
Signaling Pathways Regulating the Expression of the Glioblastoma Invasion Factor TENM1

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Migration and Invasion of GBM
3. ODZ1 and Its Role in Migration
3.1. ODZ1 Expression Is Induced by Microenvironmental Signals through a Stat3-Dependent Transcriptional Pathway
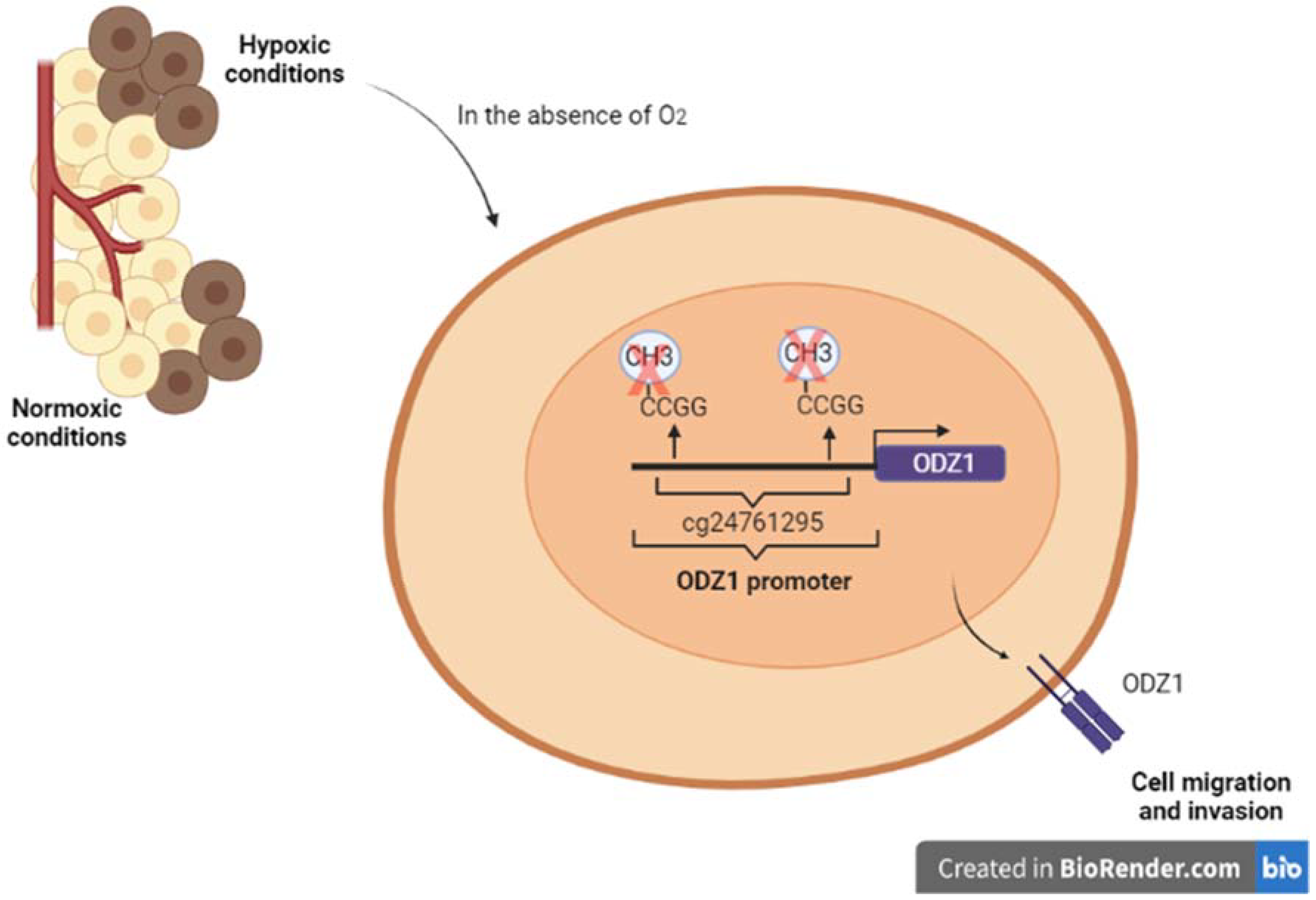
3.2. ODZ1 Expression Is Induced under Hypoxic Conditions
3.2.1. HIF Signaling
3.2.2. Epigenetic Regulation
4. ODZ1 as a Potential Therapeutic Target
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nerv-ous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 12 (Suppl 2), III1–III105. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Soffietti, R.; Baumert, B.G.; Bello, L.; Von Deimling, A.; Duffau, H.; Frénay, M.; Grisold, W.; Grant, R.; Graus, F.; Hoang-Xuan, K.; et al. Guidelines on management of low-grade gliomas: Report of an EFNS-EANO Task Force. Eur. J. Neurol. 2010, 17, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, S.; Yan, J.-L.; Torheim, T.; Boonzaier, N.R.; Sinha, R.; Matys, T.; Markowetz, F.; Price, S.J. Characterizing tumor invasiveness of glioblastoma using multiparametric magnetic resonance imaging. J. Neurosurg. 2020, 132, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Garcia, H.; Alvarado-Estrada, K.; Schiapparelli, P.; Quinones-Hinojosa, A.; Trifiletti, D.M. Engineering Three-Dimensional Tumor Models to Study Glioma Cancer Stem Cells and Tumor Microenvironment. Front. Cell. Neurosci. 2020, 14, 558381. [Google Scholar] [CrossRef]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef] [PubMed]
- Talamillo, A.; Grande, L.; Ruiz-Ontañon, P.; Velasquez, C.; Mollinedo, P.; Torices, S.; Sanchez-Gomez, P.; Aznar, A.; Esparis-Ogando, A.; Lopez-Lopez, C.; et al. ODZ1 allows glioblastoma to sustain invasiveness through a Myc-dependent transcriptional upregulation of RhoA. Oncogene 2016, 36, 1733–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, F.; Le Rhun, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Frank Winkler, F.; Kienast, Y.; Fuhrmann, M.; Von Baumgarten, L.; Burgold, S.; Mitteregger, G.; Kretzschmar, H.; Herms, J. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia 2009, 57, 1306–1315. [Google Scholar] [CrossRef]
- Jayaram, S.; Balakrishnan, L.; Singh, M.; Zabihi, A.; Ganesh, R.A.; Mangalaparthi, K.K.; Sonpatki, P.; Gupta, M.K.; Amaresha, C.B.; Prasad, K.; et al. Identification of a Novel Splice Variant of Neural Cell Adhesion Molecule in Glioblastoma Through Proteogenomics Analysis. OMICS A J. Integr. Biol. 2018, 22, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, C.; Mansouri, S.; Mora, C.; Nassiri, F.; Suppiah, S.; Martino, J.; Zadeh, G.; Fernández-Luna, J.L. Molecular and Clinical Insights into the Invasive Capacity of Glioblastoma Cells. J. Oncol. 2019, 2019, 1740763. [Google Scholar] [CrossRef] [Green Version]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, F.; Brotchi, J.; Kiss, R. Possible Future Issues in the Treatment of Glioblastomas: Special Emphasis on Cell Migration and the Resistance of Migrating Glioblastoma Cells to Apoptosis. J. Clin. Oncol. 2005, 23, 2411–2422. [Google Scholar] [CrossRef]
- So, J.S.; Kim, H.; Han, K.S. Mechanisms of Invasion in Glioblastoma: Extracellular Matrix, Ca2+ Signaling, and Glutamate. Front. Cell. Neurosci. 2021, 15, 663092. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2017, 33, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Wu, H.; Li, Y.; Shen, L.; Yu, R.; Yin, H.; Sun, T.; Sun, C.; Zhou, Y.; Du, Z. SALL4 suppresses PTEN expression to promote glioma cell proliferation via PI3K/AKT signaling pathway. J. Neuro-Oncol. 2017, 135, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, S.; Wides, R. Discovery of Teneurins. Front. Neurosci. 2019, 13, 230. [Google Scholar] [CrossRef] [PubMed]
- Peppino, G.; Ruiu, R.; Arigoni, M.; Riccardo, F.; Iacoviello, A.; Barutello, G.; Quaglino, E. Teneurins: Role in Cancer and Potential Role as Diagnostic Biomarkers and Targets for Therapy. Int. J. Mol. Sci. 2021, 22, 2321. [Google Scholar] [CrossRef]
- Ziegler, A.; Corvalán, A.; Roa, I.; Brañes, J.A.; Wollscheid, B. Teneurin protein family: An emerging role in human tumorigenesis and drug resistance. Cancer Lett. 2012, 326, 1–7. [Google Scholar] [CrossRef]
- Schöler, J.; Ferralli, J.; Thiry, S.; Chiquet-Ehrismann, R. The intracellular domain of teneurin-1 induces the activity of microphthal-mia-associated transcription factor (MITF) by binding to transcriptional repressor HINT1. J. Biol. Chem. 2015, 290, 8154–8165. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Rotzinger, S.; Al Chawaf, A.; Elias, C.F.; Baršytė-Lovejoy, D.; Qian, X.; Wang, N.-C.; De Cristofaro, A.; Belsham, D.; Bittencourt, J.C.; et al. Teneurin proteins possess a carboxy terminal sequence with neuromodulatory activity. Mol. Brain Res. 2005, 133, 253–265. [Google Scholar] [CrossRef]
- Lovejoy, D.A.; Al Chawaf, A.; Cadinouche, M.A. Teneurin C-terminal associated peptides: An enigmatic family of neuropeptides with structural similarity to the corticotropin-releasing factor and calcitonin families of peptides. Gen. Comp. Endocrinol. 2006, 148, 299–305. [Google Scholar] [CrossRef]
- Chand, D.; Song, L.; de Lannoy, L.; Barsyte-Lovejoy, D.; Ackloo, S.; Boutros, P.C.; Evans, K.; Belsham, D.D.; Lovejoy, D.A. C-Terminal region of teneurin-1 co-localizes with dystroglycan and modulates cytoskeletal organization through an ex-tracellular signal-regulated kinase-dependent stathmin- and filamin A-mediated mechanism in hippocampal cells. Neuroscience 2012, 219, 255–270. [Google Scholar] [CrossRef]
- Husić, M.; Barsyte-Lovejoy, D.; Lovejoy, D.A. Teneurin C-Terminal Associated Peptide (TCAP)-1 and Latrophilin Interaction in HEK293 Cells: Evidence for Modulation of Intercellular Adhesion. Front. Endocrinol. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-P.; Chen, M.-J.; Chien, M.-N.; Lin, C.-H.; Lee, J.-J.; Liu, C.-L. Overexpression of teneurin transmembrane protein 1 is a potential marker of disease progression in papillary thyroid carcinoma. Clin. Exp. Med. 2016, 17, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-H.; Liu, Z.-F.; Yang, B.-B.; Yu, B. MicroRNA-486 inhibits cell proliferation, invasion and migration via down-regulating the TENM1 expressions and affecting ERK and Akt signaling pathways and epithelial-to-mesenchymal transition in papillary thyroid carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8429–8439. [Google Scholar] [PubMed]
- Ruchong, P.; Haiping, T.; Xiang, W. A Five-Gene Prognostic Nomogram Predicting Disease-Free Survival of Differentiated Thyroid Cancer. Dis. Markers 2021, 2021, 5510780. [Google Scholar] [CrossRef]
- Wang, Y.; Song, B.; Zhu, L.; Zhang, X. Long non-coding RNA, LINC01614 as a potential biomarker for prognostic prediction in breast cancer. PeerJ 2019, 7, e7976. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad—Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Bartolini, S.; Brandes, A.A. Glioblastoma Microenvironment: From an Inviolable Defense to a Therapeutic Chance. Front. Oncol. 2022, 12, 852950. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Hu, C.; Lal, B.; Zhou, W.; Ma, Y.; Ying, M.; Prinos, P.; Quiñones-Hinojosa, A.; Lim, M.; Laterra, J.; et al. Reprogramming Transcription Factors Oct4 and Sox2 Induce a BRD-Dependent Immunosuppressive Transcriptome in GBM-Propagating Cells. Cancer Res. 2021, 81, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion—An inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, V.; Gutierrez, O.; Talamillo, A.; Velasquez, C.; Fernandez-Luna, J.L. Glioblastoma invasion factor ODZ1 is induced by microenvironmental signals through activation of a Stat3-dependent transcriptional pathway. Sci. Rep. 2021, 11, 16196. [Google Scholar] [CrossRef]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting Interleukin 6 Signaling Suppresses Glioma Stem Cell Survival and Tumor Growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [Green Version]
- Ouédraogo, Z.G.; Biau, J.; Kemeny, J.L.; Morel, L.; Verrelle, P.; Chautard, E. Role of STAT3 in Genesis and Progression of Human Malignant Gliomas. Mol. Neurobiol. 2017, 54, 5780–5797. [Google Scholar] [CrossRef]
- Tan, M.S.Y.; Sandanaraj, E.; Chong, Y.K.; Lim, S.W.; Koh, L.W.H.; Ng, W.H.; Tan, N.S.; Tan, P.; Ang, B.T.; Tang, C. A STAT3-based gene signature stratifies glioma patients for targeted therapy. Nat. Commun. 2019, 10, 3601. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yu, X.; Sun, L.; Zheng, Y.; Chen, L.; Xu, X.; Jin, J.; Lan, Q.; Chen, C.C.; Li, M. GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene 2020, 39, 5042–5055. [Google Scholar] [CrossRef]
- McFarland, B.C.; Hong, S.W.; Rajbhandari, R.; Twitty, G.B., Jr.; Gray, G.K.; Yu, H.; Benveniste, E.N.; Nozell, S.E. NF-κB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS ONE 2013, 8, e78728. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Kloss, J.M.; Tuncali, S.; Tran, N.L.; Loftus, J.C. TROY signals through JAK1-STAT3 to promote glioblastoma cell migration and resistance. Neoplasia 2020, 22, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Levesque, J.; Bousquet-Gagnon, N.; Béliveau, R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp. Cell Res. 2012, 318, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Jiao, H.; Teng, G.; Wang, W.; Zhang, R.; Wang, Y.; Qiao, L. Embelin reduces colitis-associated tumorigenesis through limiting IL-6/STAT3 signaling. Mol. Cancer Ther. 2014, 13, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Gatto, L.; Di Nunno, V.; Franceschi, E.; Brandes, A.A. Chimeric antigen receptor macrophage for glioblastoma immunotherapy: The way forward. Immunotherapy 2021, 13, 879–883. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; Dunnen, W.F.D.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α–ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ding, X.; Ye, H.; Wang, J.; Shao, J.; Huang, T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. NeuroReport 2018, 29, 1578–1585. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, L.; Gong, S.; Xiong, S.; Wang, J.; Zou, D.; Liao, B. HIF1α/HIF2α-Sox2/Klf4 promotes the malignant progression of glioblastoma via the EGFR-PI3K/AKT signalling pathway with positive feedback under hypoxia. Cell Death Dis. 2021, 12, 312. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Simon, M.C. Biology of hypoxia-inducible factor-2α in development and disease. Cell Death Differ. 2008, 15, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, E.; Yarchoan, R. Differential Gene Up-Regulation by Hypoxia-Inducible Factor-1α and Hypoxia-Inducible Factor-2α in HEK293T Cells. Cancer Res. 2005, 65, 3299–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Nusblat, L.M.; Tanna, S.; Roth, C.M. Gene silencing of HIF-2α disrupts glioblastoma stem cell phenotype. Cancer Drug Resist. 2020, 3, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Carcelén, M.; Velásquez, C.; Vidal, V.; Gutierrez, O.; Fernandez-Luna, J.L. HIF2α Upregulates the Migration Factor ODZ1 under Hypoxia in Glioblastoma Stem Cells. Int. J. Mol. Sci. 2022, 23, 741. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Q.; Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immuno-therapy. Cancers 2022, 14, 1176. [Google Scholar]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Aberrant Epigenetic Landscape in Cancer: How Cellular Identity Goes Awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Kim, H.; Kim, S.H.; Joe, E.H.; Jou, I. Epigenetic downregulation of STAT6 increases HIF-1α expression via mTOR/S6K/S6, leading to enhanced hypoxic viability of glioma cells. Acta Neuropathol. Commun. 2019, 7, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Hermans, E. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Velásquez, C.; Mansouri, S.; Gutiérrez, O.; Mamatjan, Y.; Mollinedo, P.; Karimi, S.; Singh, O.; Teran, N.; Martino, J.; Zadeh, G.; et al. Hypoxia Can Induce Migration of Glioblastoma Cells Through a Methylation-Dependent Control of ODZ1 Gene Expression. Front. Oncol. 2019, 9, 1036. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Useros, J.; Martin-Galan, M.; Florez-Cespedes, M.; Garcia-Foncillas, J. Epigenetics of Most Aggressive Solid Tumors: Path-ways, Targets and Treatments. Cancers 2021, 13, 3209. [Google Scholar] [CrossRef]
- Cui, Y.; Li, Y.-Y.; Li, J.; Zhang, H.-Y.; Wang, F.; Bai, X.; Li, S.-S. STAT3 regulates hypoxia-induced epithelial mesenchymal transition in oesophageal squamous cell cancer. Oncol. Rep. 2016, 36, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Kadioglu, O.; Saeed, M.E.; Mahmoud, N.; Azawi, S.; Mrasek, K.; Liehr, T.; Efferth, T. Identification of novel drug resistance mechanisms by genomic and transcriptomic profiling of glioblastoma cells with mutation-activated EGFR. Life Sci. 2021, 284, 119601. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carcelen, M.; Velasquez, C.; Vidal, V.; Gutiérrez, O.; Fernández-Luna, J.L. Signaling Pathways Regulating the Expression of the Glioblastoma Invasion Factor TENM1. Biomedicines 2022, 10, 1104. https://doi.org/10.3390/biomedicines10051104
Carcelen M, Velasquez C, Vidal V, Gutiérrez O, Fernández-Luna JL. Signaling Pathways Regulating the Expression of the Glioblastoma Invasion Factor TENM1. Biomedicines. 2022; 10(5):1104. https://doi.org/10.3390/biomedicines10051104
Chicago/Turabian StyleCarcelen, María, Carlos Velasquez, Verónica Vidal, Olga Gutiérrez, and José L. Fernández-Luna. 2022. "Signaling Pathways Regulating the Expression of the Glioblastoma Invasion Factor TENM1" Biomedicines 10, no. 5: 1104. https://doi.org/10.3390/biomedicines10051104
APA StyleCarcelen, M., Velasquez, C., Vidal, V., Gutiérrez, O., & Fernández-Luna, J. L. (2022). Signaling Pathways Regulating the Expression of the Glioblastoma Invasion Factor TENM1. Biomedicines, 10(5), 1104. https://doi.org/10.3390/biomedicines10051104