
Oncolytic Vaccinia Virus Augments T Cell Factor 1-Positive Stem-like CD8+ T Cells, Which Underlies the Efficacy of Anti-PD-1 Combination Immunotherapy

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Cell Lines
2.3. Oncolytic Virus
2.4. Tumor Models and Treatment Regimens
2.5. Multicolor Flow Cytometry Analysis of Tumor-Associated Immune Cells
2.6. Immunofluorescence Microscopy
2.7. Statistical Analysis
3. Results
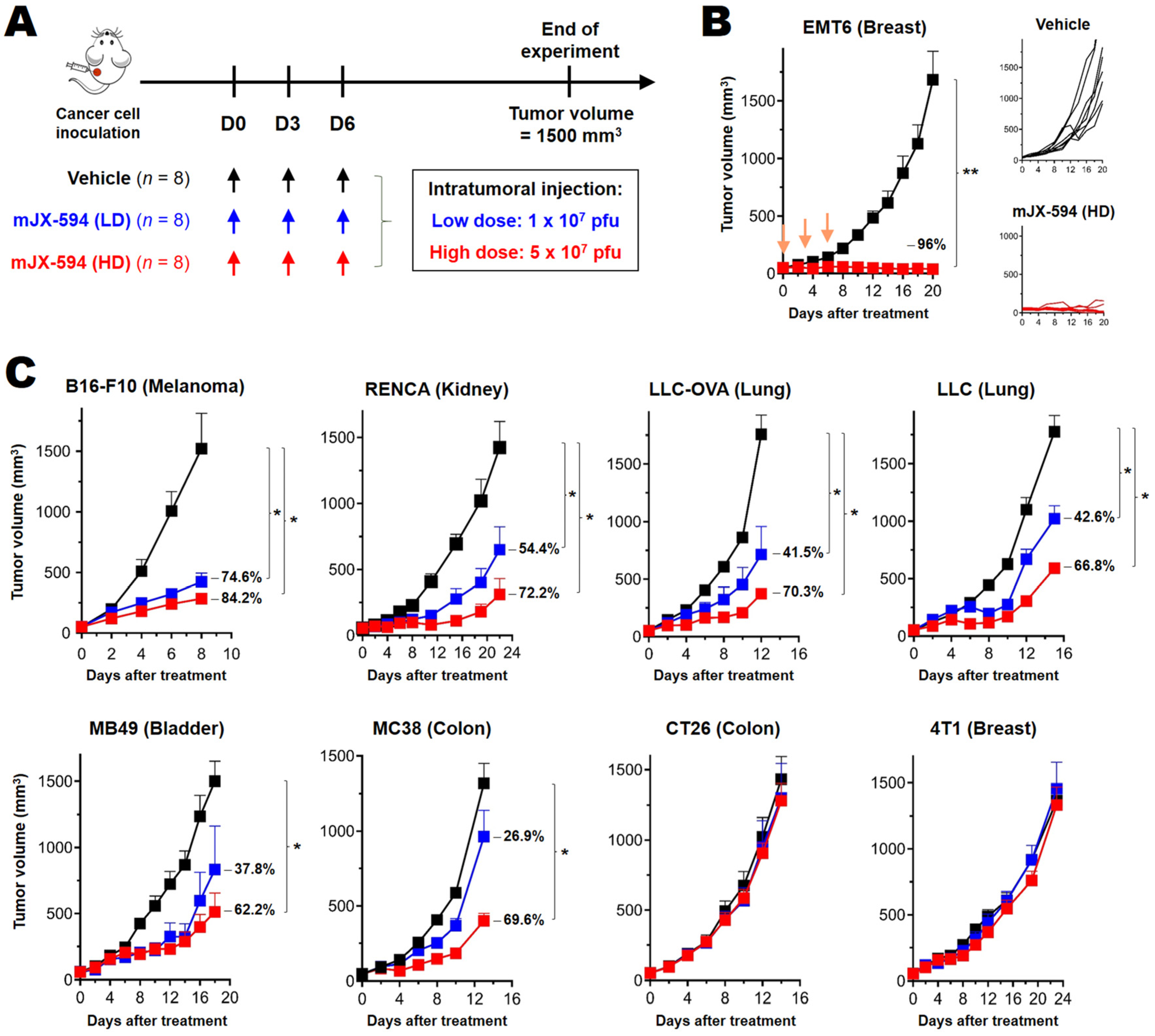
3.1. Differential Treatment Efficacy of mJX-594 in Syngeneic Murine Cancer Models
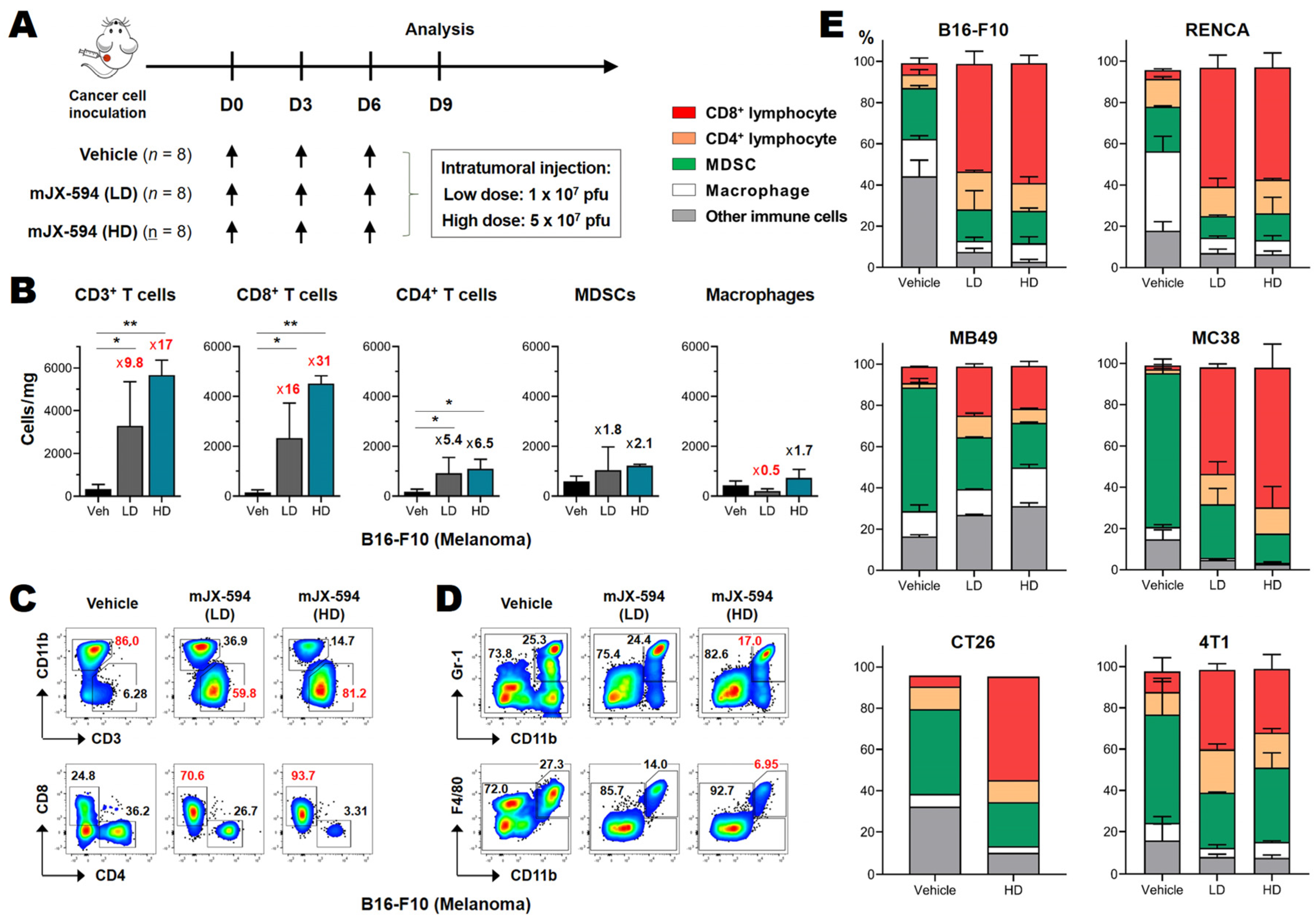
3.2. Marked CD8+ T Cell Recruitment in the Tumor following mJX-594 Treatment
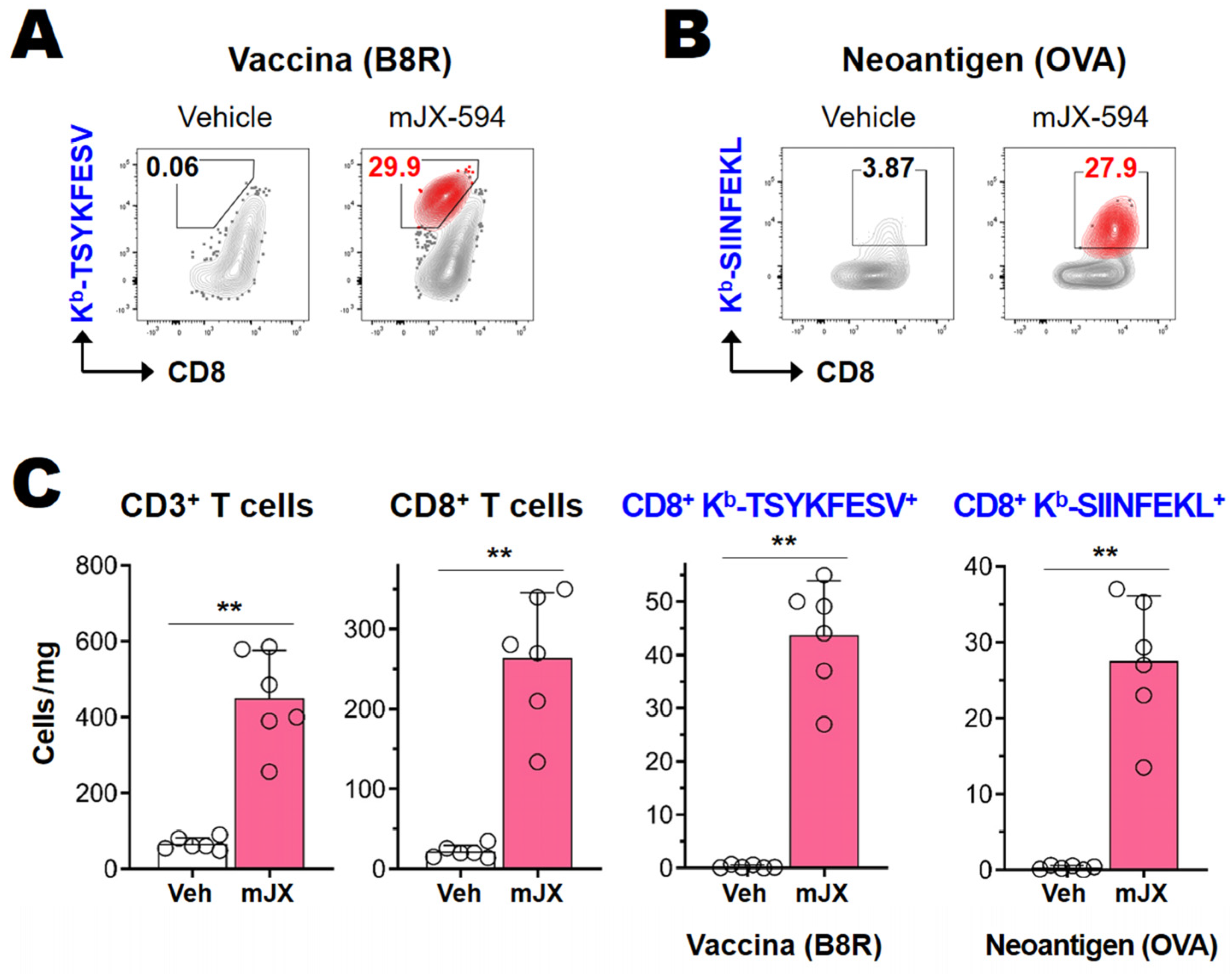
3.3. Cancer-Specific CD8+ T Cells as Well as Vaccinia Virus-Specific CD8+ T Cells Are Efficiently Activated and Recruited to Tumor Tissue
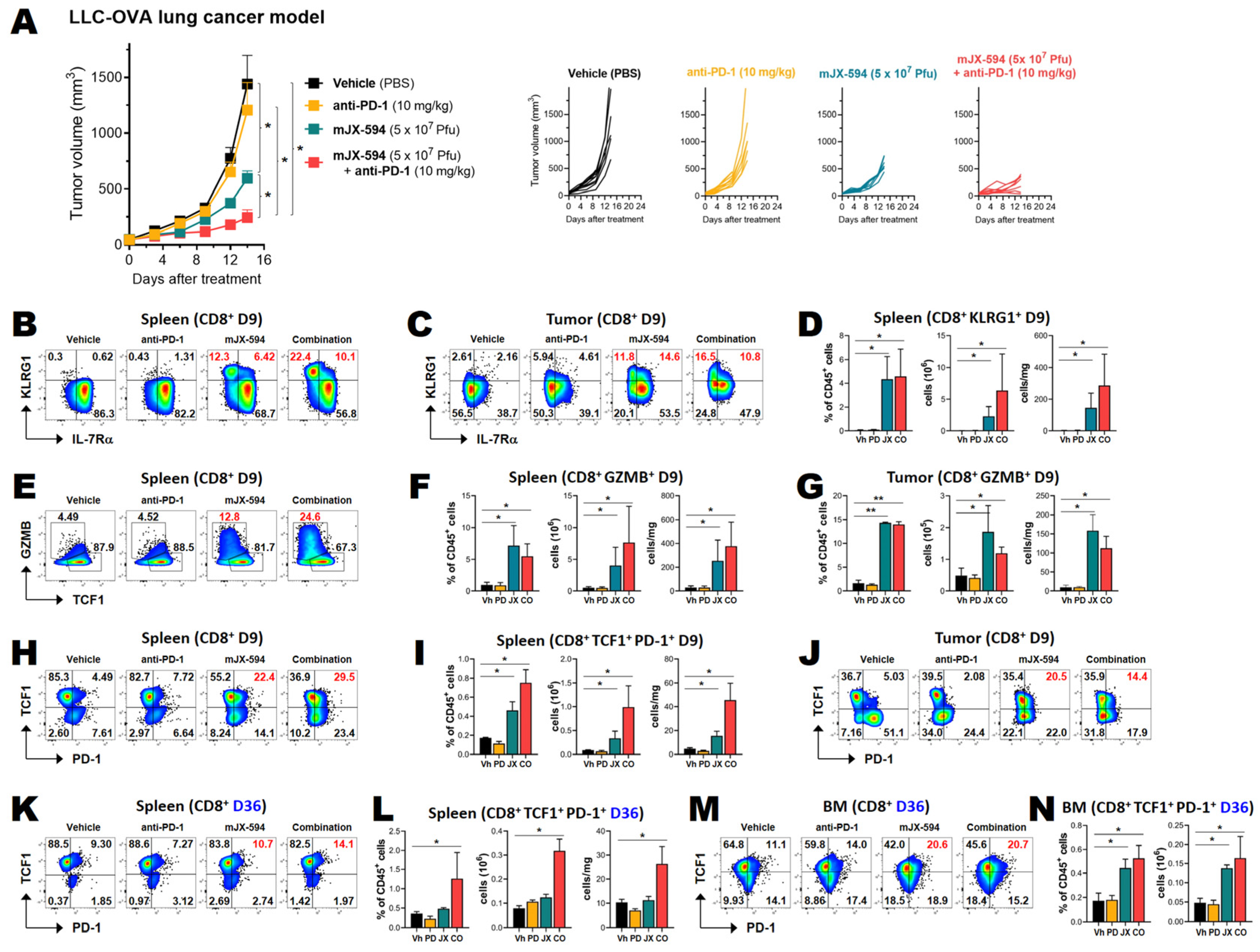
3.4. Anti-PD-1 Combination Therapy Increases Treatment Efficacy and Intratumoral Infiltration of T Cells in Two Murine Cancer Models
3.5. Among Cancer Antigen-Specific T Cells, TCF1+ Stem-like CD8+ T Cells Are Increased by mJX-594 and Further Increased by Anti-PD-1 Antibody Combination Treatment
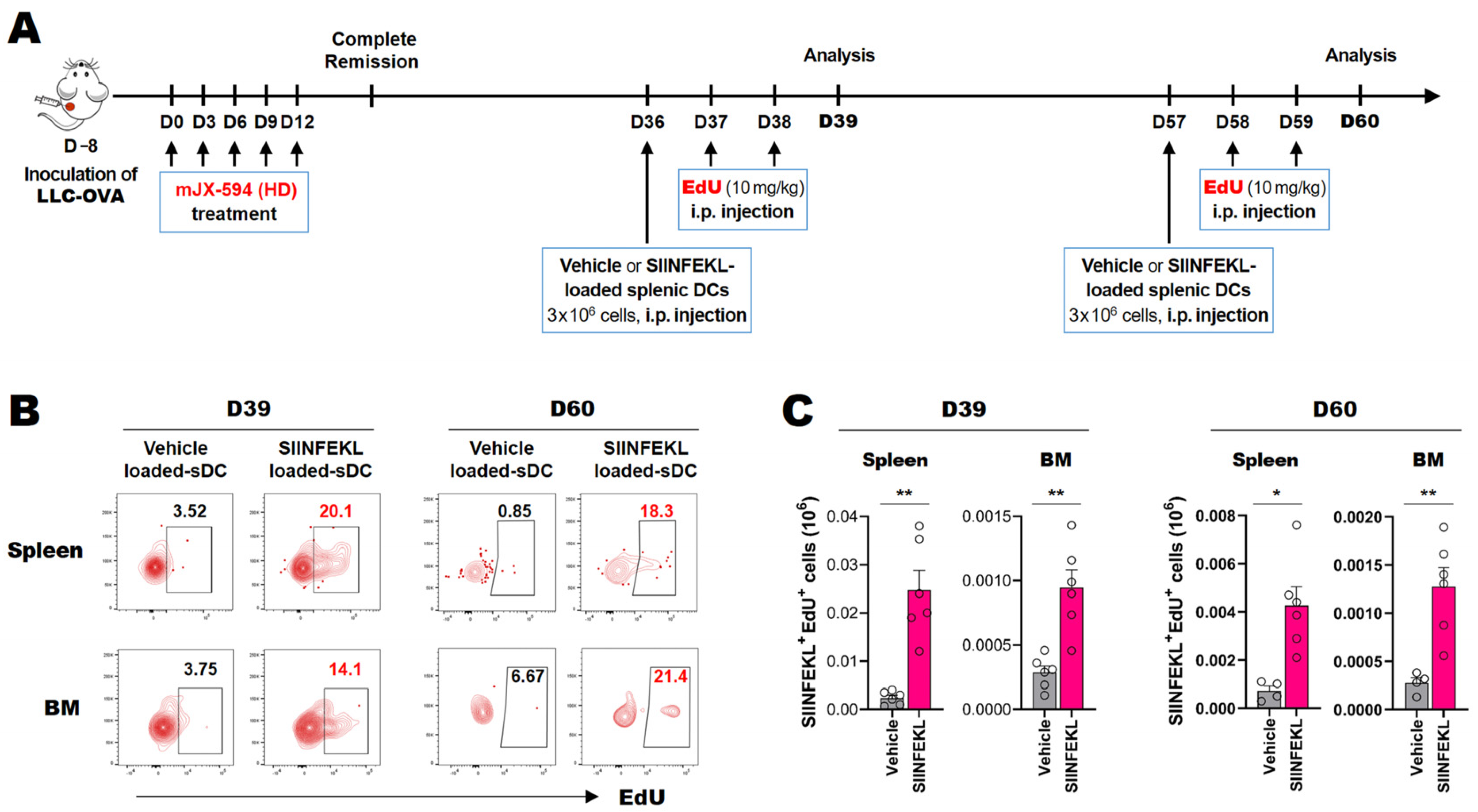
3.6. Cancer Neoantigen-Specific CD8+ T Cells That Survive for Extended Periods in the Spleen and Bone Marrow Proliferate in Response to Antigen-Loaded DCs In Situ
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| ATCC | American Type Culture Collection |
| CR | complete remission |
| DC | dendritic cells |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GZMB | granzyme B |
| HD | high-dose |
| ICB | immune checkpoint blockade |
| IFN-γ | interferon gamma |
| JX-594 | pexastimogene devacirepvec |
| LD | low-dose |
| LLC-OVA | LLC cell line expressing OVA |
| MDSC | myeloid-derived suppressor cell |
| mJX-594 | a mouse variant of JX-594 |
| mJX | mJX-594 |
| OV | oncolytic viruses |
| OVA | ovalbumin |
| PBS | phosphate-buffered saline |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed death-ligand 1 |
| Pexa-vec | pexastimogene devacirepvec |
| pfu | plaque-forming units |
| SEM | standard error of the mean |
| TAA | tumor-associated antigen |
| TCF1 | T cell factor 1 |
| TK | thymidine kinase |
| TME | tumor microenvironment |
| VACV | vaccinia virus |
| Veh | Vehicle |
References
- Russell, S.J.; Peng, K.H.; John, C.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Anandappa, A.J.; Wu, C.J.; Ott, P.A. Directing Traffic: How to Effectively Drive T Cells into Tumors. Cancer Discov. 2020, 10, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [Green Version]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2021, 21, 116–128. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahé, E.A.; Cooper, R.; Woodcock, V.; Traill, Z.; et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.A.; Kuchroo, M.; Venkat, A.; Khatun, A.; Wang, J.; William, I.; Hornick, N.I.; Fitzgerald, B.L.; Damo, M.; Kasmani, M.Y.; et al. A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 2021, 6, eabg7836. [Google Scholar] [CrossRef] [PubMed]
- Baharom, F.; Ramirez-Valdez, R.A.; Tobin, K.K.S.; Yamane, H.; Dutertre, C.A.; Khalilnezhad, A.; Reynoso, G.V.; Coble, V.L.; Lynn, G.M.; Mulè, M.P.; et al. Intravenous nanoparticle vaccination generates stem-like TCF1+ neoantigen-specific CD8+ T cells. Nat. Immunol. 2021, 22, 41–52. [Google Scholar] [CrossRef]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gurusamy, D.; et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Avidal, L.M.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef]
- Chon, H.J.; Lee, W.S.; Yang, H.; Kong, S.J.; Lee, N.K.; Moon, E.S.; Choi, J.; Han, E.C.; Kim, J.H.; Ahn, J.B.; et al. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin. Cancer Res. 2019, 25, 1612–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Lee, W.S.; Kim, C.W.; Lee, S.J.; Yang, H.; Kong, S.J.; Ning, J.; Yang, K.M.; Kang, B.; Kim, W.R.; et al. Oncolytic vaccinia virus reinvigorates peritoneal immunity and cooperates with immune checkpoint inhibitor to suppress peritoneal carcinomatosis in colon cancer. J. Immunother. Cancer 2020, 8, e000857. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Lee, M.E.; Jang, W.S.; Kim, J.; Park, S.M.; Oh, K.; Lee, N.; Ham, W.S. Systemic Injection of Oncolytic Vaccinia Virus Suppresses Primary Tumor Growth and Lung Metastasis in Metastatic Renal Cell Carcinoma by Remodeling Tumor Microenvironment. Biomedicines 2022, 10, 173. [Google Scholar] [CrossRef]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giehl, E.; Kosaka, H.; Liu, Z.; Feist, M.; Kammula, U.S.; Lotze, M.T.; Ma, C.; Guo, Z.S.; Bartlett, D.L. In Vivo Priming of Peritoneal Tumor-Reactive Lymphocytes with a Potent Oncolytic Virus for Adoptive Cell Therapy. Front. Immunol. 2021, 12, 610042. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nitschké, M.; Sennino, B.; Murer, P.; Schriver, B.J.; Bell, A.; Subramanian, A.; McDonald, C.E.; Wang, J.; Cha, H.; et al. Amplification of Oncolytic Vaccinia Virus Widespread Tumor Cell Killing by Sunitinib through Multiple Mechanisms. Cancer Res. 2018, 78, 922–937. [Google Scholar] [CrossRef] [Green Version]
- Jeannet, G.; Boudousquié, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 2010, 107, 9777–9782. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, L.L.; Bonasio, R.; Mazo, I.B.; Halin, C.; Cheng, G.; van der Velden, A.W.; Cariappa, A.; Chase, C.; Russell, P.; Starnbach, M.N.; et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat. Immunol. 2005, 6, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Oh, K.; Shon, S.Y.; Seo, M.W.; Lee, H.M.; Oh, J.E.; Choi, E.Y.; Lee, D.S.; Park, K.S. Murine Sca1+Lin− bone marrow contains an endodermal precursor population that differentiates into hepatocytes. Exp. Mol. Med. 2015, 47, e187. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Ferreira, D.P.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.B.A.G.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Khatwani, N.; Searles, T.G.; Turk, M.J.; Angeles, C.V. Memory CD8+ T cell responses to cancer. Semin. Immunol. 2020, 49, 101435. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Sherry, R.M.; Morton, K.E.; Scharfman, W.J.; Yang, J.C.; Topalian, S.L.; Royal, R.E.; Kammula, U.; Restifo, N.P.; Hughes, M.S.; et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J. Immunol. 2005, 175, 6169–6176. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.; Feng, Z.; Luo, J.; He, Z.; Liu, J.; Wu, J.; Rong, P. Tertiary Lymphoid Structures in Cancer: The Double-Edged Sword Role in Antitumor Immunity and Potential Therapeutic Induction Strategies. Front. Immunol. 2021, 12, 689270. [Google Scholar] [CrossRef]
- Im, S.J.; Konieczny, B.T.; Hudson, W.H.; Masopust, D.; Ahmed, R. PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 4292–4299. [Google Scholar] [CrossRef]
- Chang, H.D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef]
- Masopust, D.; Soerens, A.G. Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol. 2019, 37, 521–546. [Google Scholar] [CrossRef]
- Park, S.L.; Gebhardt, T.; Mackay, L.K. Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends Immunol. 2019, 40, 735–747. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, Y.-H.; Lee, N.; Yoo, J.; Won, S.; Shin, S.-k.; Kim, K.-H.; Park, J.-G.; Kim, M.-G.; Kim, H.-R.; Oh, K.; et al. Oncolytic Vaccinia Virus Augments T Cell Factor 1-Positive Stem-like CD8+ T Cells, Which Underlies the Efficacy of Anti-PD-1 Combination Immunotherapy. Biomedicines 2022, 10, 805. https://doi.org/10.3390/biomedicines10040805
Jeon Y-H, Lee N, Yoo J, Won S, Shin S-k, Kim K-H, Park J-G, Kim M-G, Kim H-R, Oh K, et al. Oncolytic Vaccinia Virus Augments T Cell Factor 1-Positive Stem-like CD8+ T Cells, Which Underlies the Efficacy of Anti-PD-1 Combination Immunotherapy. Biomedicines. 2022; 10(4):805. https://doi.org/10.3390/biomedicines10040805
Chicago/Turabian StyleJeon, Yun-Hui, Namhee Lee, Jiyoon Yoo, Solchan Won, Suk-kyung Shin, Kyu-Hwan Kim, Jun-Gyu Park, Min-Gang Kim, Hang-Rae Kim, Keunhee Oh, and et al. 2022. "Oncolytic Vaccinia Virus Augments T Cell Factor 1-Positive Stem-like CD8+ T Cells, Which Underlies the Efficacy of Anti-PD-1 Combination Immunotherapy" Biomedicines 10, no. 4: 805. https://doi.org/10.3390/biomedicines10040805
APA StyleJeon, Y.-H., Lee, N., Yoo, J., Won, S., Shin, S.-k., Kim, K.-H., Park, J.-G., Kim, M.-G., Kim, H.-R., Oh, K., & Lee, D.-S. (2022). Oncolytic Vaccinia Virus Augments T Cell Factor 1-Positive Stem-like CD8+ T Cells, Which Underlies the Efficacy of Anti-PD-1 Combination Immunotherapy. Biomedicines, 10(4), 805. https://doi.org/10.3390/biomedicines10040805