
Inhibitory Effectiveness in Delayed-Rectifier Potassium Current Caused by Vortioxetine, Known to Be a Novel Antidepressant


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs, Chemicals and Solutions
2.2. Cell Preparations
2.3. Electrophysiological Measurements
2.4. Data Recordings
2.5. Analyses of IK(DR) Evoked by Membrane Depolarization
2.6. Statistical Analyses
3. Results
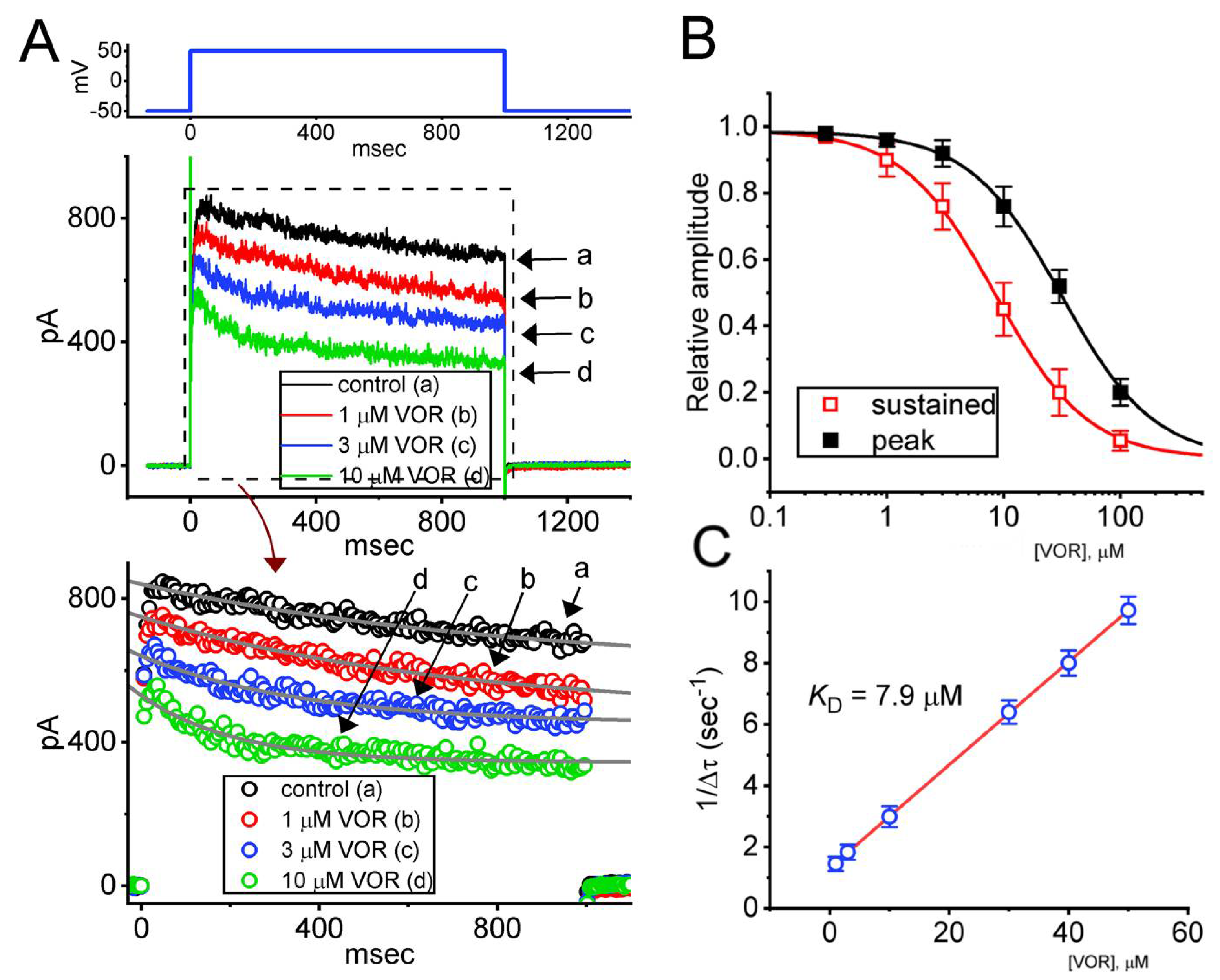
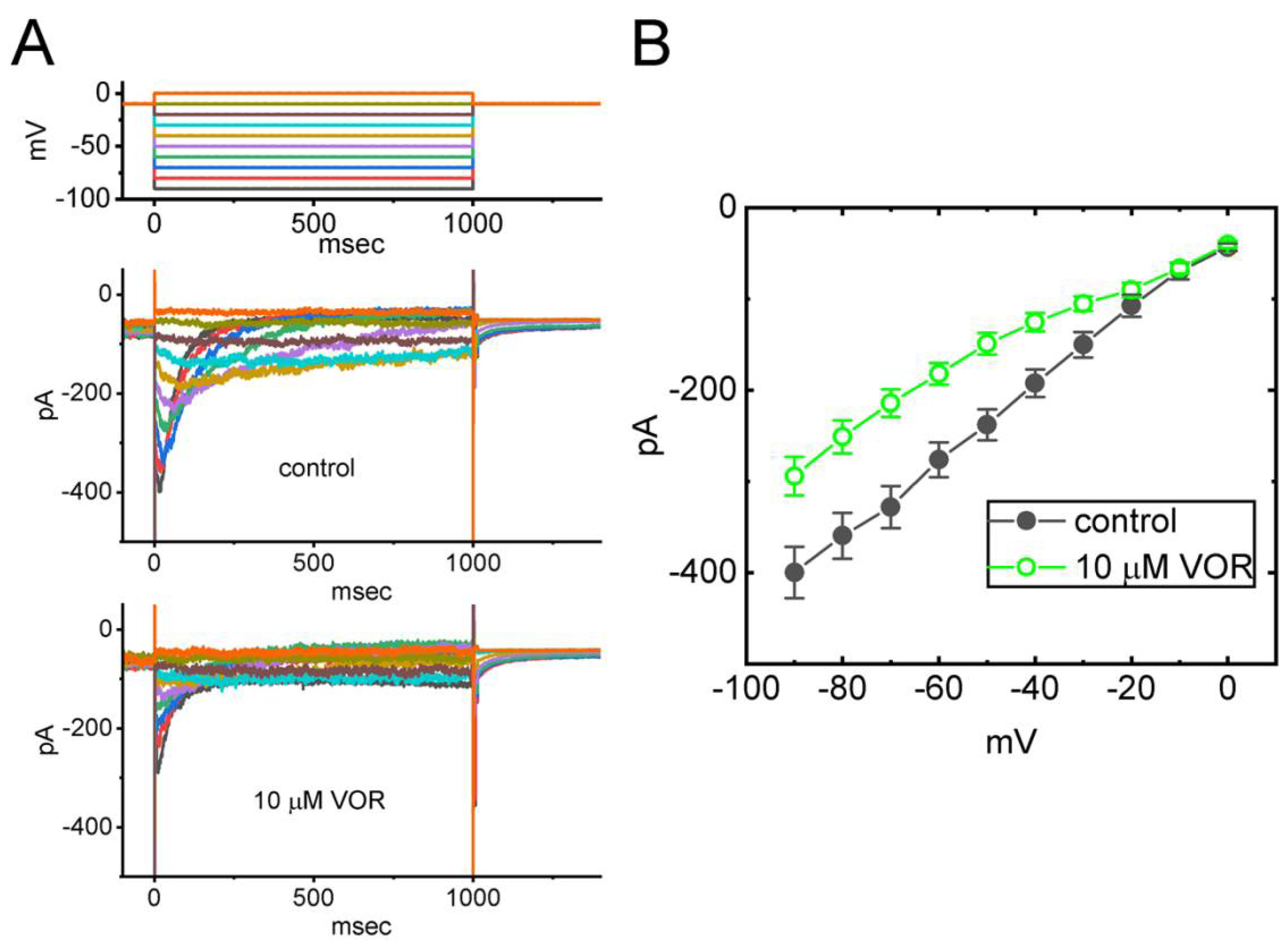
3.1. Effect of VOR on Delayed-Rectifier K+ Current (IK(DR)) Recorded from Pituitary GH3 Cells
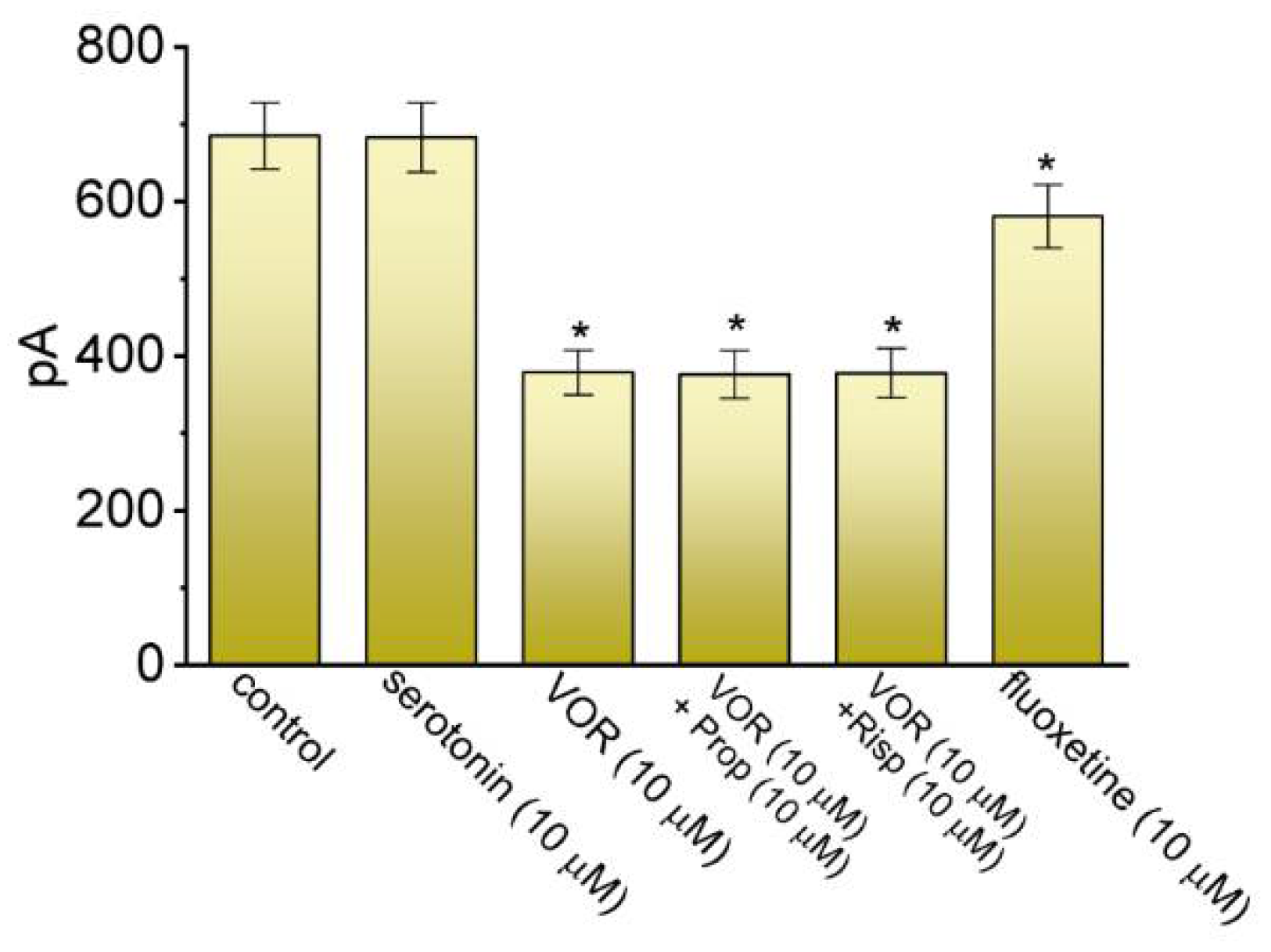
3.2. Comparison in Effects on IK(DR) Caused by Serotonin, VOR, VOR plus Propranolol, VOR plus Risperidone, and Fluoxetine
3.3. Mean Current-Voltage (I-V) Relationship of Peak and Sustained Components of IK(DR) Caused by VOR
3.4. Steady-State Inactivation Curve of IK(DR) Produced by the Presence of VOR
3.5. Change in Current Inactivation Time Course Produced by VOR
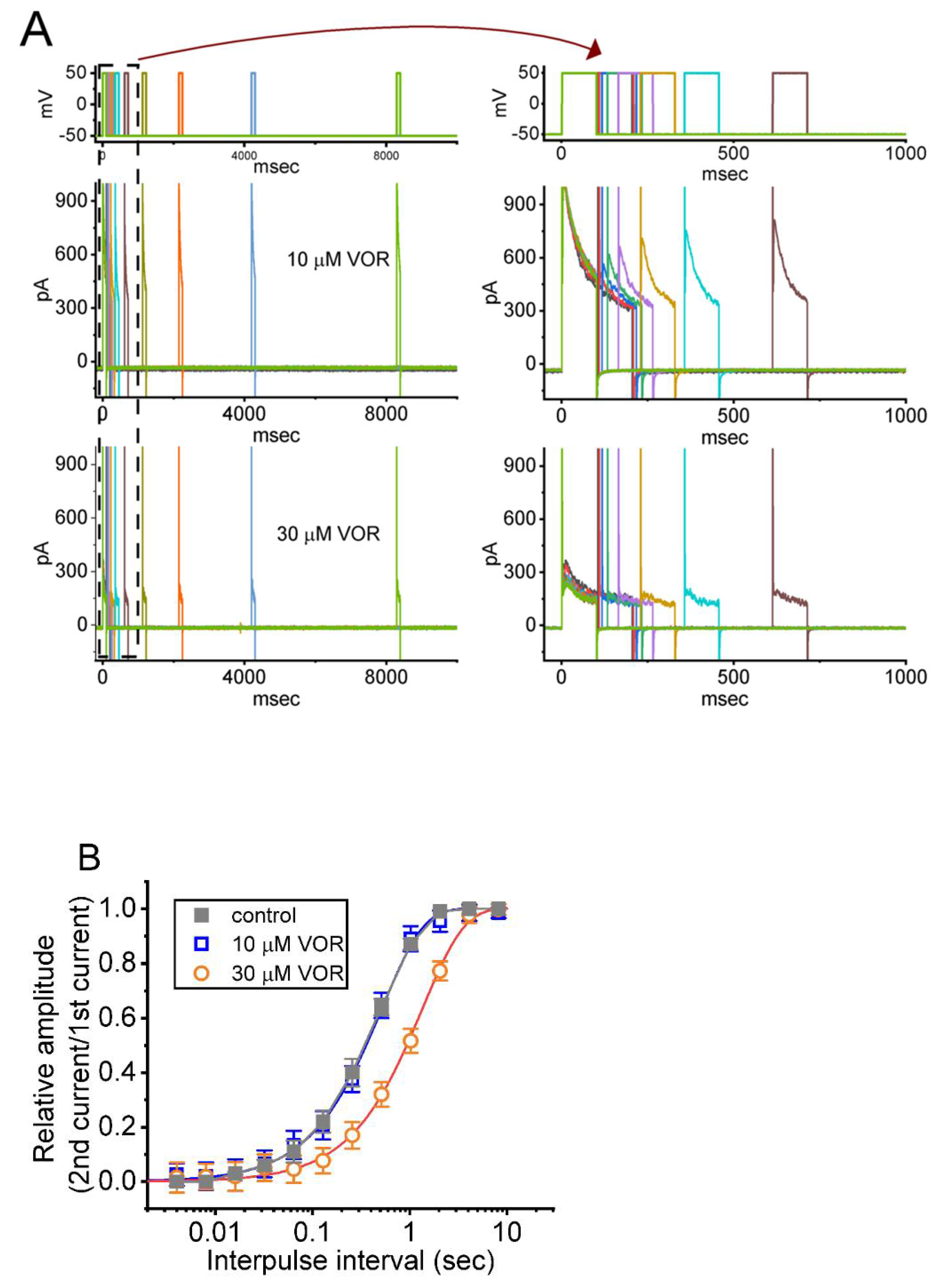
3.6. IK(DR) Recovery from Inactivation in the Presence of VOR
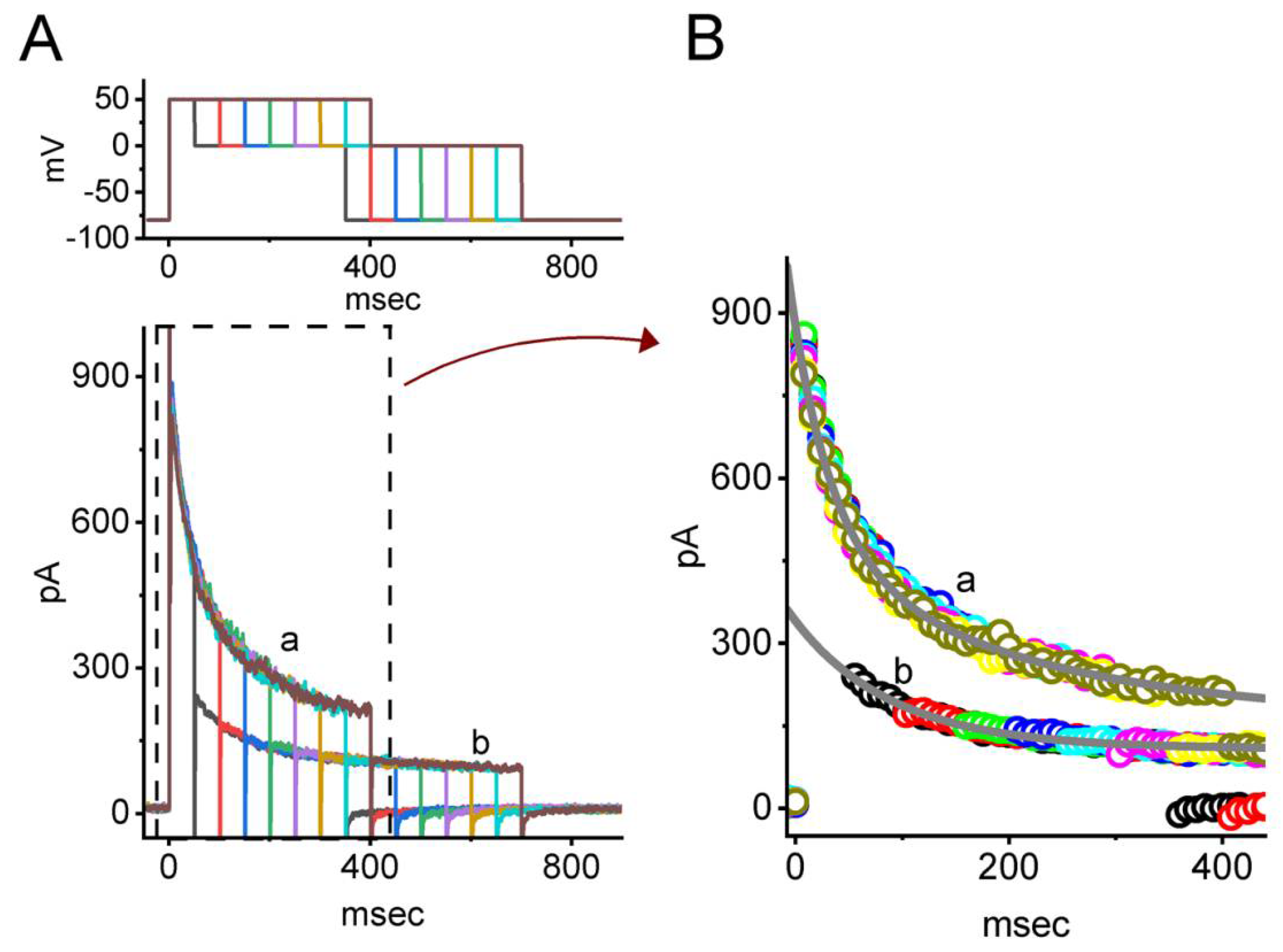
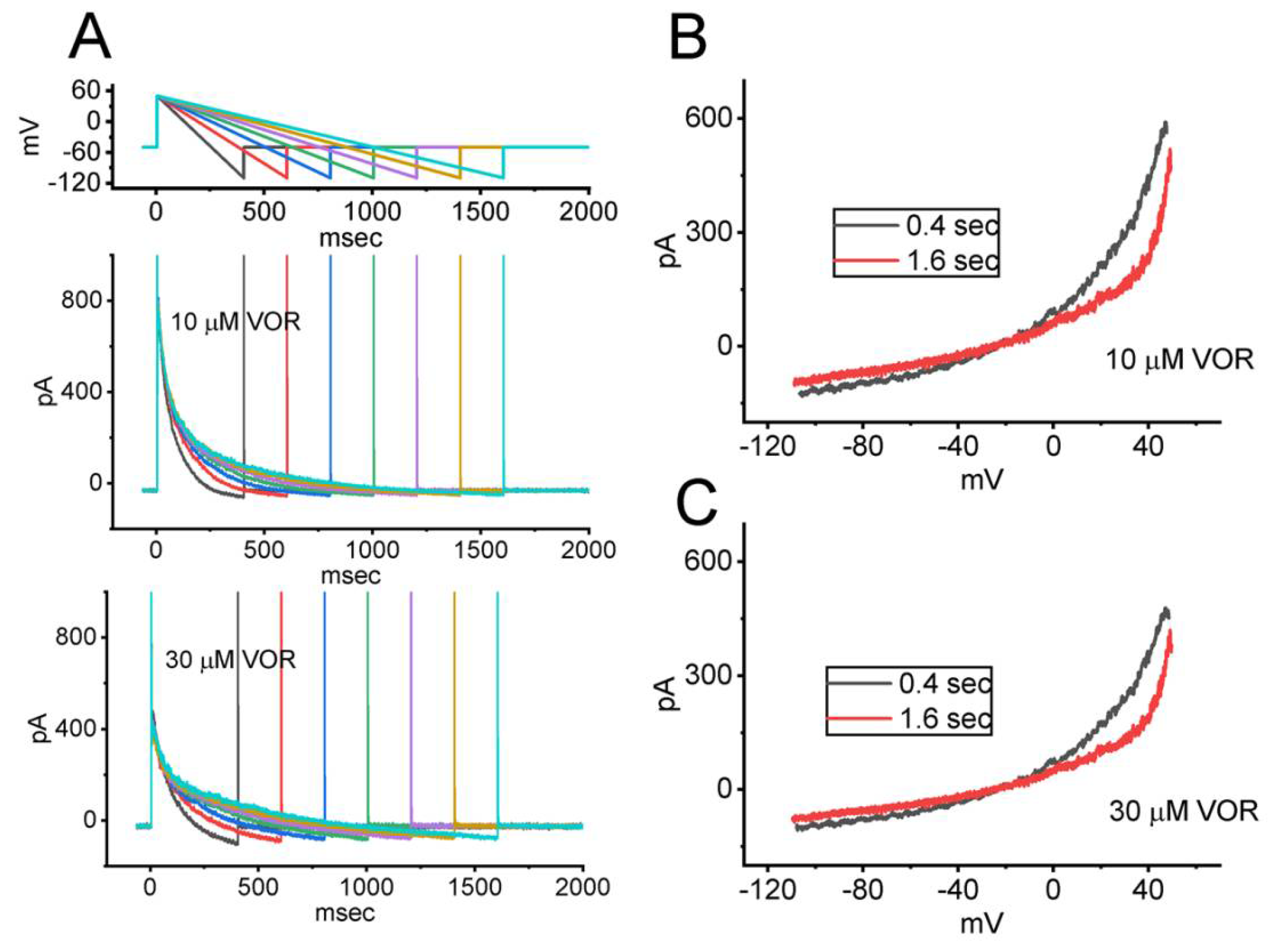
3.7. Effect of VOR on IK(DR) Activated by Varying Downsloping Ramp Pulse
3.8. VOR-Induced Increase in Cumulative Inhibition of IK(DR) Inactivation
3.9. Effect of VOR on Erg-Mediated K+ Current (IK(erg)) in GH3 Cells
3.10. Inhibitory Effect of VOR on IK(DR) Residing in Neuro-2a Neuroblastoma Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bang-Andersen, B.; Ruhland, T.; Jørgensen, M.; Smith, G.; Frederiksen, K.; Gjervig-Jensen, K.; Zhong, H.; Møller-Nielsen, S.; Hogg, S.; Mørk, A.; et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A novel multimodal compound for the treatment of major depressive disorder. J. Med. Chem. 2011, 54, 3206–3221. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Perez, V.; Artigas, F. Pharmacology and clinical potential of vortioxetine in the treatment of major depressive disorder. Neuropsychiatr. Dis. Treat. 2014, 10, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Sarvaiya, J. The efficacy of vortioxetine for the treatment of major depressive disorder. Expert Rev. Neurother. 2014, 14, 1349–1363. [Google Scholar] [CrossRef]
- Pae, C.U.; Wang, S.M.; Han, C.; Lee, S.J.; Patkar, A.A.; Masand, P.S.; Serretti, A. Vortioxetine, a multimodal antidepressant for generalized anxiety disorder: A systematic review and meta-analysis. J. Psychiatr. Res. 2015, 64, 88–98. [Google Scholar] [CrossRef]
- Zhang, J.; Mathis, M.V.; Sellers, J.W.; Kordzakhia, G.; Jackson, A.J.; Dow, A.; Yang, P.; Fossom, L.; Zhu, H.; Patel, H.; et al. The US Food and Drug Administration’s perspective on the new antidepressant vortioxetine. J. Clin. Psychiatry 2015, 76, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Koesters, M.; Ostuzzi, G.; Guaiana, G.; Breilmann, J.; Barbui, C. Vortioxetine for Depression in Adults. Cochrane Database Syst. Rev. 2017, 7, Cd011520. [Google Scholar] [CrossRef] [PubMed]
- Paolucci, S. Advances in antidepressants for treating post-stroke depression. Expert Opin. Pharmacother. 2017, 18, 1011–1017. [Google Scholar] [CrossRef]
- Beyer, J.L.; Johnson, K.G. Advances in Pharmacotherapy of Late-Life Depression. Curr. Psychiatry Rep. 2018, 20, 34. [Google Scholar] [CrossRef]
- Gonda, X.; Sharma, S.R.; Tarazi, F.I. Vortioxetine: A novel antidepressant for the treatment of major depressive disorder. Expert Opin. Drug Discov. 2019, 14, 81–89. [Google Scholar] [CrossRef]
- Long, J.D. Vortioxetine for Depression in Adults. Issues Ment. Health Nurs. 2019, 40, 819–820. [Google Scholar] [CrossRef]
- Ostuzzi, G.; Gastaldon, C.; Barbato, A.; D’Avanzo, B.; Tettamanti, M.; Monti, I.; Aguglia, A.; Aguglia, E.; Alessi, M.C.; Amore, M.; et al. Tolerability and efficacy of vortioxetine versus SSRIs in elderly with major depression. Study protocol of the VESPA study: A pragmatic, multicentre, open-label, parallel-group, superiority, randomized trial. Trials 2020, 21, 695. [Google Scholar] [CrossRef] [PubMed]
- Marchiafava, M.; Piccirilli, M.; Bedetti, C.; Baglioni, A.; Menna, M.; Elisei, S. Effectiveness of serotonergic drugs in the management of problem behaviors in patients with neurodevelopmental disorders. Psychiatr. Danub. 2018, 30 (Suppl. S7), 644–647. [Google Scholar] [PubMed]
- Lochmann, D.; Richardson, T. Selective Serotonin Reuptake Inhibitors. Handb. Exp. Pharmacol. 2019, 250, 135–144. [Google Scholar]
- Keks, N.A.; Hope, J.; Culhane, C. Vortioxetine: A multimodal antidepressant or another selective serotonin reuptake inhibitor? Australas. Psychiatry 2015, 23, 210–213. [Google Scholar] [CrossRef]
- Kirilly, E.; Gonda, X.; Bagdy, G. Antidepressants, stressors and the serotonin 1A receptor. Neuropsychopharmacol. Hung. 2015, 17, 81–89. [Google Scholar]
- Franklin, M.; Hlavacova, N.; Li, Y.; Bermudez, I.; Csanova, A.; Sanchez, C.; Jezova, D. Contrasting effects of vortioxetine and paroxetine on pineal gland biochemistry in a tryptophan-depletion model of depression in female rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 499–502. [Google Scholar] [CrossRef]
- Brivio, P.; Corsini, G.; Riva, M.A.; Calabrese, F. Chronic vortioxetine treatment improves the responsiveness to an acute stress acting through the ventral hippocampus in a glucocorticoid-dependent way. Pharmacol. Res. 2019, 142, 14–21. [Google Scholar] [CrossRef]
- Mago, R.; Tripathi, N.; Andrade, C. Cardiovascular adverse effects of newer antidepressants. Expert Rev. Neurother. 2014, 14, 539–551. [Google Scholar] [CrossRef]
- Behlke, L.M.; Lenze, E.J.; Carney, R.M. The Cardiovascular Effects of Newer Antidepressants in Older Adults and Those with or At High Risk for Cardiovascular Diseases. CNS Drugs 2020, 34, 1133–1147. [Google Scholar] [CrossRef]
- Wulfsen, I.; Hauber, H.P.; Schiemann, D.; Bauer, C.K.; Schwarz, J.R. Expression of mRNA for voltage-dependent and inward-rectifying K channels in GH3/B6 cells and rat pituitary. J. Neuroendocrinol. 2000, 12, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Figueroa, D.J.; Austin, C.P.; Liu, Y.; Bugianesi, R.M.; Slaughter, R.S.; Kaczorowski, G.J.; Kohler, M. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes 2004, 53, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, P.A.; Sherman, A.; Stojilkovic, S.S. Common and diverse elements of ion channels and receptors underlying electrical activity in endocrine pituitary cells. Mol. Cell. Endocrinol. 2018, 463, 23–36. [Google Scholar] [CrossRef]
- Kuo, P.C.; Yang, C.J.; Lee, Y.C.; Chen, P.C.; Liu, Y.C.; Wu, S.N. The comprehensive electrophysiological study of curcuminoids on delayed-rectifier K(+) currents in insulin-secreting cells. Eur. J. Pharmacol. 2018, 819, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Chang, W.T.; Wu, S.N. Characterization of the Synergistic Inhibition of I(K(erg)) and I(K(DR)) by Ribociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor. Int. J. Mol. Sci. 2020, 21, 8078. [Google Scholar] [CrossRef]
- Hsiao, H.T.; Lu, G.L.; Liu, Y.C.; Wu, S.N. Effective Perturbations of the Amplitude, Gating, and Hysteresis of I(K(DR)) Caused by PT-2385, an HIF-2α Inhibitor. Membranes 2021, 11, 636. [Google Scholar] [CrossRef]
- Marom, S.; Goldstein, S.A.; Kupper, J.; Levitan, I.B. Mechanism and modulation of inactivation of the Kv3 potassium channel. Recept Channels 1993, 1, 81–88. [Google Scholar] [PubMed]
- Lin, M.W.; Wang, Y.J.; Liu, S.I.; Lin, A.A.; Lo, Y.C.; Wu, S.N. Characterization of aconitine-induced block of delayed rectifier K+ current in differentiated NG108-15 neuronal cells. Neuropharmacology 2008, 54, 912–923. [Google Scholar] [CrossRef]
- Wu, S.N.; Chen, B.S.; Lin, M.W.; Liu, Y.C. Contribution of slowly inactivating potassium current to delayed firing of action potentials in NG108-15 neuronal cells: Experimental and theoretical studies. J. Theor. Biol. 2008, 252, 711–721. [Google Scholar] [CrossRef]
- Labro, A.J.; Priest, M.F.; Lacroix, J.J.; Snyders, D.J.; Bezanilla, F. Kv3.1 uses a timely resurgent K(+) current to secure action potential repolarization. Nat. Commun. 2015, 6, 10173. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kaczmarek, L.K. Kv3.3 potassium channels and spinocerebellar ataxia. J. Physiol. 2016, 594, 4677–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, L.K.; Zhang, Y. Kv3 Channels: Enablers of Rapid Firing, Neurotransmitter Release, and Neuronal Endurance. Physiol. Rev. 2017, 97, 1431–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, N.; Linley, D.; Richardson, A.; Anderson, M.; Robinson, S.W.; Marra, V.; Ciampani, V.; Walter, S.M.; Kopp-Scheinpflug, C.; Steinert, J.R. Kv3.1 and Kv3.3 subunits differentially contribute to Kv3 channels and action potential repolarization in principal neurons of the auditory brainstem. J. Physiol. 2020, 598, 2199–2222. [Google Scholar] [CrossRef] [PubMed]
- Simasko, S.M. Evidence for a delayed rectifier-like potassium current in the clonal rat pituitary cell line GH3. Am. J. Physiol. 1991, 261 Pt 1, E66–E75. [Google Scholar] [CrossRef]
- Chang, W.T.; Liu, P.Y.; Gao, Z.H.; Lee, S.W.; Lee, W.K.; Wu, S.N. Evidence for the Effectiveness of Remdesivir (GS-5734), a Nucleoside-Analog Antiviral Drug in the Inhibition of I (K(M)) or I (K(DR)) and in the Stimulation of I (MEP). Front. Pharmacol. 2020, 11, 1091. [Google Scholar] [CrossRef]
- Parekh, P.K.; Sidor, M.M.; Gillman, A.; Becker-Krail, D.; Bettelini, L.; Arban, R.; Alvaro, G.S.; Zambello, E.; Mutinelli, C.; Huang, Y.; et al. Antimanic Efficacy of a Novel Kv3 Potassium Channel Modulator. Neuropsychopharmacology 2018, 43, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Hibino, H.; Lossin, C.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007, 1178, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Furutani, K.; Ohno, Y.; Inanobe, A.; Hibino, H.; Kurachi, Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol. Pharmacol. 2009, 75, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Ohno, Y.; Lossin, C.; Hibino, H.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J. Pharmacol. Exp. Ther. 2007, 320, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.K.; Park, M.H.; Lee, B.H.; Jo, S.H. Block of the human ether-a-go-go-related gene (hERG) K+ channel by the antidepressant desipramine. Biochem. Biophys. Res. Commun. 2010, 394, 536–541. [Google Scholar] [CrossRef]
- Staudacher, I.; Wang, L.; Wan, X.; Obers, S.; Wenzel, W.; Tristram, F.; Koschny, R.; Staudacher, K.; Kisselbach, J.; Koelsch, P. hERG K+ channel-associated cardiac effects of the antidepressant drug desipramine. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.E.; Li, H.; An, J.R.; Seo, M.S.; Na, S.H.; Jung, W.K.; Firth, A.L.; Ha, K.S.; Han, E.T.; Hong, S.H.; et al. Inhibition of the Voltage-Dependent K(+) Current by the Tricyclic Antidepressant Desipramine in Rabbit Coronary Arterial Smooth Muscle Cells. Cardiovasc. Toxicol. 2018, 18, 252–260. [Google Scholar] [CrossRef] [PubMed]
- An, J.R.; Kang, H.; Li, H.; Seo, M.S.; Jung, H.S.; Jung, W.K.; Choi, I.W.; Ryu, S.W.; Park, H.; Bae, Y.M.; et al. Protriptyline, a tricyclic antidepressant, inhibits voltage-dependent K+ channels in rabbit coronary arterial smooth muscle cells. Acta Biochim. Biophys. Sin. 2020, 52, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.N.; Lo, Y.K.; Li, H.F.; Shen, A.Y. Functional coupling of voltage-dependent L-type Ca2+ current to Ca2+-activated K+ current in pituitary GH3 cells. Chin. J. Physiol. 2001, 44, 161–167. [Google Scholar] [PubMed]
- Wu, C.C.; Chen, Y.C.; Chang, Y.C.; Wang, L.W.; Lin, Y.C.; Chiang, Y.L.; Ho, C.J.; Huang, C.C. Human umbilical vein endothelial cells protect against hypoxic-ischemic damage in neonatal brain via stromal cell-derived factor 1/C-X-C chemokine receptor type 4. Stroke 2013, 44, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.N.; Wu, Y.H.; Chen, B.S.; Lo, Y.C.; Liu, Y.C. Underlying mechanism of actions of tefluthrin, a pyrethroid insecticide, on voltage-gated ion currents and on action currents in pituitary tumor (GH3) cells and GnRH-secreting (GT1-7) neurons. Toxicology 2009, 258, 70–77. [Google Scholar] [CrossRef]
- Lai, M.C.; Wu, S.N.; Huang, C.W. Telmisartan, an Antagonist of Angiotensin II Receptors, Accentuates Voltage-Gated Na(+) Currents and Hippocampal Neuronal Excitability. Front. Neurosci. 2020, 14, 902. [Google Scholar] [CrossRef]
- Chuang, T.H.; Cho, H.Y.; Wu, S.N. Effective Accentuation of Voltage-Gated Sodium Current Caused by Apocynin (4’-Hydroxy-3’-methoxyacetophenone), a Known NADPH-Oxidase Inhibitor. Biomedicines 2021, 9, 1146. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Asin, K.E.; Artigas, F. Vortioxetine, a novel antidepressant with multimodal activity: Review of preclinical and clinical data. Pharmacol. Ther. 2015, 145, 43–57. [Google Scholar] [CrossRef]
- Mørk, A.; Pehrson, A.; Brennum, L.T.; Nielsen, S.M.; Zhong, H.; Lassen, A.B.; Miller, S.; Westrich, L.; Boyle, N.J.; Sánchez, C.; et al. Pharmacological effects of Lu AA21004: A novel multimodal compound for the treatment of major depressive disorder. J. Pharmacol. Exp. Ther. 2012, 340, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Pehrson, A.L.; Cremers, T.; Bétry, C.; van der Hart, M.G.; Jørgensen, L.; Madsen, M.; Haddjeri, N.; Ebert, B.; Sanchez, C. Lu AA21004, a novel multimodal antidepressant, produces regionally selective increases of multiple neurotransmitters—A rat microdialysis and electrophysiology study. Eur. Neuropsychopharmacol. 2013, 23, 133–145. [Google Scholar] [CrossRef]
- Tinajero, J.C.; Fabbri, A.; Dufau, M.L. Serotonergic inhibition of rat Leydig cell function by propranolol. Endocrinology 1993, 133, 257–264. [Google Scholar] [CrossRef]
- Vanhauwe, J.F.; Fraeyman, N.; Francken, B.J.; Luyten, W.H.; Leysen, J.E. Comparison of the ligand binding and signaling properties of human dopamine D(2) and D(3) receptors in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 1999, 290, 908–916. [Google Scholar]
- Wang, Y.; Nomikos, G.G.; Karim, A.; Munsaka, M.; Serenko, M.; Liosatos, M.; Harris, S. Effect of Vortioxetine on Cardiac Repolarization in Healthy Adult Male Subjects: Results of a Thorough QT/QTc Study. Clin. Pharmacol. Drug Dev. 2013, 2, 298–309. [Google Scholar] [CrossRef]
- Rochester, M.P.; Kane, A.M.; Linnebur, S.A.; Fixen, D.R. Evaluating the risk of QTc prolongation associated with antidepressant use in older adults: A review of the evidence. Ther. Adv. Drug Saf. 2018, 9, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.N.; Yang, W.H.; Yeh, C.C.; Huang, H.C. The inhibition by di(2-ethylhexyl)-phthalate of erg-mediated K+ current in pituitary tumor (GH3) cells. Arch. Toxicol. 2012, 86, 713–723. [Google Scholar] [CrossRef]
- Hsu, H.T.; Lo, Y.C.; Wu, S.N. Characterization of Convergent Suppression by UCL-2077 (3-(Triphenylmethylaminomethyl)pyridine), Known to Inhibit Slow Afterhyperpolarization, of erg-Mediated Potassium Currents and Intermediate-Conductance Calcium-Activated Potassium Channels. Int. J. Mol. Sci. 2020, 21, 1441. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Højer, A.M.; Areberg, J.; Nomikos, G. Vortioxetine: Clinical Pharmacokinetics and Drug Interactions. Clin. Pharmacokinet. 2018, 57, 673–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.; Dart, C. Pharmacokinetics and Safety of Vortioxetine in the Chinese Population. Adv. Ther. 2019, 36, 3134–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, E.S.; Dart, C. Neuronal and Cardiovascular Potassium Channels as Therapeutic Drug Targets: Promise and Pitfalls. J. Biomol. Screen. 2015, 20, 1055–1073. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, H.-T.; Wang, J.C.-F.; Wu, S.-N. Inhibitory Effectiveness in Delayed-Rectifier Potassium Current Caused by Vortioxetine, Known to Be a Novel Antidepressant. Biomedicines 2022, 10, 1318. https://doi.org/10.3390/biomedicines10061318
Hsiao H-T, Wang JC-F, Wu S-N. Inhibitory Effectiveness in Delayed-Rectifier Potassium Current Caused by Vortioxetine, Known to Be a Novel Antidepressant. Biomedicines. 2022; 10(6):1318. https://doi.org/10.3390/biomedicines10061318
Chicago/Turabian StyleHsiao, Hung-Tsung, Jeffrey Chi-Fei Wang, and Sheng-Nan Wu. 2022. "Inhibitory Effectiveness in Delayed-Rectifier Potassium Current Caused by Vortioxetine, Known to Be a Novel Antidepressant" Biomedicines 10, no. 6: 1318. https://doi.org/10.3390/biomedicines10061318
APA StyleHsiao, H.-T., Wang, J. C.-F., & Wu, S.-N. (2022). Inhibitory Effectiveness in Delayed-Rectifier Potassium Current Caused by Vortioxetine, Known to Be a Novel Antidepressant. Biomedicines, 10(6), 1318. https://doi.org/10.3390/biomedicines10061318