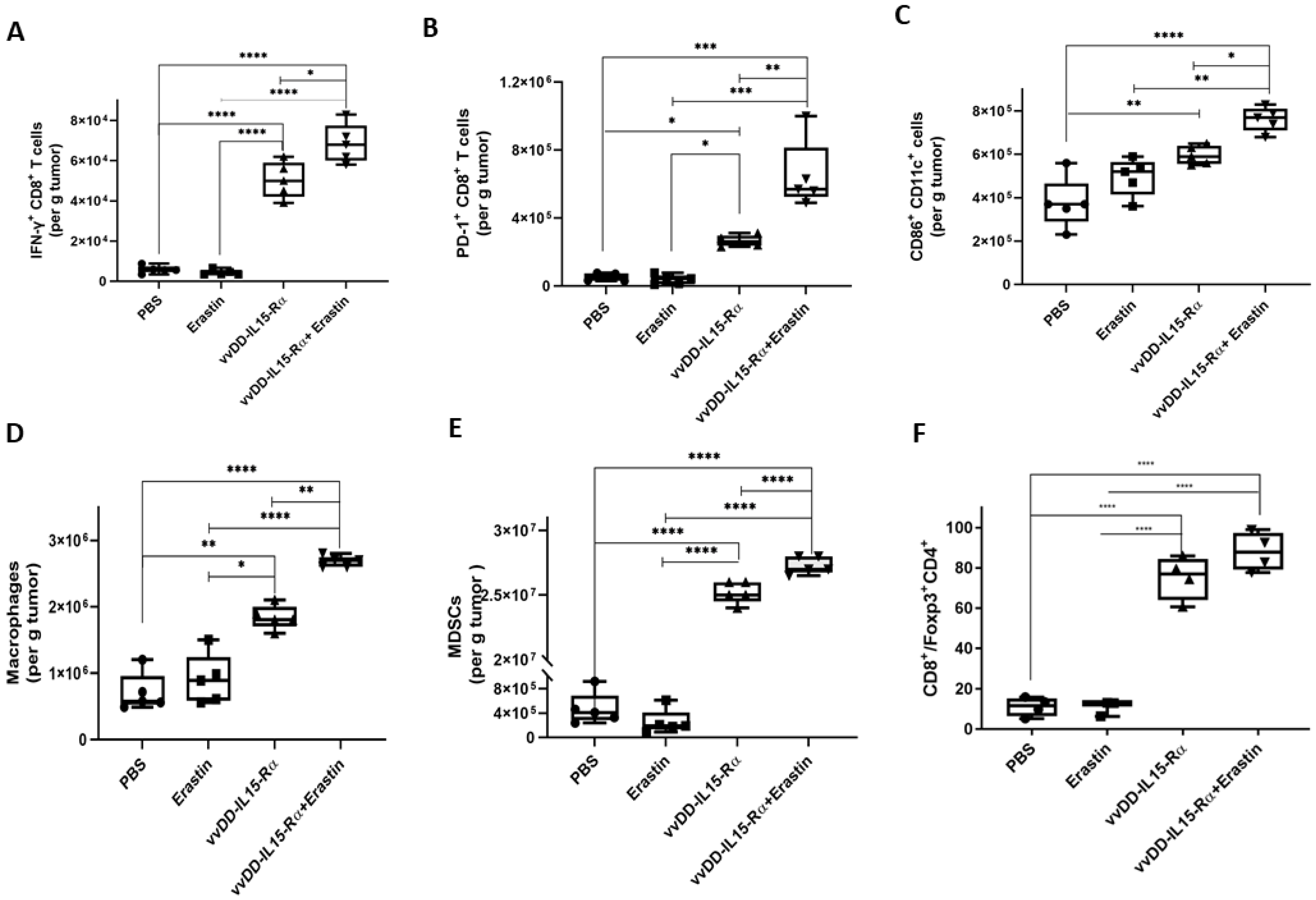
3.4. The Dual Therapy Induced More Potent Antitumor Immunity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xie, D.; Yang, L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Van Vloten, J.P.; Workenhe, S.T.; Wootton, S.K.; Mossman, K.L.; Bridle, B.W. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J. Immunol. 2018, 200, 450–458. [Google Scholar] [CrossRef]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef] [PubMed]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, J.; Chard, L.S.; Yuan, M.; Wang, J.; Howells, A.; Li, Y.; Li, H.; Zhang, Z.; Lu, S.; Gao, D.; et al. A new oncolytic Vaccinia virus augments antitumor immune responses to prevent tumor recurrence and metastasis after surgery. J. Immunother. Cancer 2020, 8, e000415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Alemany, R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef]
- Pelin, A.; Boulton, S.; Tamming, L.A.; Bell, J.C.; Singaravelu, R. Engineering vaccinia virus as an immunotherapeutic battleship to overcome tumor heterogeneity. Expert Opin. Biol. Ther. 2020, 20, 1083–1097. [Google Scholar] [CrossRef]
- Marelli, G.; Chard Dunmall, L.S.; Yuan, M.; Di Gioia, C.; Miao, J.; Cheng, Z.; Zhang, Z.; Liu, P.; Ahmed, J.; Gangeswaran, R.; et al. A systemically deliverable Vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J. Immunother. Cancer 2021, 9, e001624. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, M.E.; Jang, W.S.; Kim, J.; Park, S.M.; Oh, K.; Lee, N.; Ham, W.S. Systemic Injection of Oncolytic Vaccinia Virus Suppresses Primary Tumor Growth and Lung Metastasis in Metastatic Renal Cell Carcinoma by Remodeling Tumor Microenvironment. Biomedicines 2022, 10, 173. [Google Scholar] [CrossRef]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 8627. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.H.; Lee, N.; Yoo, J.; Won, S.; Shin, S.K.; Kim, K.H.; Park, J.G.; Kim, M.G.; Kim, H.R.; Oh, K.; et al. Oncolytic Vaccinia Virus Augments T Cell Factor 1-Positive Stem-like CD8(+) T Cells, Which Underlies the Efficacy of Anti-PD-1 Combination Immunotherapy. Biomedicines 2022, 10, 805. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e629. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012 e1005. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Bai, L.; Qu, C.; Dai, E.; Liu, J.; Kang, R.; Zhou, D.; Tang, D.; Zhao, Y. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem. Biophys. Res. Commun. 2021, 576, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef]
- Shi, L.; Liu, Y.; Li, M.; Luo, Z. Emerging roles of ferroptosis in the tumor immune landscape: From danger signals to anti-tumor immunity. FEBS J. 2022. [Google Scholar] [CrossRef]
- Corbett, T.H.; Griswold, D.P., Jr.; Roberts, B.J.; Peckham, J.C.; Schabel, F.M., Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975, 35, 2434–2439. [Google Scholar]
- Guo, Z.S.; Parimi, V.; O’Malley, M.E.; Thirunavukarasu, P.; Sathaiah, M.; Austin, F.; Bartlett, D.L. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther. 2010, 17, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar]
- Liu, W.; Dai, E.; Liu, Z.; Ma, C.; Guo, Z.S.; Bartlett, D.L. In Situ Therapeutic Cancer Vaccination with an Oncolytic Virus Expressing Membrane-Tethered IL-2. Mol. Ther. Oncolyt. 2020, 17, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.S.; Naik, A.; O’Malley, M.E.; Popovic, P.; Demarco, R.; Hu, Y.; Yin, X.; Yang, S.; Zeh, H.J.; Moss, B.; et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005, 65, 9991–9998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whilding, L.M.; Archibald, K.M.; Kulbe, H.; Balkwill, F.R.; Oberg, D.; McNeish, I.A. Vaccinia virus induces programmed necrosis in ovarian cancer cells. Mol. Ther. 2013, 21, 2074–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, X.; Yin, Y.; Song, C.; Tan, L.; Qiu, X.; Liao, Y.; Liu, W.; Meng, S.; Sun, Y.; Ding, C. Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. iScience 2021, 24, 102837. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, A.; Fiegler, N.; Brunner-Weinzierl, M.; Cerwenka, A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-gamma production in response to mature dendritic cells. J. Immunol. 2014, 192, 4184–4191. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feist, M.; Zhu, Z.; Dai, E.; Ma, C.; Liu, Z.; Giehl, E.; Ravindranathan, R.; Kowalsky, S.J.; Obermajer, N.; Kammula, U.S.; et al. Oncolytic virus promotes tumor-reactive infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther. 2021, 28, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Milenova, I.; Lopez Gonzalez, M.; Quixabeira, D.C.A.; Santos, J.M.; Cervera-Carrascon, V.; Dong, W.; Hemminki, A.; van Beusechem, V.W.; van de Ven, R.; de Gruijl, T.D. Oncolytic Adenovirus ORCA-010 Activates Proinflammatory Myeloid Cells and Facilitates T Cell Recruitment and Activation by PD-1 Blockade in Melanoma. Hum. Gene Ther. 2021, 32, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, W.; Zhang, Y.; Dong, X.; Liu, C.; Yi, J.; Zhang, S.; Wen, C.; Zheng, L.; Wang, H. Oncolytic adenoviruses synergistically enhance anti-PD-L1 and anti-CTLA-4 immunotherapy by modulating the tumour microenvironment in a 4T1 orthotopic mouse model. Cancer Gene Ther. 2022, 29, 456–465. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Chen, H.; Zhu, Z.; Liu, Z.; Ma, C.; Lee, Y.J.; Bartlett, D.L.; Guo, Z.-S. Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy. Biomedicines 2022, 10, 1425. https://doi.org/10.3390/biomedicines10061425
Liu W, Chen H, Zhu Z, Liu Z, Ma C, Lee YJ, Bartlett DL, Guo Z-S. Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy. Biomedicines. 2022; 10(6):1425. https://doi.org/10.3390/biomedicines10061425
Chicago/Turabian StyleLiu, Weilin, Hongqi Chen, Zhi Zhu, Zuqiang Liu, Congrong Ma, Yong J. Lee, David L. Bartlett, and Zong-Sheng Guo. 2022. "Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy" Biomedicines 10, no. 6: 1425. https://doi.org/10.3390/biomedicines10061425
APA StyleLiu, W., Chen, H., Zhu, Z., Liu, Z., Ma, C., Lee, Y. J., Bartlett, D. L., & Guo, Z.-S. (2022). Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy. Biomedicines, 10(6), 1425. https://doi.org/10.3390/biomedicines10061425