
The Vascular Microenvironment in Glioblastoma: A Comprehensive Review

 , , and
, , and
Abstract
1. Introduction
2. The Perivascular Niche
3. Microvascular Patterns
3.1. Microvascular Sprouting
3.2. Vascular Cluster
3.3. Vascular Garland
3.4. Glomeruloid Vascular Proliferation
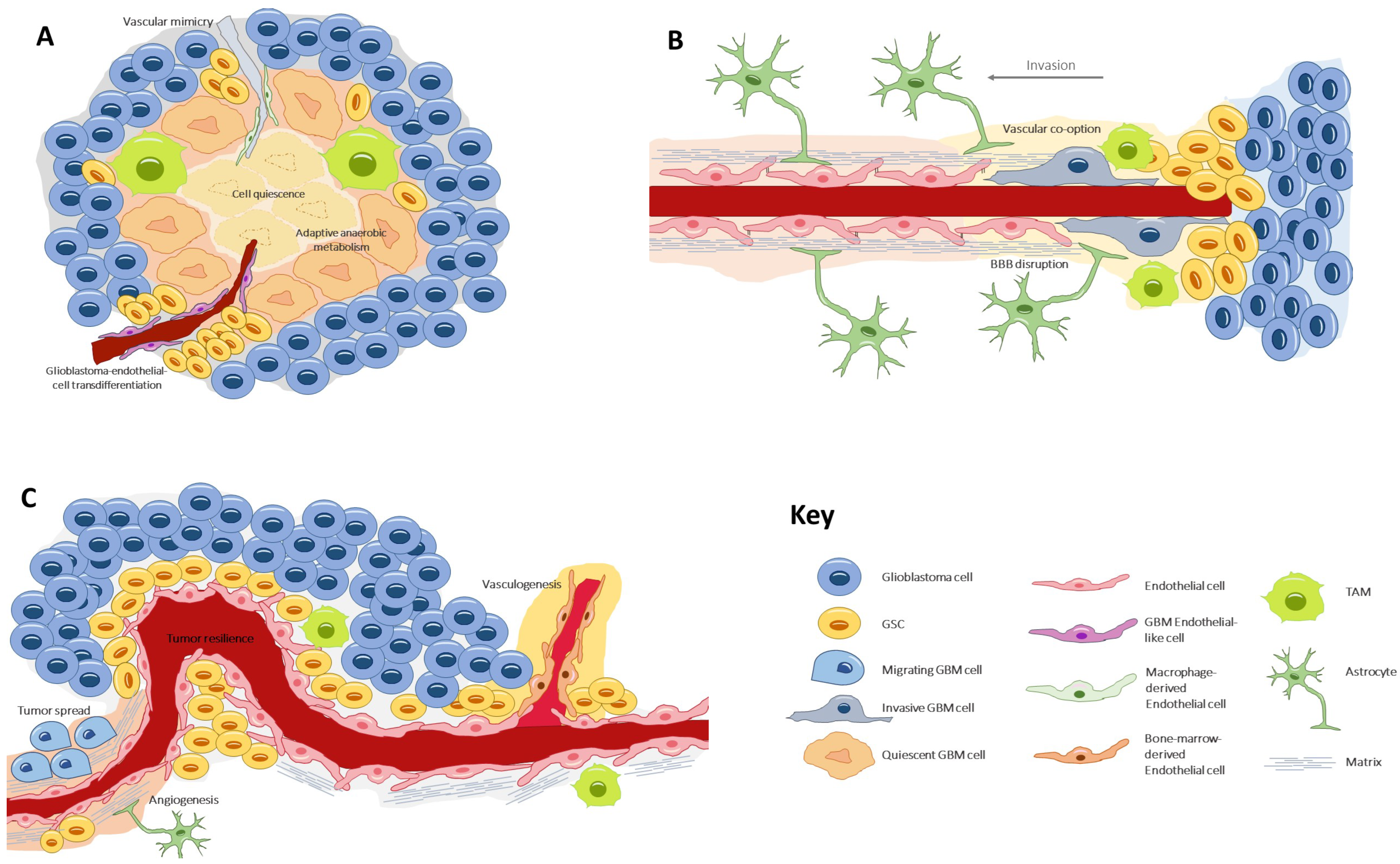
4. Vascular Generation and Related Processes
4.1. Angiogenesis
4.2. Vasculogenesis
4.3. Vascular Mimicry
4.4. Glioblastoma-Endothelial-Cell Transdifferentiation
4.5. Vascular Co-Option (or Angiotropism or Perivascular Migration)
5. Endothelial–Tumor Cell Crosstalk
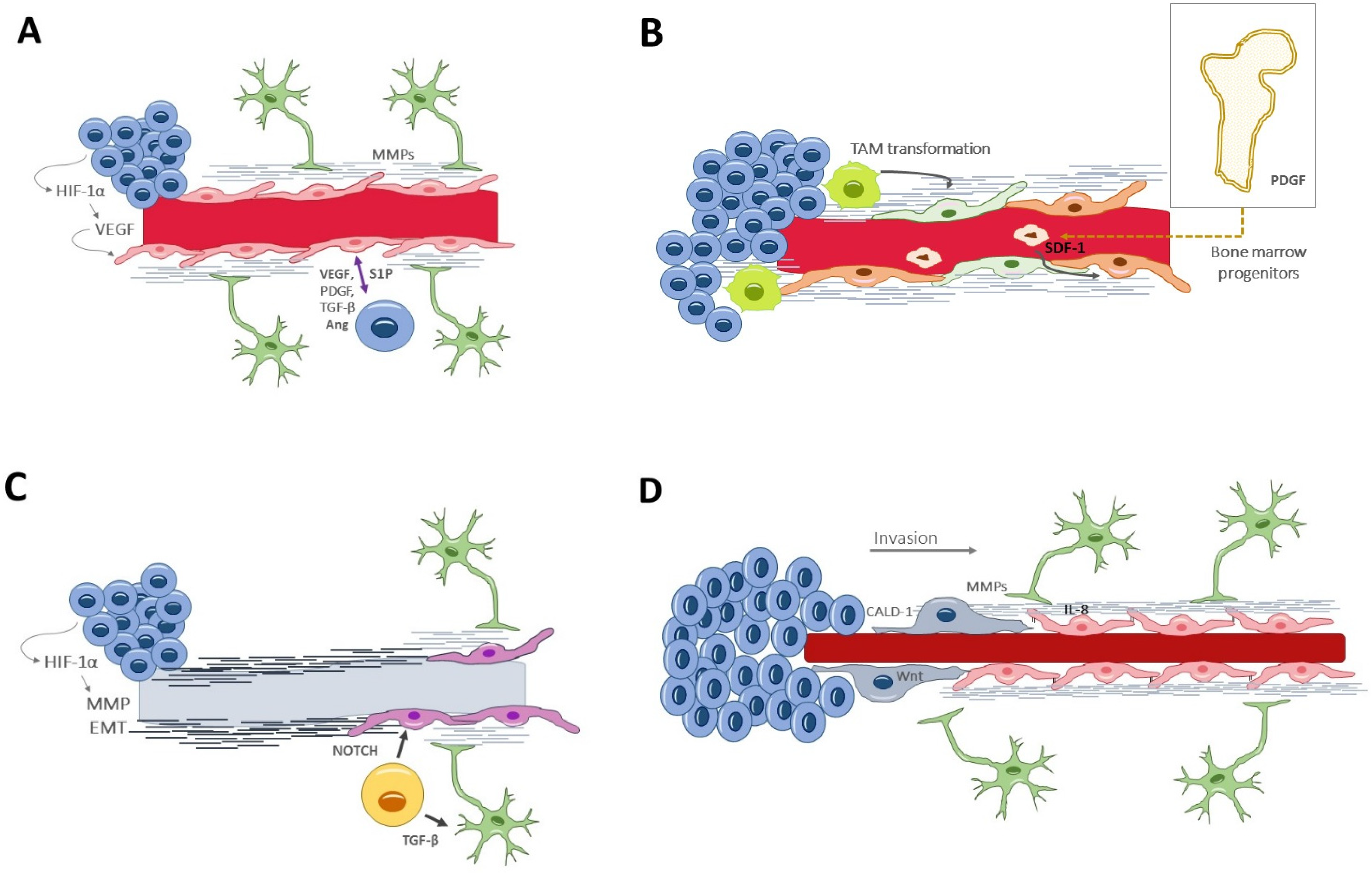
6. Signaling Pathways Involved in Neovascularization
- (i)
- Vascular endothelial growth factor (VEGF) [81] is the paramount signaling molecule in angiogenesis, particularly the VEGF-A isoform. VEGF secretion is induced under hypoxic conditions by HIF-1α. VEGF can act alone or in synergic collaboration with other proangiogenic factors, such as fibroblast growth factor 2 (FGF-2) and platelet-derived growth factor BB (PDGF-BB) [82]. VEGF stimulates endothelial cells to breakdown the extracellular matrix, to proliferate and migrate, and it also increases the vascular permeability.
- (ii)
- Fibroblast growth factor (FGF) [83], in the early stages of angiogenesis it mediates proteolysis of the matrix and endothelial migration; in the later stages, it favors capillary conformation and basal membrane synthesis.
- (iii)
- Platelet-derived growth factor (PDGF) acts locally, by stimulating endothelial proliferation, and distally, by inducing extramedullary hematopoiesis.
- (iv)
- (v)
- Hepatocyte growth factor/scatter factor (HGF/SF) [85] directly promotes endothelial proliferation, migration, and organization into capillary tubes. Indirectly, HGF/SF promotes angiogenesis by upregulating VEGF and suppressing thrombospondin 1 (TSP-1).
- (vi)
- Angiopoietin 1 (Ang-1) [86] induces vessel formation and stabilization by interacting with the endothelial cell kinase 2 (Tie-2) receptor at the tunica interna.
- (vii)
- Angiopoietin 2 (Ang-2) [51,87] also acts on the Tie-2 receptor, but with opposing effects. Ang-2 favors VEGF-mediated endothelial proliferation and migration. Paradoxically, in the absence of VEGF, Ang-2 is an antagonist of Ang-1 and thus promotes tumor vessel regression and leakiness.Initially, during the tumor development, co-opted vessels express both Ang-2 and Ang-1, which favors pericyte recruitment and vessel integrity. However, as the tumor progresses, the upregulation of Ang-2 leads to vessel destabilization and disruption. In the presence of Ang-2, VEGF promotes endothelial proliferation and migration, resulting in angiogenesis. The final stages of angiogenesis involve capillary morphogenesis, mediated largely by integrins [88], and the endothelial cells secrete PDGF, which recruits pericytes to the new vessel.
- (viii)
- Angiopoietin 4 (Ang-4) [89] is a strong proangiogenic that exerts its function by upregulating the transcription of VEGF.
- (ix)
- Matrix metalloproteinases (MMPs) [90] degrade the extracellular matrix but also facilitate the proliferation and migration of pericytes for vessel stabilization.
- (x)
- Stromal-derived factor-1α (SDF-1α) [91] is a hypoxia-induced factor that recruits bone marrow-derived CXCR4+ endothelial and pericyte progenitors to form new vessels. Both the SDF-1a/CXCR4 and the ANG-2/TIE-2 pathways seem relevant in promoting vasculogenesis.
- (xi)
- Sphingosine-1-phosphate (S1P) [92] is a pleiotropic angiogenic factor, regulating both the early and the late stages of angiogenesis. It drives endothelial proliferation, migration and tubular vessel conformation. It is secreted by endothelial cells and is increased by the presence of glioma cells.
- (xii)
- Caldesmon 1 (CALD1) [93] modulates actin filaments of the cytoskeleton, controlling cell morphology and movement. CALD1 expression is augmented in both neoplastic and vascular cells, and it confers worse clinical outcomes.
- (xiii)
- Annexin A2 (AnxA2) [94] is a ubiquitous membrane-binding protein whose function varies according to its location. During inflammation, AnxA2 limits vascular permeability by maintaining cadherin endothelial junctions; it modulates the inflammasome, and it enables angiogenesis and tissue healing. However, sustained inflammation might lead to AnxA2 upregulation and excessive pathological angiogenesis [95].
- (xiv)
- Chemokines and pro-inflammatory factors are also overexpressed in the tumor microenvironment and have been shown to play an essential role in the neovascularization processes. Importantly, these molecules act as interconnectors between the vascular and immune tumor responses [96]. Among chemokines, IL-8 is a paradigmatic example, acting over two cell-surface G-protein-coupled receptors (CXCR1 and CXCR2). Endothelial cells secrete IL-8 and other chemotactic factors to activate GBM invasion and vascular mimicry [52,97]. Chemokines have also been implicated in the tumor resistance to anti-angiogenic drugs. For instance, Vatalanib, a small molecule protein kinase inhibitor, collaterally increases the expression of various chemokines and their receptors at the invasive front of GBM; the subsequent increase in myeloid cells and activated endothelial cells could be responsible for the resistance to the anti-angiogenic effect of Vatalanib [98].
7. Perivascular Satellitosis
8. Sustained Vascularization and Immunomodulation
9. Preclinical Models
9.1. BBB Spheroid Models
9.2. Vascularized Organoids
9.3. Perfusable 3D Models (Cancers-on-Chip)
9.4. Matrix-Based Models
9.5. Bioprinted Models
9.6. The Chick Embryo Chorioallantoic Membrane (CAM)
9.7. Major Challenges in Model Development
10. Anti-Angiogenic Therapeutic Approaches
10.1. Bevacizumab: State of the Art and Reasons for Failure
10.2. Novel Anti-Angiogenic Therapies
10.3. Vascular Normalization and Immune Checkpoint Inhibitors
10.4. Clinical Trials of Anti-Angiogenic Drugs
11. Future Directions
12. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Algeiet, A.; Fisher, B.; Belander, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, M.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of bevacizumab in the treatment of glioblastoma. J. Neurooncol. 2017, 133, 455–467. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro-Oncology 2010, 12, 233–242. [Google Scholar] [CrossRef]
- Xue, W.; Du, X.; Wu, H.; Liu, H.; Xie, T.; Tong, H.; Cheng, X.; Guo, Y.; Zhang, W. Aberrant glioblastoma neovascularization patterns and their correlation with DCE-MRI-derived parameters following temozolomide and bevacizumab treatment. Sci. Rep. 2017, 7, 1389. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, Y.J.; Lee, S.H.; Kim, T.M.; Choi, S.H.; Kim, D.W.; Park, C.K.; Kim, I.H.; Kim, J.H.; Kim, E.; et al. Post-bevacizumab clinical outcomes and the impact of early discontinuation of bevacizumab in patients with recurrent malignant glioma. Cancer Res. Treat. 2017, 49, 129–140. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma stem cells: Driving resilience through chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- Ishii, A.; Kimura, T.; Sadahiro, H.; Kawano, H.; Takubo, K.; Suzuki, M.; Ikeda, E. Histological Characterization of the Tumorigenic "Peri-Necrotic Niche" Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma. PLoS ONE 2016, 11, e0147366. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-mediated mechanisms associated with antiangiogenic treatment resistance in glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef]
- Rosińska, S.; Gavard, J. Tumor vessels fuel the fire in glioblastoma. Int. J. Mol. Sci. 2021, 22, 6514. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Holland, E.C. The perivascular niche microenvironment in brain tumor progression. Cell Cycle 2010, 9, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma stem cell niches in human glioblastoma are periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef]
- Hira, V.V.; Ploegmakers, K.J.; Grevers, F.; Verbovsek, U.; Silvestre-Roig, C.; Aronica, E.; Tigchelaar, W.; Turnšek, T.L.; Molenaar, R.J.; Van Noorden, C.J. CD133+ and nestin+ glioma stem-like cells reside around CD31+arterioles in niches that express SDF-1alpha, CXCR4, osteopontin and cathepsin K. J. Histochem. Cytochem. 2015, 63, 481–493. [Google Scholar] [CrossRef]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-tumoral metabolic zonation and resultant phenotypic diversification are dictated by blood vessel proximity. Cell Metab. 2019, 30, 201–211. [Google Scholar] [CrossRef]
- Allen, E.; Miéville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.W.; Hanahan, D. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR Signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and niche concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef]
- Birner, P.; Piribauer, M.; Fischer, I.; Gatterbauer, B.; Marosi, C.; Ambros, P.F.; Ambros, I.M.; Bredel, M.; Oberhuber, G.; Rössler, K.; et al. Vascular patterns in glioblastoma influence clinical outcome and associate with variable expression of angiogenic proteins: Evidence for distinct angiogenic subtypes. Brain Pathol. 2003, 13, 133–143. [Google Scholar] [CrossRef]
- Chen, J.; Mao, S.; Li, H.; Zheng, M.; Yi, L.; Lin, J.M.; Lin, Z.X. The pathological structure of the perivascular niche in different microvascular patterns of glioblastoma. PLoS ONE 2017, 12, e0182183. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.C.; Cantu Gutierrez, M.; Lozzi, B.; Huang-Hobbs, E.; Turner, W.D.; Tepe, B.; Zhang, Y.; Herman, A.M.; Rao, G.; Creighton, C.J.; et al. Identification of diverse tumor endothelial cell populations in malignant glioma. Neuro-Oncology 2021, 23, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.; Chapouly, C.; Andrique, L.; Bikfalvi, A.; Daubon, T. The normal and brain tumor vasculature: Morphological and functional characteristics and therapeutic targeting. Front. Physiol. 2021, 12, 622615. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Cauwenberghs, S.; Carmeliet, P. Vessel abnormalization: Another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr. Opin. Genet. Dev. 2011, 21, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Brem, S. The role of vascular proliferation in the growth of brain tumors. Clin. Neurosurg. 1976, 23, 440–453. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Sawamiphak, S.; Seidel, S.; Essmann, C.L.; Wilkinson, G.A.; Pitulescu, M.E.; Acker, T.; Acker-Palmer, A. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 2010, 465, 487–491. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor development and angiogenesis in adult brain tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [PubMed]
- Döme, B.; Hendrix, M.J.; Paku, S.; Tóvári, J.; Tímár, J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; de Palma, M.; Naldini, L. Tie2-expressing monocytes and tumor angiogenesis: Regulation by hypoxia and angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef] [PubMed]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef]
- Liu, X.M.; Zhang, Q.P.; Mu, Y.G.; Zhang, X.H.; Sai, K.; Pang, J.C.; Ng, H.K.; Chen, Z.P. Clinical significance of vasculogenic mimicry in human gliomas. J. Neurooncol. 2011, 105, 173–179. [Google Scholar] [CrossRef]
- Tso, C.L.; Shintaku, P.; Chen, J.; Liu, Q.; Liu, J.; Chen, Z.; Yoshimoto, K.; Mischel, P.S.; Cloughesy, T.F.; Liau, L.M.; et al. Primary glioblastomas express mesenchymal stem-like properties. Mol. Cancer Res. 2006, 4, 607–619. [Google Scholar] [CrossRef]
- Cui, C.; Chen, X.; Liu, Y.; Cao, B.; Xing, Y.; Liu, C.; Yang, F.; Li, Y.; Yang, T.; Hua, L.; et al. β1,4-galactosyltransferase V activates notch1 signaling in glioma stem-like cells and promotes their transdifferentiation into endothelial cells. J. Biol. Chem. 2018, 293, 2219–2230. [Google Scholar] [CrossRef]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, P.; Valiente, M. Vascular co-option in brain metastasis. Angiogenesis 2020, 23, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef]
- Kulahin, N.; Li, S.; Hinsby, A.; Kiselyov, V.; Berezin, V.; Bock, E. Fibronectin type III (FN3) modules of the neuronal cell adhesion molecule L1 interact directly with the fibroblast growth factor (FGF) receptor. Mol. Cell. Neurosci. 2008, 37, 528–536. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef]
- Pezzella, F.; Gatter, K. Non-angiogenic tumours unveil a new chapter in cancer biology. J. Pathol. 2015, 235, 381–383. [Google Scholar] [CrossRef]
- Șovrea, A.S.; Boșca, B.; Melincovici, C.S.; Constantin, A.M.; Crintea, A.; Mărginean, M.; Dronca, E.; Jianu, M.E.; Suflețel, R.; Gonciar, D.; et al. Multiple faces of the glioblastoma microenvironment. Int. J. Mol. Sci. 2022, 23, 595. [Google Scholar] [CrossRef]
- Yuen, T.J.; Silbereis, J.C.; Griveau, A.; Chang, S.M.; Daneman, R.; Fancy, S.P.J.; Zahed, H.; Maltepe, E.; Rowitch, D.H. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 2014, 158, 383–396. [Google Scholar] [CrossRef]
- Tsai, H.H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 2016, 351, 379–384. [Google Scholar] [CrossRef]
- Umans, R.A.; Pollock, C.; Mills, W.A., 3rd; Clark, K.C.; Pan, Y.A.; Sontheimer, H. Using zebrafish to elucidate glial-vascular interactions during CNS development. Front. Cell Dev. Biol. 2021, 9, 654338. [Google Scholar] [CrossRef] [PubMed]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K. A glial signature and Wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell 2018, 33, 874–889. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef]
- Mei, X.; Chen, Y.S.; Chen, F.R.; Xi, S.Y.; Chen, Z.P. Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro-Oncology 2017, 19, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauvé, C.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic activation of WNT5A drives glioblastoma stem cell differentiation and invasive growth. Cell 2016, 167, 1281–1295.e18. [Google Scholar] [CrossRef] [PubMed]
- Donier, E.; Gomez-Sanchez, J.A.; Grijota-Martinez, C.; Lakomá, J.; Baars, S.; Garcia-Alonso, L.; Cabedo, H. L1CAM binds ErbB receptors through Ig-like domains coupling cell adhesion and neuregulin signalling. PLoS ONE 2012, 7, e40674. [Google Scholar] [CrossRef]
- Burgett, M.E.; Lathia, J.D.; Roth, P.; Nowacki, A.S.; Galileo, D.S.; Pugacheva, E.; Huang, P.; Vasanji, A.; Li, M.; Byzova, T.; et al. Direct contact with perivascular tumor cells enhances integrin αvβ3 signaling and migration of endothelial cells. Oncotarget 2016, 7, 43852–43867. [Google Scholar] [CrossRef]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; de Carvalho, A.C.; Mikkelsen, T.; Castro, M.G. Lowenstein PR CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef]
- Lindberg, O.R.; McKinney, A.; Engler, J.R.; Koshkakaryan, G.; Gong, H.; Robinson, A.E.; Ewald, A.J.; Huillard, E.; David James, C.; Molinaro, A.M.; et al. GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget 2016, 7, 79101–79116. [Google Scholar] [CrossRef][Green Version]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Iser, I.C.; Ceschini, S.M.; Onzi, G.R.; Bertoni, A.P.; Lenz, G.; Wink, M.R. Conditioned medium from adipose-derived stem cells (ADSCs) promotes epithelial-to-mesenchymal-like transition (EMT-like) in glioma cells in vitro. Mol. Neurobiol. 2016, 53, 7184–7199. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling nanotubes and tumor microtubes in cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef]
- Sharma, M.; Schilero, C.; Peereboom, D.M.; Hobbs, B.P.; Elson, P.; Stevens, G.H.J.; McCrae, K.; Nixon, A.B.; Ahluwalia, M.S. Phase II study of dovitinib in recurrent glioblastoma. J. Neurooncol. 2019, 144, 359–368. [Google Scholar] [CrossRef]
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib alone or in combination with bevacizumab in patients with recurrent glioblastoma: Phase I/II clinical trial data. Neurooncol. Adv. 2021, 3, vdab142. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Brenner, A.; de Groot, J.F.; Butowski, N.A.; Zach, L.; Campian, J.L.; Ellingson, B.M.; Freedman, L.S.; Cohen, Y.C.; Lowenton-Spier, N.; et al. A randomized controlled phase III study of VB-111 combined with bevacizumab vs bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro-Oncology 2020, 22, 705–717. [Google Scholar] [CrossRef]
- Brenner, A.J.; Floyd, J.; Fichtel, L.; Michalek, J.; Kanakia, K.P.; Huang, S.; Reardon, D.; Wen, P.Y.; Lee, E.Q. Phase 2 trial of hypoxia activated evofosfamide (TH302) for treatment of recurrent bevacizumab-refractory glioblastoma. Sci. Rep. 2021, 11, 2306. [Google Scholar] [CrossRef]
- Kuang, R.; Jahangiri, A.; Mascharak, S.; Nguyen, A.; Chandra, A.; Flanigan, P.M.; Yagnik, G.; Wagner, J.R.; De Lay, M.; Carrera, D.; et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2017, 2, e88815. [Google Scholar] [CrossRef]
- A Phase II/III Study of High-Dose, Intermittent Sunitinib in Patients with Recurrent Glioblastoma Multiforme (STELLAR). Available online: https://clinicaltrials.gov/ct2/show/NCT03025893?term=antiangiogenic&type=Intr&cond=Glioblastoma&phase=123&draw=2&rank=7 (accessed on 24 April 2022).
- Combined Treatment of Camrelizumab and Bevacizumab for Adult Patients with Recurrent Glioblastoma (GBM). Available online: https://clinicaltrials.gov/ct2/show/study/NCT04952571?term=antiangiogenic&type=Intr&cond=Glioblastoma&phase=123&draw=3&rank=17 (accessed on 24 April 2022).
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef]
- Cao, R.; Brakenhielm, E.; Pawliuk, R.; Wariaro, D.; Post, M.J.; Wahlberg, E.; Leboulch, P.; Cao, Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat. Med. 2003, 9, 604–613. [Google Scholar] [CrossRef]
- Kanda, S.; Landgren, E.; Ljungstrom, M.; Claesson-Welsh, L. Fibroblast growth factor receptor 1-induced differentiation of endothelial cell line established from ts, A58 large T transgenic mice. Cell Growth Differ. 1996, 7, 383–395. [Google Scholar] [PubMed]
- Platten, M.; Wick, W.; Wild-Bode, C.; Aulwurm, S.; Dichgans, J.; Weller, M. Transforming growth factors beta(1) (TGF-beta(1)) and TGF-beta(2) promote glioma cell migration via up-regulation of alpha(V)beta(3) integrin expression. Biochem. Biophys. Res. Commun. 2000, 268, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Gherardi, E.; Sellers, L.A.; Wood, J.M.; Sasisekharan, R.; Fan, T.P. Hepatocyte growth factor/scatter factor can induce angiogenesis independently of vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; McClain, J.; Thurston, G.; McDonald, D.M.; Zhou, H.; Oldmixon, E.H.; Sato, T.N.; Yancopoulos, G.D. Increased vascularization in mice overexpressing angiopoietin-1. Science 1998, 282, 468–471. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Brunckhorst, M.K.; Wang, H.; Lu, R.; Yu, Q. Angiopoietin-4 promotes glioblastoma progression by enhancing tumor cell viability and angiogenesis. Cancer Res. 2010, 70, 7283–7293. [Google Scholar] [CrossRef]
- Chantrain, C.F.; Henriet, P.; Jodele, S.; Emonard, H.; Feron, O.; Courtoy, P.J.; DeClerck, Y.A.; Marbaix, E. Mechanisms of pericyte recruitment in tumour angiogenesis: A new role formetalloproteinases. Eur. J. Cancer 2006, 42, 310–318. [Google Scholar] [CrossRef]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: An emerging sensitizer for anticancer therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef]
- Abdel Hadi, L.; Anelli, V.; Guarnaccia, L.; Navone, S.; Beretta, M.; Moccia, F. A bidirectional crosstalk between glioblastoma and brain endothelial cells potentiates the angiogenic and proliferative signaling of sphingosine-1-phosphate in the glioblastoma microenvironment. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1179–1192. [Google Scholar] [CrossRef]
- Cheng, Q.; Tang, A.; Wang, Z.; Fang, N.; Zhang, Z.; Zhang, L.; Li, C.; Zeng, Y. CALD1 modulates gliomas progression via facilitating tumor angiogenesis. Cancers 2021, 13, 2705. [Google Scholar] [CrossRef] [PubMed]
- Dallacasagrande, V.; Hajjar, K.A. Annexin A2 in inflammation and host defense. Cells 2020, 9, 1499. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hajjar, K.A. The annexin A2 system and angiogenesis. Biol. Chem. 2016, 397, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappalà, A.; Cammarata, F.P.; et al. The hallmarks of glioblastoma: Heterogeneity, intercellular crosstalk and molecular signature of invasiveness and progression. Biomedicines 2022, 10, 806. [Google Scholar] [CrossRef]
- Piantino, M.; Figarol, A.; Matsusaki, M. Three-dimensional in vitro models of healthy and tumor brain microvasculature for drug and toxicity screening. Front. Toxicol. 2021, 3, 656254. [Google Scholar] [CrossRef]
- Arbab, A.S.; Rashid, M.H.; Angara, K.; Borin, T.F.; Lin, P.C.; Jain, M.; Achyut, B.R. Major challenges and potential microenvironment-targeted therapies in glioblastoma. Int. J. Mol. Sci. 2017, 18, 2732. [Google Scholar] [CrossRef]
- De Gooijer, M.C.; Guillén Navarro, M.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An experimenter’s guide to glioblastoma invasion pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef]
- Kosty, J.; Lu, F.; Kupp, R.; Mehta, S.; Lu, Q.R. Harnessing OLIG2 function in tumorigenicity and plasticity to target malignant gliomas. Cell Cycle 2017, 16, 1654–1660. [Google Scholar] [CrossRef]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Forstreuter, F.; Lucius, R.; Mentlein, R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J. Neuroimmunol. 2002, 132, 93–98. [Google Scholar] [CrossRef]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The cornerstone of glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Chen, Y.Y.; Muthana, M.; Welford, A.F.; Tal, A.O.; Scholz, A.; Plate, K.H.; Reiss, Y.; Murdoch, C.; De Palma, M.; et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J. Immunol. 2011, 186, 4183–4190. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune evasion strategies of glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Urich, E.; Patsch, C.; Aigner, S.; Graf, M.; Iacone, R.; Freskgård, P.O. Multicellular self-assembled spheroidal model of the blood brain barrier. Sci. Rep. 2013, 3, 1500. [Google Scholar] [CrossRef]
- Nashimoto, Y.; Okada, R.; Hanada, S.; Arima, Y.; Nishiyama, K.; Miura, T.; Yokokawa, R. Vascularized cancer on a chip: The effect of perfusion on growth and drug delivery of tumor spheroid. Biomaterials 2020, 229, 119547. [Google Scholar] [CrossRef]
- Cho, C.F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Gómez-Oliva, R.; Domínguez-García, S.; Carrascal, L.; Abalos-Martínez, J.; Pardillo-Díaz, R.; Verástegui, C.; Castro, C.; Nunez-Abades, P.; Geribaldi-Doldán, N. Evolution of experimental models in the study of glioblastoma: Toward finding efficient treatments. Front. Oncol. 2021, 10, 614295. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Zhang, C.; Jin, M.; Zhao, J.; Chen, J.; Jin, W. Organoid models of glioblastoma: Advances, applications and challenges. Am. J. Cancer Res. 2020, 10, 2242–2257. [Google Scholar]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Yamaguchi, Y.; Usui, S.; Nashimoto, Y.; Hanada, S.; Kiyokawa, E.; Uemura, A.; Yokokawa, R.; Nishiyama, K.; Miura, T. A new perfusion culture method with a self-organized capillary network. PLoS ONE 2020, 15, e0240552. [Google Scholar] [CrossRef] [PubMed]
- Cucullo, L.; Hossain, M.; Tierney, W.; Janigro, D. A new dynamic in vitro modular capillaries-venules modular system: Cerebrovascular physiology in a box. BMC Neurosci. 2013, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.T.; Harley, B.A. The influence of hyaluronic acid and glioblastoma cell coculture on the formation of endothelial cell networks in gelatin hydrogels. Adv. Healthc. Mater. 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Xie, Q.; Gimple, R.C.; Zhong, Z.; Tam, T.; Tian, J.; Kidwell, R.L.; Wu, Q.; Prager, B.C.; Qiu, Z.; et al. Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 2020, 30, 833–853. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kim, S.; Chen, Z.; Shin, H.K.; Lee, S.Y.; Moon, H.E.; Paek, S.H.; Park, S. 3D bioprinted vascularized tumour for drug testing. Int. J. Mol. Sci. 2020, 21, 2993. [Google Scholar] [CrossRef]
- Yi, H.G.; Jeong, Y.H.; Kim, Y.; Choi, Y.J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane as an experimental model to study in vivo angiogenesis in glioblastoma multiforme. Brain Res. Bull. 2022, 182, 26–29. [Google Scholar] [CrossRef]
- Valiulytė, I.; Curkūnavičiūtė, R.; Ribokaitė, L.; Kazlauskas, A.; Vaitkevičiūtė, M.; Skauminas, K.; Valančiūtė, A. The anti-tumorigenic activity of Sema3C in the chick embryo chorioallantoic membrane model. Int. J. Mol. Sci. 2019, 20, 5672. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, S.; Harsh, G.; Recht, L. Bevacizumab improves quality of life in patients with recurrent glioblastoma. Chemother. Res. Pract. 2011, 2011, 602812. [Google Scholar] [CrossRef]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, E.; London, N.; Fuja, D.; Logie, J.; Tyrrell, J.A.; Kamoun, W.; Munn, L.L.; Jain, R.K. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS ONE 2009, 4, e5123. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Ramezani, S.; Vousooghi, N.; Joghataei, M.T.; Chabok, S.Y. The role of kinase signaling in resistance to bevacizumab therapy for glioblastoma multiforme. Cancer Biother. Radiopharm. 2019, 34, 345–354. [Google Scholar] [CrossRef]
- Wei, Q.; Singh, O.; Ekinci, C.; Gill, J.; Li, M.; Mamatjan, Y.; Karimi, S.; Bunda, S.; Mansouri, S.; Aldape, K.; et al. TNFα secreted by glioma associated macrophages promotes endothelial activation and resistance against anti-angiogenic therapy. Acta Neuropathol. Commun. 2021, 9, 67. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, H.; Uyar, R.; Hossain, J.A.; Miletic, H.; Tonn, J.C.; Glass, R.; Kälin, R.E. Comparing Tumor Cell Invasion and Myeloid Cell Composition in Compatible Primary and Relapsing Glioblastoma. Cancers 2021, 13, 3636. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1a induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Eckerich, C.; Zapf, S.; Fillbrandt, R.; Loges, S.; Westphal, M.; Lamszus, K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int. J. Cancer 2007, 121, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: Contrasting, overlapping and compensatory functions. Biochim. Biophys. Acta 2010, 1803, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Zhang, P.; Wen, P.Y.; Gerstner, E.R.; Reardon, D.A.; Aldape, K.D.; deGroot, J.F.; Pan, E.; Raizer, J.J.; Kim, L.J.; et al. NRG/RTOG 1122: A phase 2, double-blinded, placebo-controlled study of bevacizumab with and without trebananib in patients with recurrent glioblastoma or gliosarcoma. Cancer 2020, 126, 2821–2828. [Google Scholar] [CrossRef]
- Meaney, C.; Rhebergen, S.; Kohandel, M. In silico analysis of hypoxia activated prodrugs in combination with anti angiogenic therapy through nanocell delivery. PLoS Comput. Biol. 2020, 16, e1007926. [Google Scholar] [CrossRef]
- Guarnaccia, L.; Marfia, G.; Masseroli, M.M.; Navone, S.E.; Balsamo, M.; Caroli, M.; Valtorta, S.; Moresco, R.M.; Campanella, R.; Garzia, E.; et al. Frontiers in anti-cancer drug discovery: Challenges and perspectives of metformin as anti-angiogenic add-on therapy in glioblastoma. Cancers 2021, 14, 112. [Google Scholar] [CrossRef]
- Ho, R.L.Y.; Ho, I.A.W. Recent advances in glioma therapy: Combining vascular normalization and immune checkpoint blockade. Cancers 2021, 13, 3686. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- He, B.; Jabouille, A.; Steri, V.; Johansson-Percival, A.; Michael, I.P.; Kotamraju, V.R.; Junckerstorff, R.; Nowak, A.K.; Hamzah, J.; Lee, G.; et al. Vascular targeting of LIGHT normalizes blood vessels in primary brain cancer and induces intratumoural high endothelial venules. J. Pathol. 2018, 245, 209–221. [Google Scholar] [CrossRef]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bähr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor vessel normalization, immunostimulatory reprogramming, and improved survival in glioblastoma with combined inhibition of PD-1, angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019, 7, 1910–1927. [Google Scholar] [CrossRef]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial Reference | Intervention | Mechanism of Action | Patient Population | Design | Primary Endpoint | PFS | Conclusion |
|---|---|---|---|---|---|---|---|
| Sharma et al. 2019 [74] | Dovitinib | Tyrosine-kinase receptor inhibitor | Recurrent or Progressive GBM | Phase II, non-randomized, parallel | PFS 6 months | PFS-6 12% in anti-angiogenic naïve and 0% in anti-angiogenic exposed | Dovitinib was not efficacious in recurrent GBM |
| Bota et al. 2021 [75] | Marizomib | Panproteanoma inhibitor + monoclonal antibody anti-VEGF | Recurrent or Progressive GBM | Phase II, non-randomized, intra-patient dose escalation | PFS 6 months | PFS-6 29.8% OS 9.1 months | No benefit of the addition of marizomib to bevacizumab |
| Cloughesy et al. 2020 [76] | VB-111 + bevacizumab | Oncolytic virus + anti-VEGF | Recurrent GBM | Phase III | OS | OS 6.8 months | No benefit and higher rates of adverse events |
| Brenner et al. 2021 [77] | Evofosfamide + bevacizumab | hypoxia activated pro-drug + anti-VEGF | Recurrent GBM | Phase II single-arm | Safety | PFS-4 31% OS 4.6 months | Deserves further investigation |
| Lee et al. 2021 [78] | Trebananib + bevacizumab | Sequester Ang1/Ang2 + anti-VEGF | Recurrent GBM | Phase II randomized | PFS 6 months | PFS-6 22.6% | Detrimental (lower PFS than bevacizumab alone) |
| STELLAR NCT03025893 [79] | Sunitinib | Tyrosine-kinase receptor inhibitor | Recurrent GBM | Phase II/III randomized, against lomustine | PFS | Ongoing | - |
| NCT04952571 [80] | Camrelizumab + Bevacizumab | Anti-PD1 + anti-VEGF monoclonal antibodies | Recurrent GBM | Phase II non-randomized, parallel | PFS | Ongoing | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosteiro, A.; Pedrosa, L.; Ferrés, A.; Diao, D.; Sierra, À.; González, J.J. The Vascular Microenvironment in Glioblastoma: A Comprehensive Review. Biomedicines 2022, 10, 1285. https://doi.org/10.3390/biomedicines10061285
Mosteiro A, Pedrosa L, Ferrés A, Diao D, Sierra À, González JJ. The Vascular Microenvironment in Glioblastoma: A Comprehensive Review. Biomedicines. 2022; 10(6):1285. https://doi.org/10.3390/biomedicines10061285
Chicago/Turabian StyleMosteiro, Alejandra, Leire Pedrosa, Abel Ferrés, Diouldé Diao, Àngels Sierra, and José Juan González. 2022. "The Vascular Microenvironment in Glioblastoma: A Comprehensive Review" Biomedicines 10, no. 6: 1285. https://doi.org/10.3390/biomedicines10061285
APA StyleMosteiro, A., Pedrosa, L., Ferrés, A., Diao, D., Sierra, À., & González, J. J. (2022). The Vascular Microenvironment in Glioblastoma: A Comprehensive Review. Biomedicines, 10(6), 1285. https://doi.org/10.3390/biomedicines10061285