
Ubiquitin-Specific Protease 6 n-Terminal-like Protein (USP6NL) and the Epidermal Growth Factor Receptor (EGFR) Signaling Axis Regulates Ubiquitin-Mediated DNA Repair and Temozolomide-Resistance in Glioblastoma


 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. GBM Cell Culture
2.2. Bioinformatic Analysis of the TCGA-GBM Database
2.3. shRNA Transfection of GBM Cells
2.4. Cell Viability Assay
2.5. Western Blotting
2.6. Immunohistochemistry Staining
2.7. Immunofluorescence Staining
2.8. Fluorescence In Situ Hybridization
2.9. Quantitative Real-Time Reverse Transcription-Quantitative Polymerase Chain Reaction
2.10. Cell Migration Assay
2.11. Cell Invasion Assay
2.12. Coupled Cell Cycle and Cell Proliferation Assay
2.13. Apoptosis Assay
2.14. Spheroid/Sphere Formation
2.15. Colony Formation Assay
2.16. Animal Studies
2.17. Statistical Analysis
3. Results
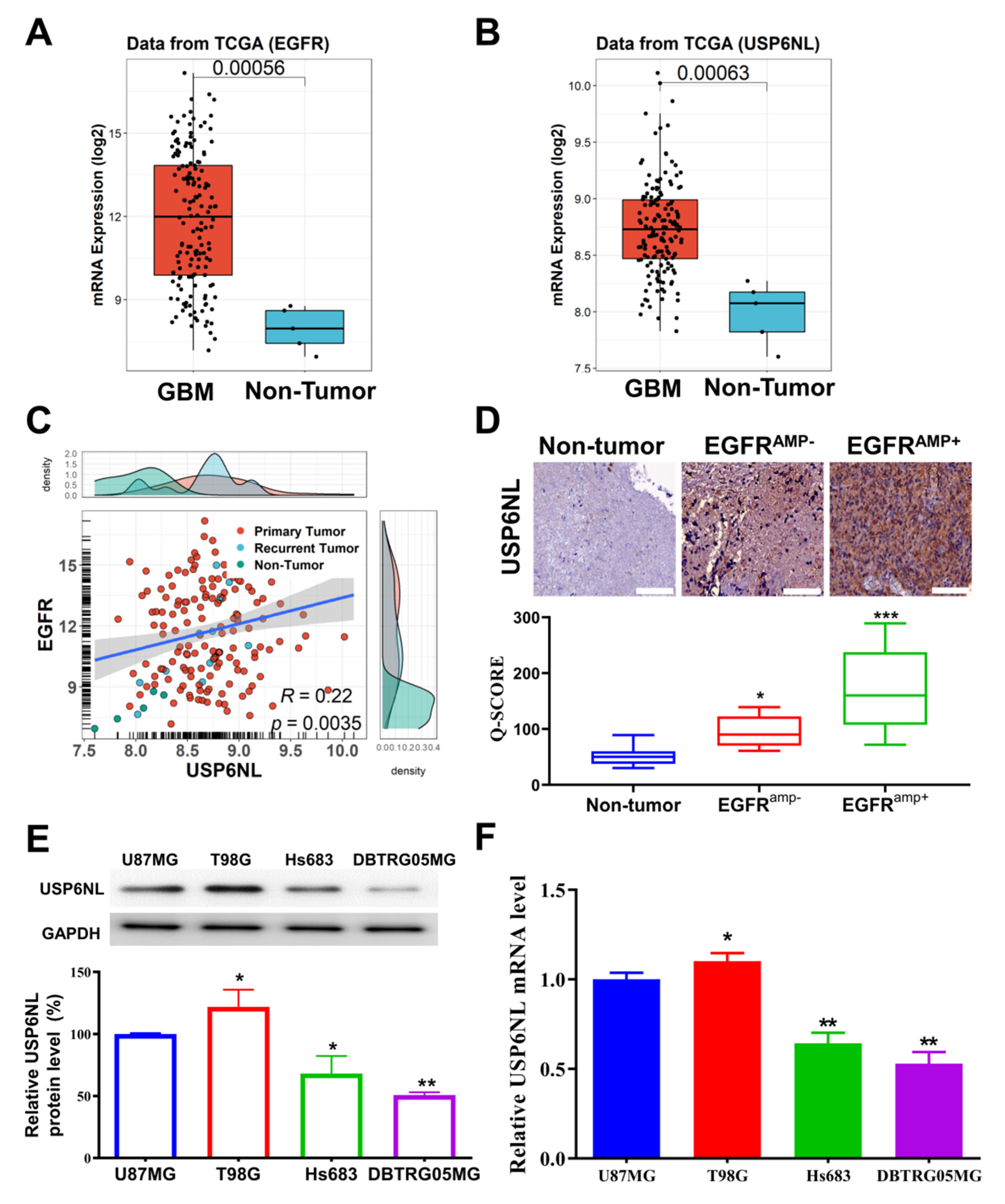
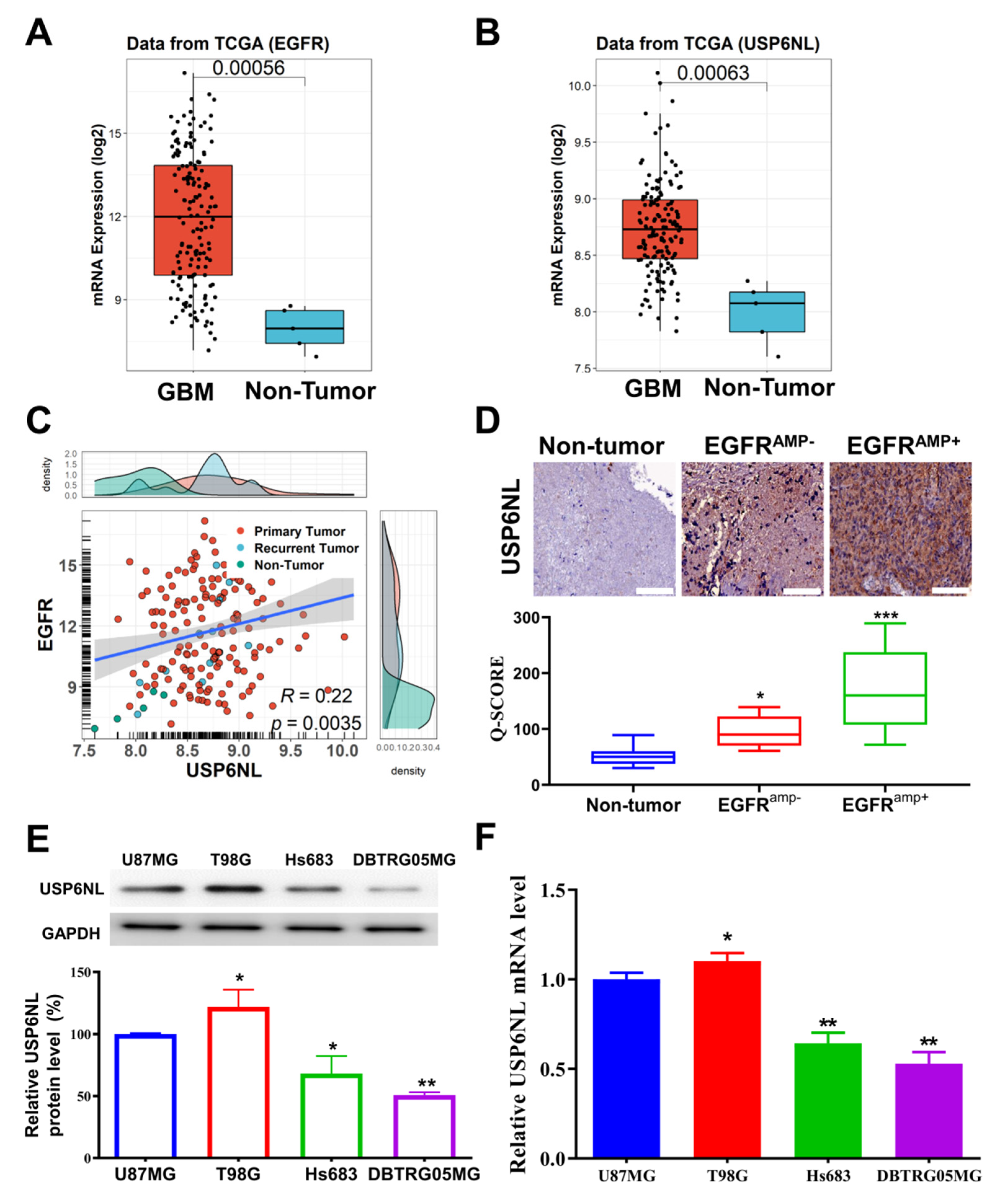
3.1. USP6NL and EGFR Overexpressed in Glioblastoma
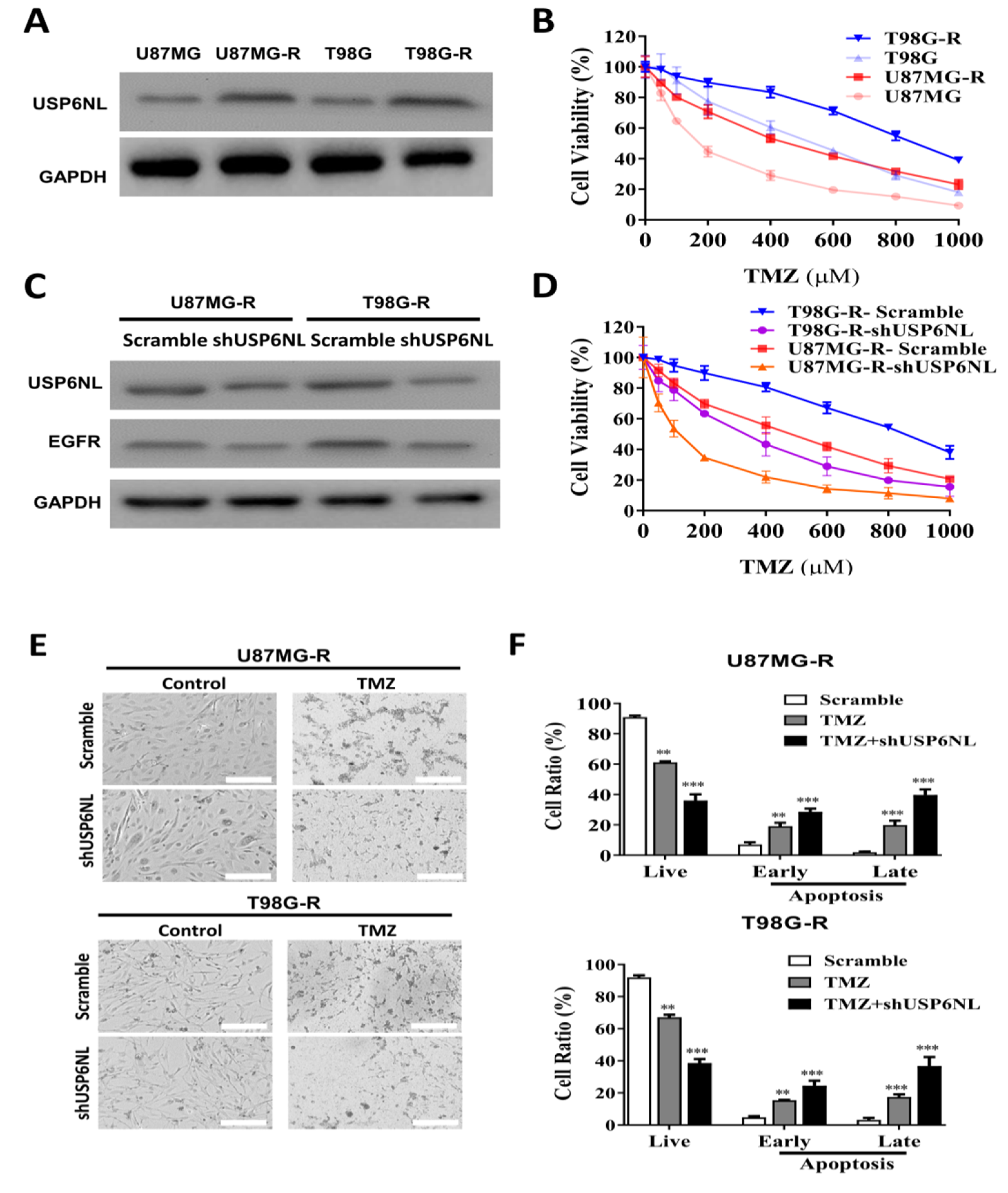
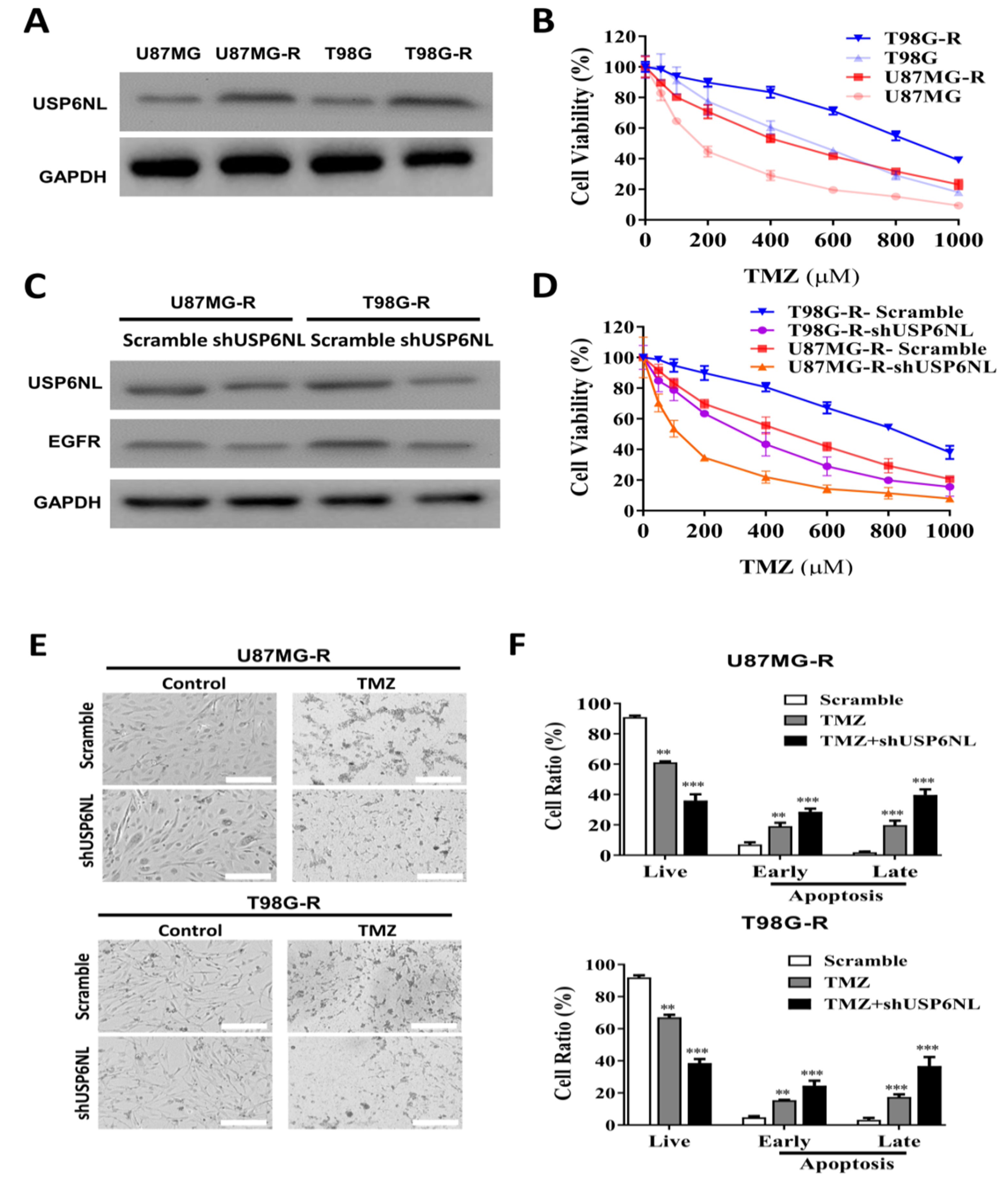
3.2. USP6NL and EGFR Positively Regulate TMZ Resistance in GBM Cells
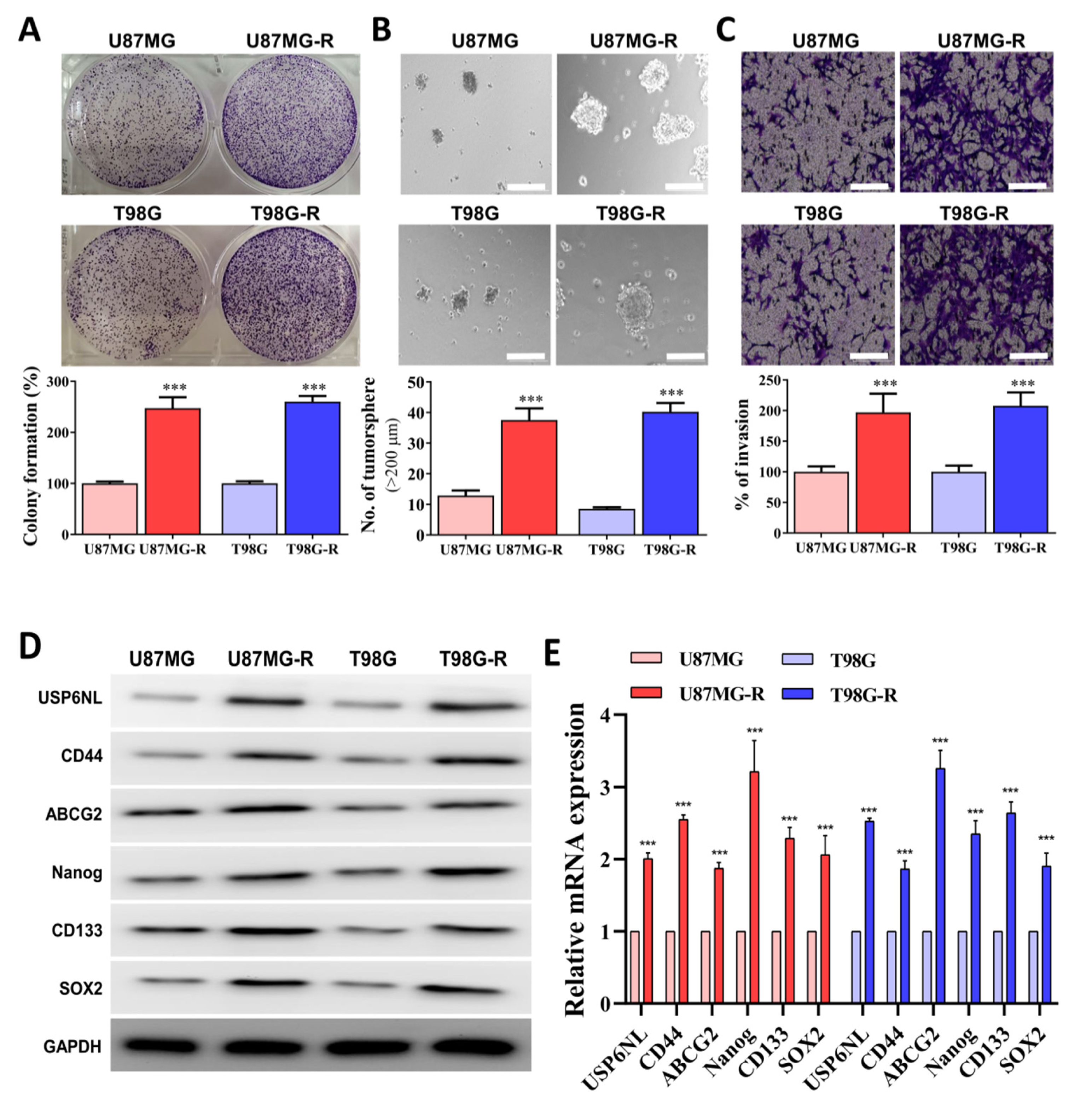
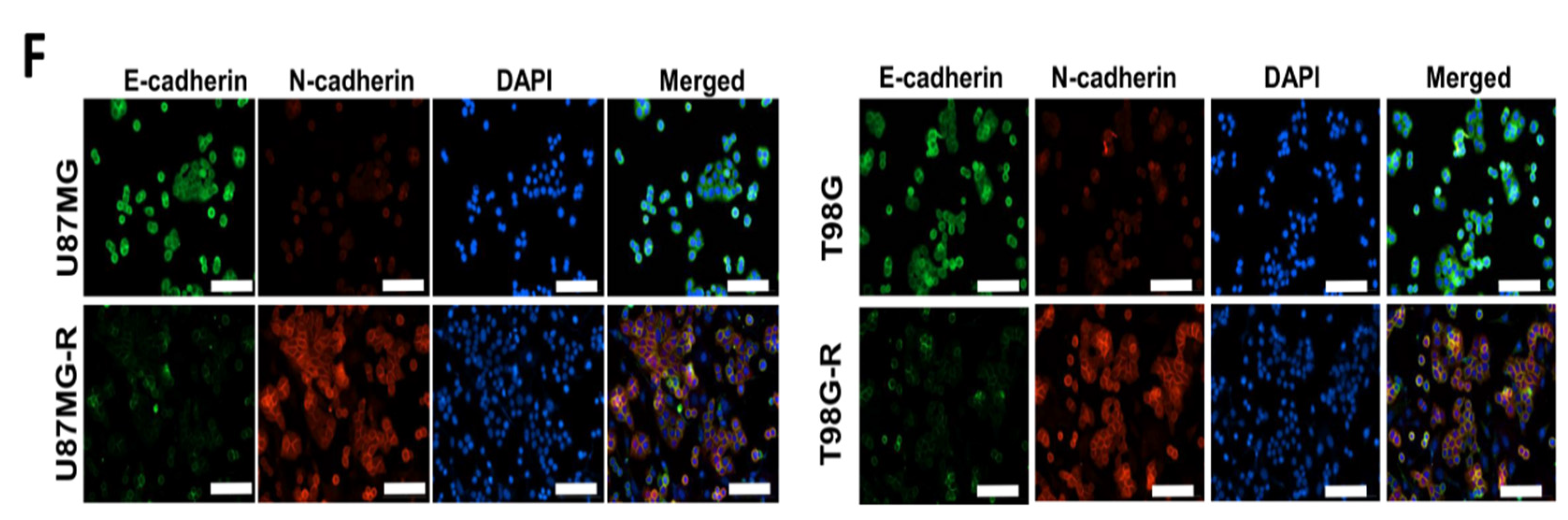
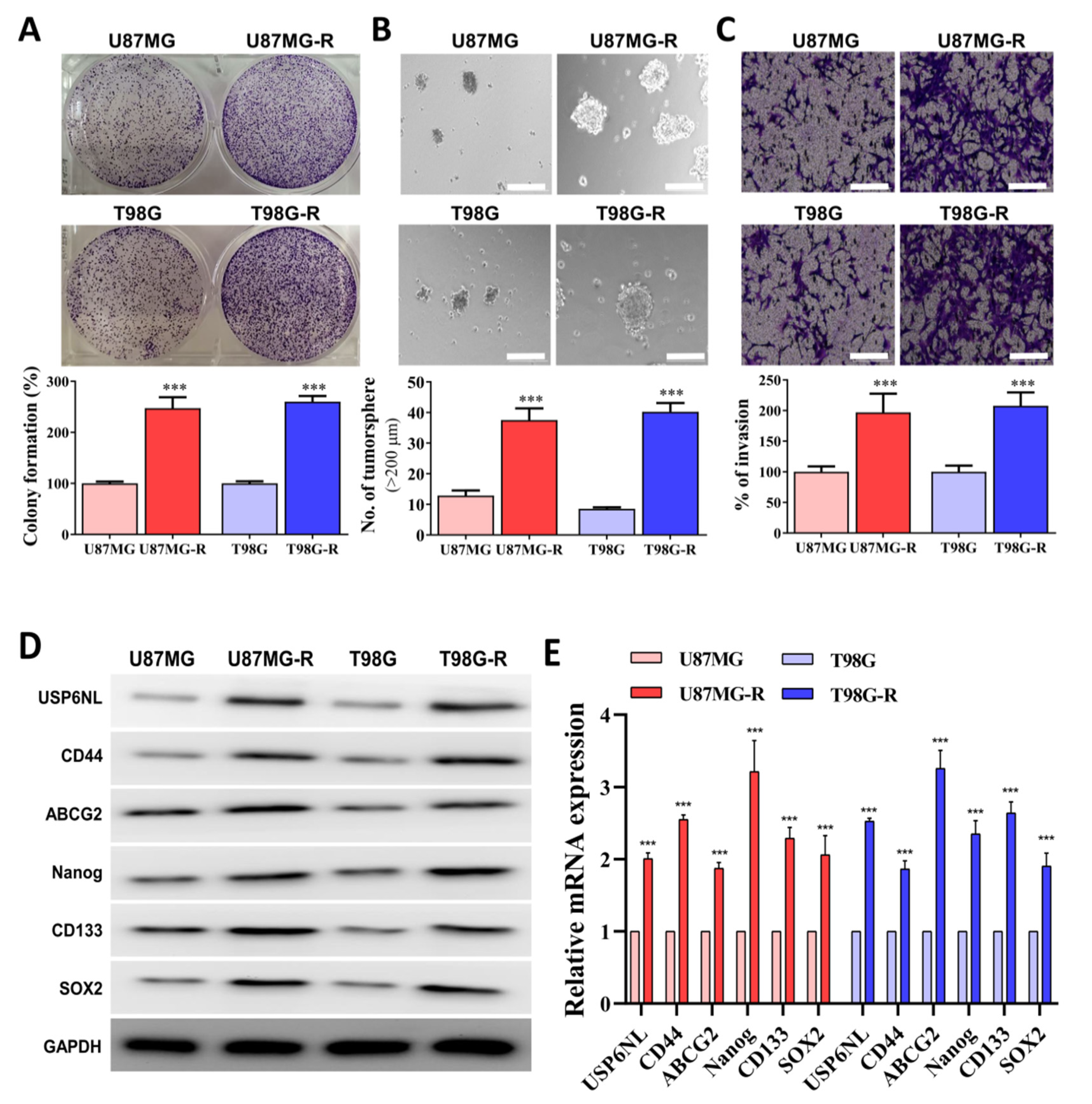
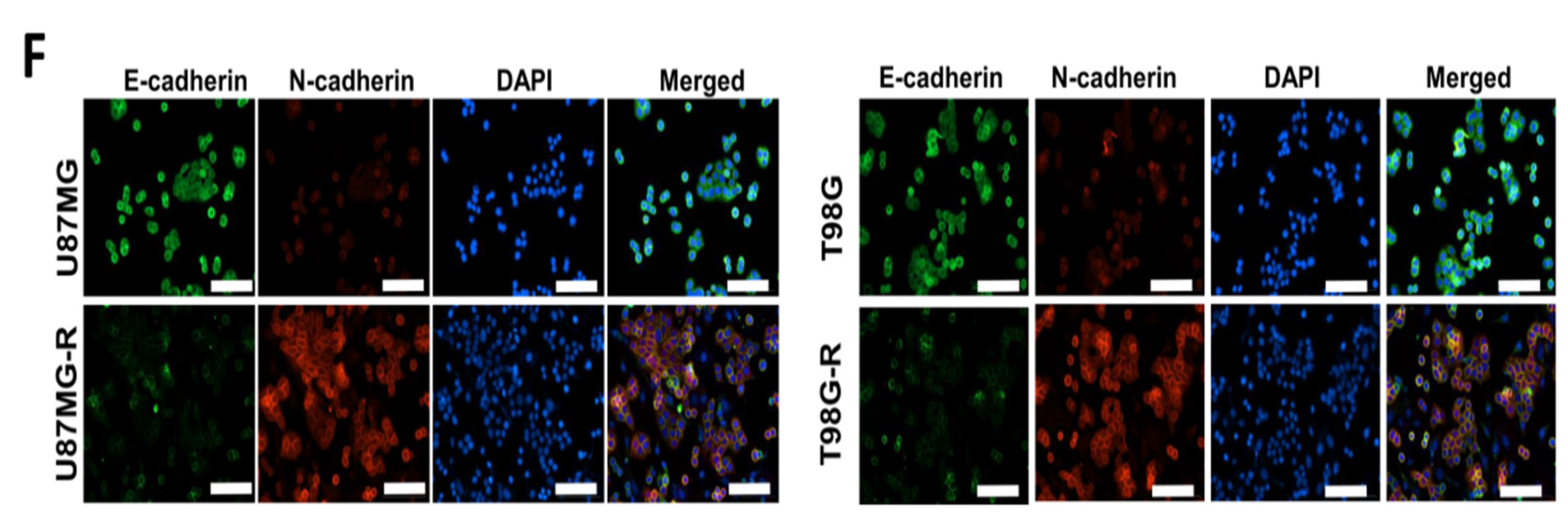
3.3. Association of Drug Resistance with High USP6NL and Cancer Stemness Properties
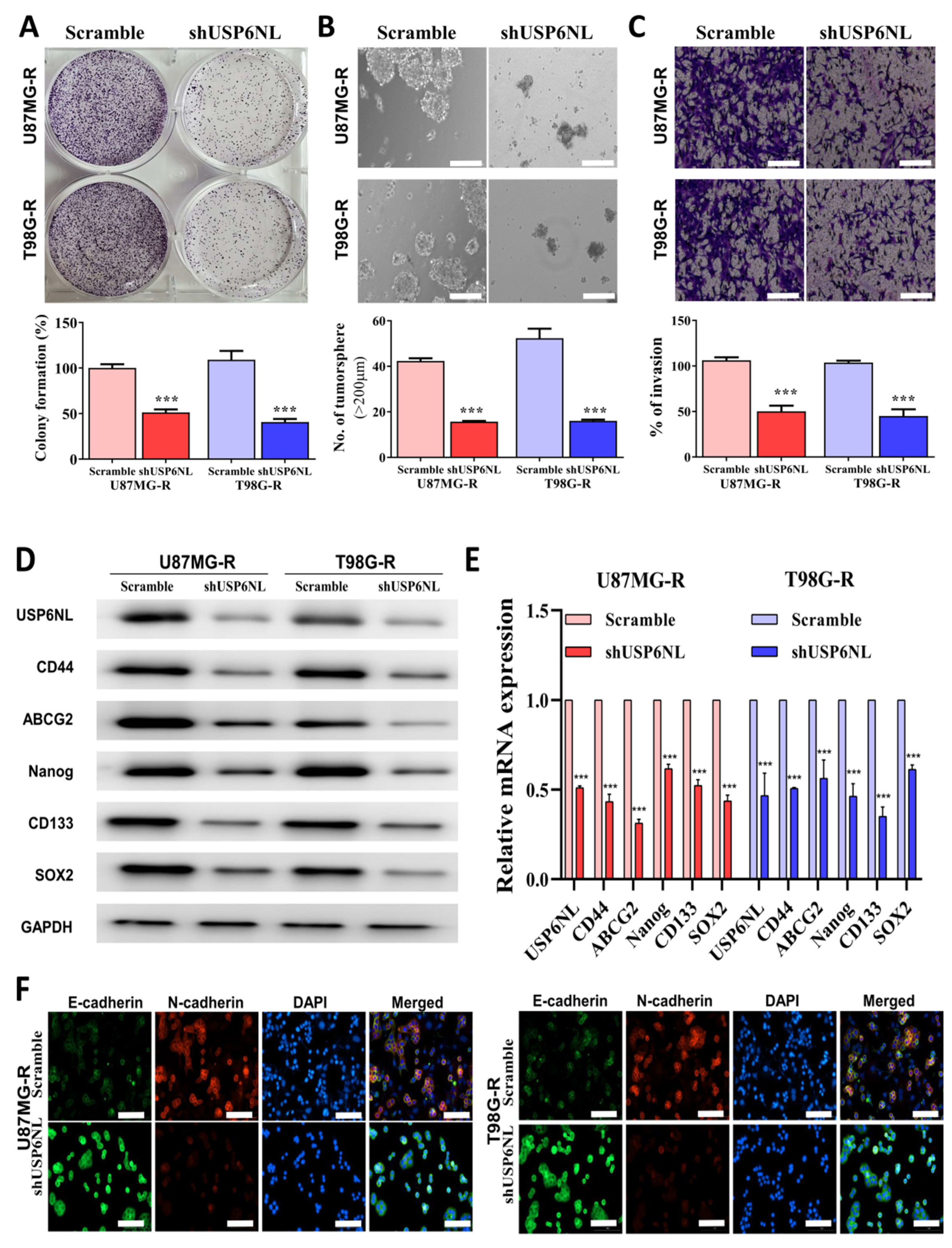
3.4. Abrogation of USP6NL Ameliorated the Stemness Phenotype of GBM Resistant Cells
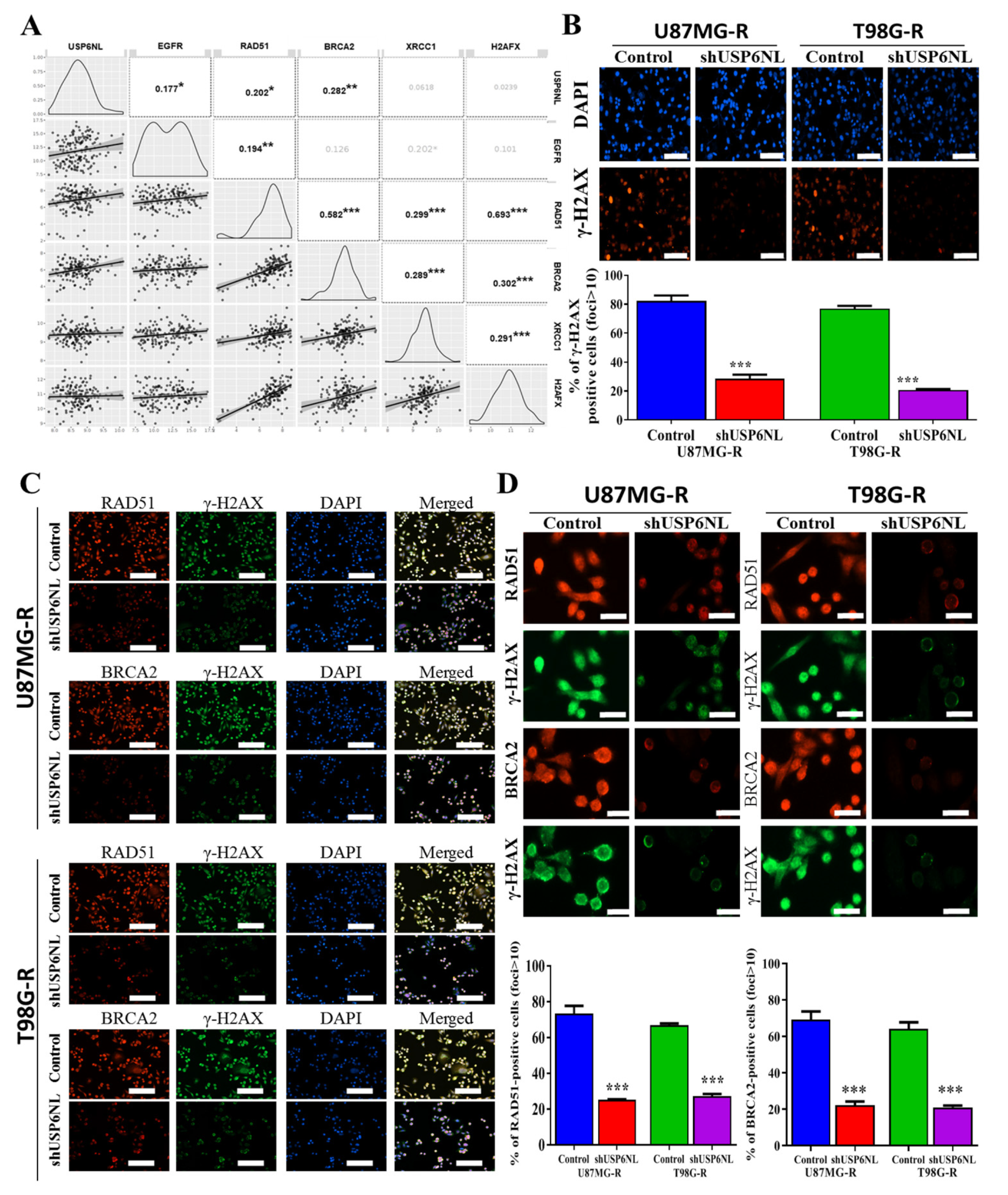
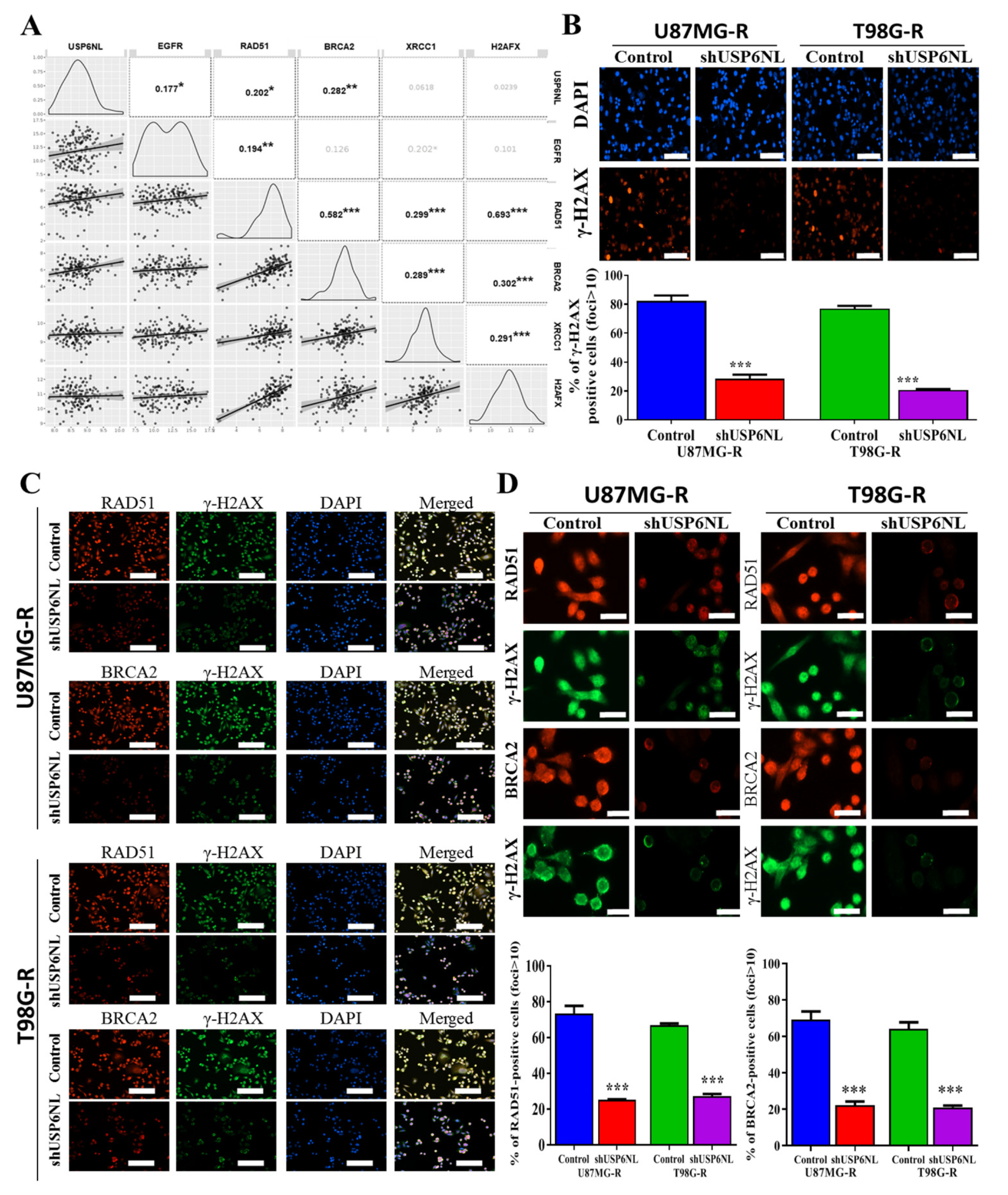
3.5. Effect of USP6NL on DNA Damage Repair Response
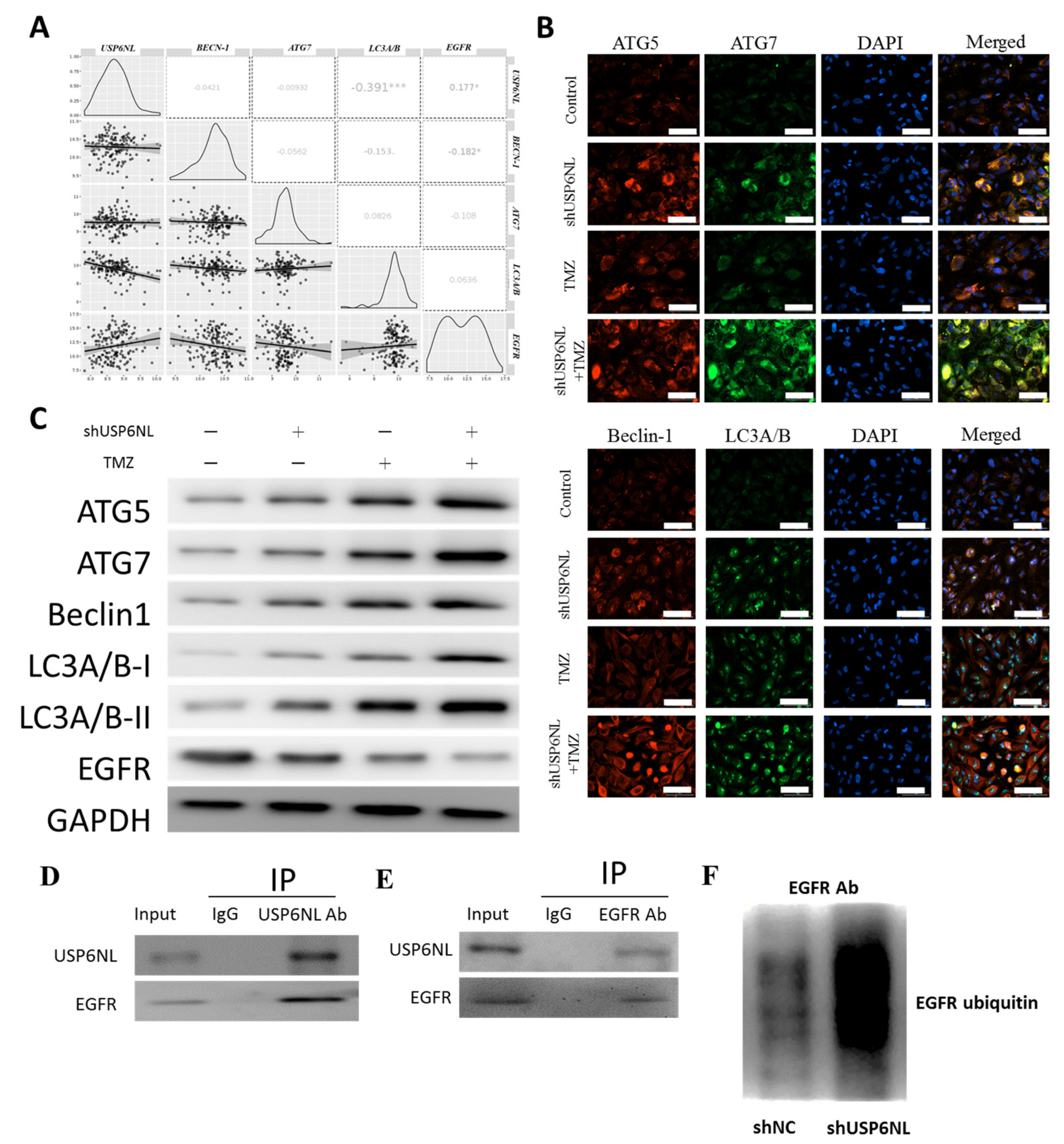
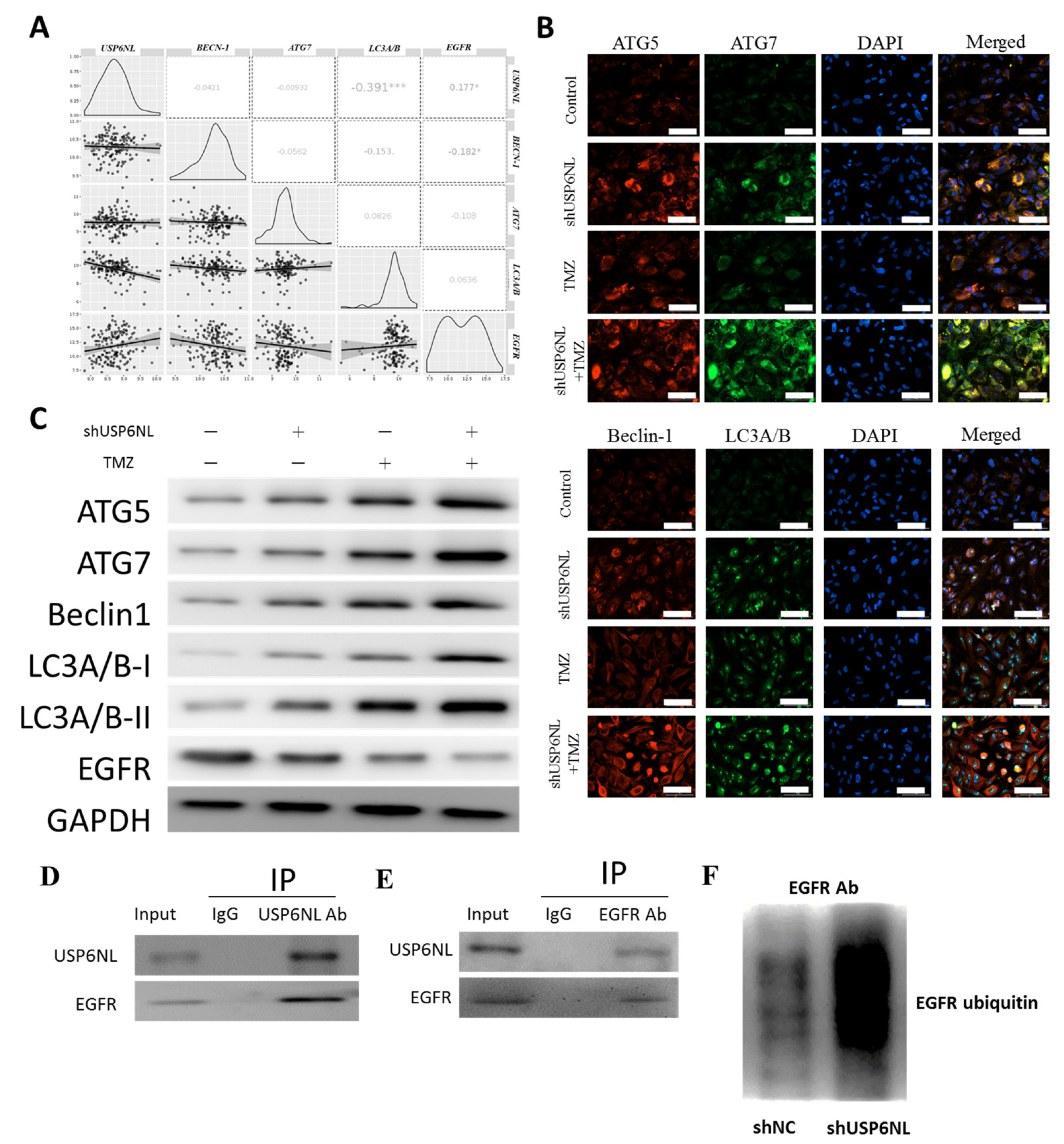
3.6. Inhibition of USP6NL Induces Autophagy of GBM Cells
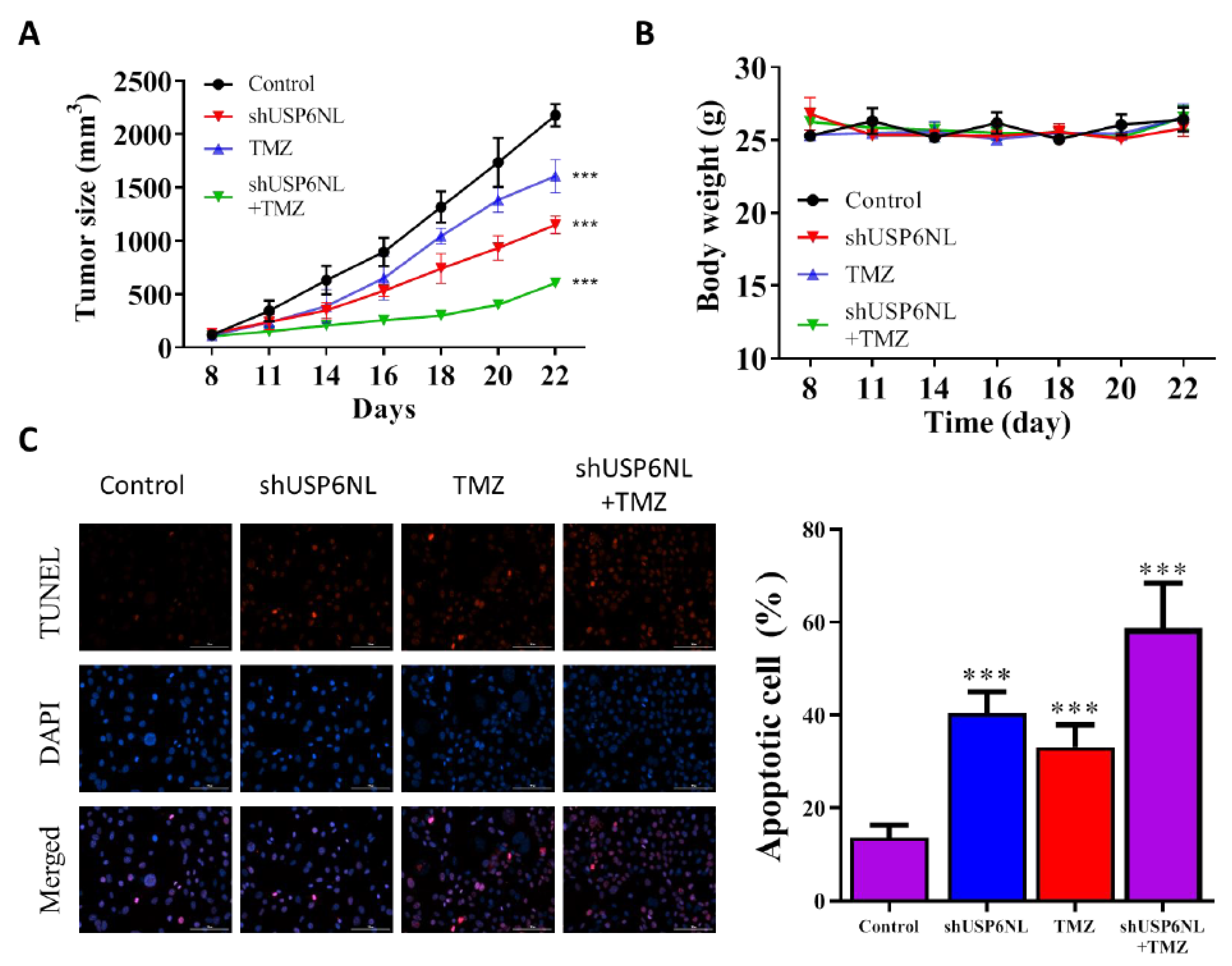
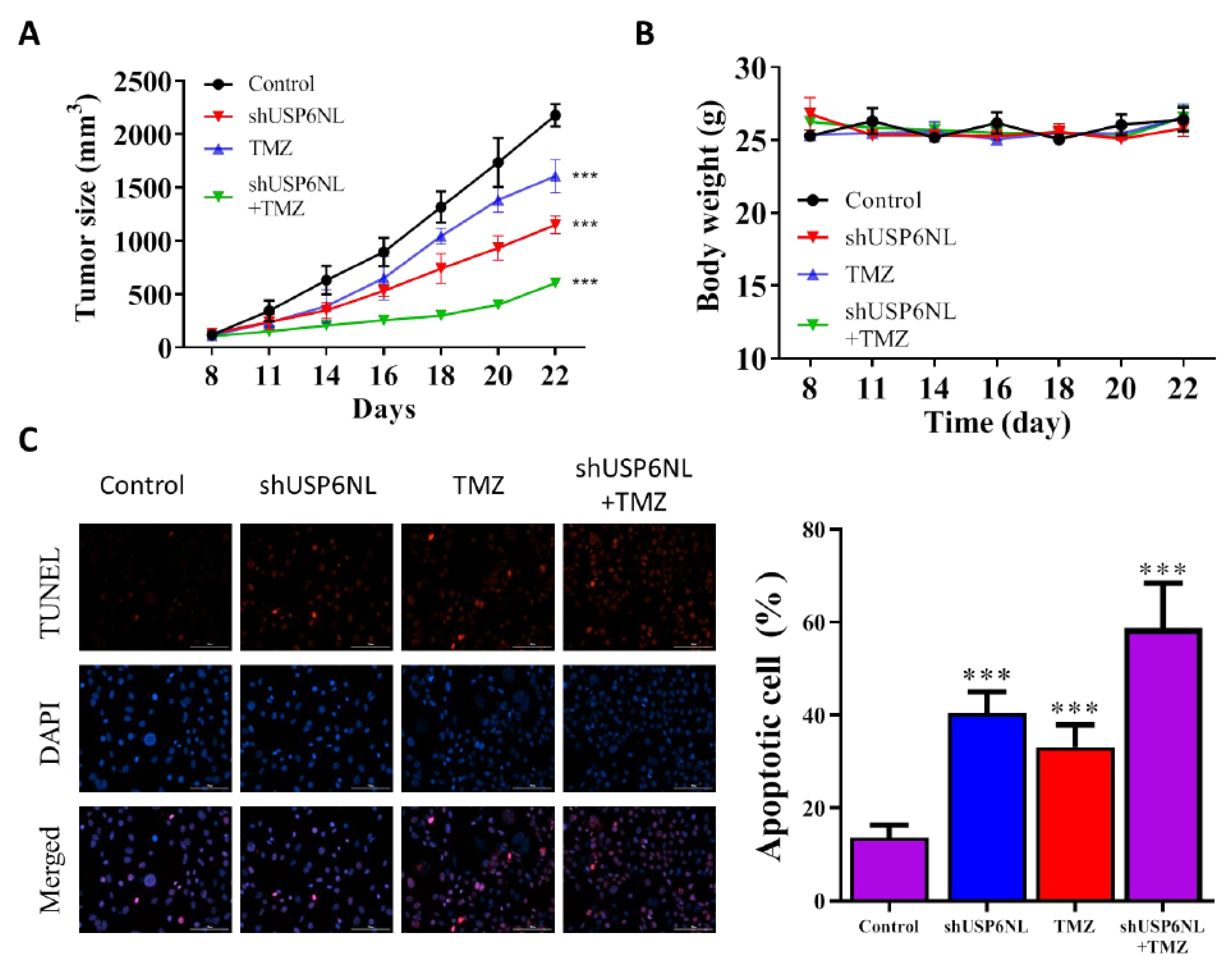
3.7. USP6NL Silencing Potentiates TMZ-Induced Cell Death In Vivo
4. Discussion
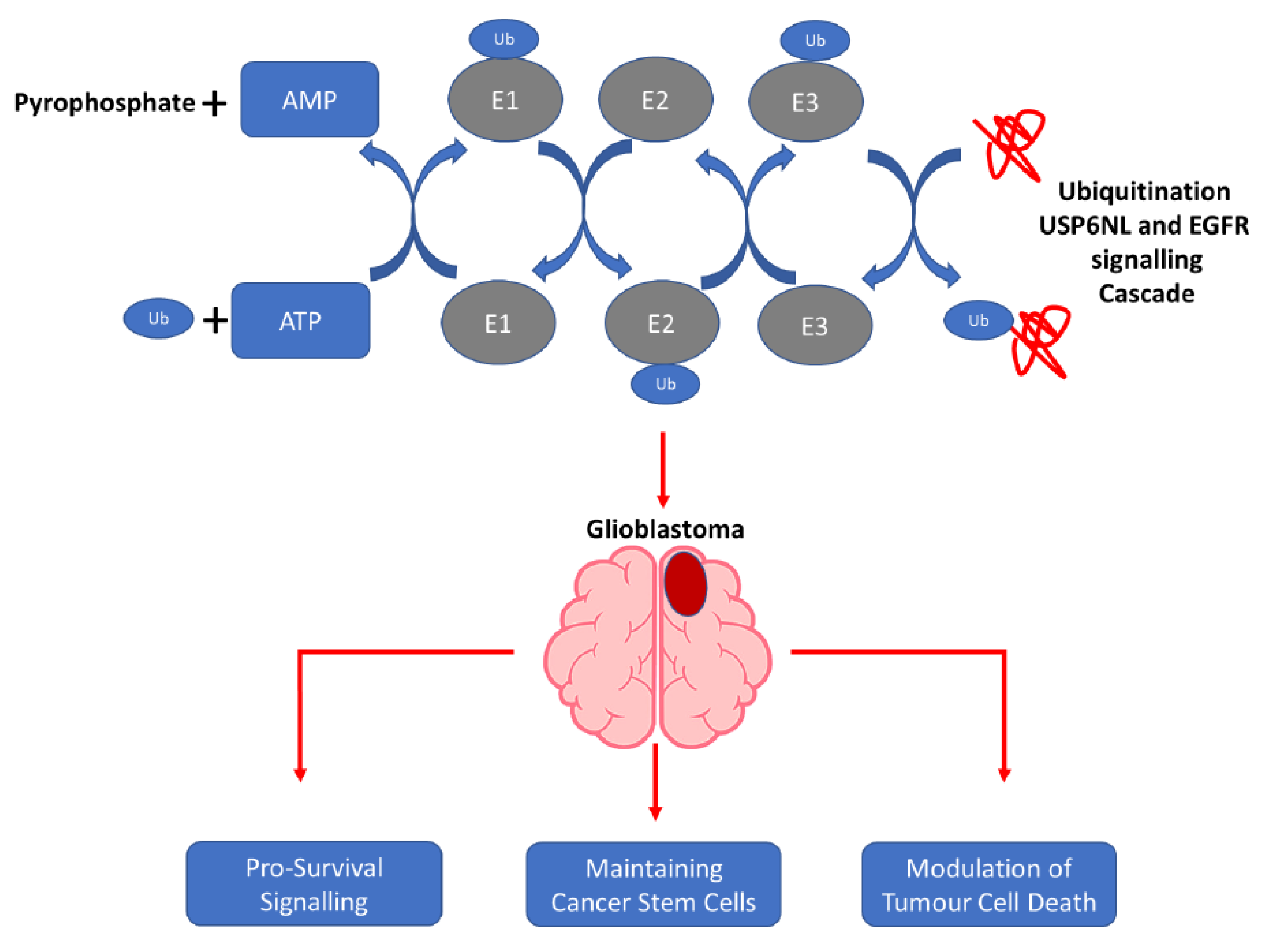
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. Codon Publ. 2017, 8, 143–153. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Devegowda, V.G.; Gupta, V.K.; Shivachar, A.; Bhosale, R.R.; Arunachalam, M.; Vaishnavi, T. Current Perspectives on Therapies, Including Drug Delivery Systems, for Managing Glioblastoma Multiforme. ACS Chem. Neurosci. 2020, 11, 2962–2977. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanson, M.; Marie, Y.; Paris, S.; Idbaih, A.; Laffaire, J.; Ducray, F.; El Hallani, S.; Boisselier, B.; Mokhtari, K.; Hoang-Xuan, K.; et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4150–4154. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.T.; Yu, L.; Lu, Y.T.; Ou, Y.H.; Li, Z.Y.; Wu, L.X.; Yao, F. IDH mutations occur frequently in Chinese glioma patients and predict longer survival but not response to concomitant chemoradiotherapy in anaplastic gliomas. Oncol. Rep. 2011, 26, 1479–1485. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Müller-Goebel, J.; Weik, A.-S.; Hoffer, K.; et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Stringer, B.W.; Kozlov, S.; Fazry, S.; Bruce, Z.C.; Ensbey, K.S.; Walker, D.G.; Boyd, A.W.; et al. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol. Oncol. 2014, 8, 1603–1615. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Voutsadakis, I.A.; Papandreou, C.N. The ubiquitin-proteasome system in glioma cell cycle control. Cell Div. 2012, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Song, L.; Luo, Z.-Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, D.; Hecker, C.M.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Scholz, N.; Kurian, K.M.; Siebzehnrubl, F.A.; Licchesi, J.D.F. Targeting the Ubiquitin System in Glioblastoma. Front. Oncol. 2020, 10, 574011. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-A.; Young, M.-J.; Wang, Y.-C.; Chen, S.-H.; Liu, C.-Y.; Lo, Y.-A.; Jen, H.-H.; Hsu, K.-C.; Hung, J.-J. USP24 promotes drug resistance during cancer therapy. Cell Death Differ. 2021, 28, 2690–2707. [Google Scholar] [CrossRef]
- Serapian, S.A.; Triveri, A.; Marchetti, F.; Castelli, M.; Colombo, G. Exploiting Folding and Degradation Machineries To Target Undruggable Proteins: What Can a Computational Approach Tell Us? ChemMedChem 2021, 16, 1593–1599. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S.J.C. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Crews, C.M. Targeting the undruggable proteome: The small molecules of my dreams. Chem. Biol. 2010, 17, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Alidousty, C.; Baar, T.; Martelotto, L.G.; Heydt, C.; Wagener, S.; Fassunke, J.; Duerbaum, N.; Scheel, A.H.; Frank, S.; Holz, B.; et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC: A central role of TP53 mutations. J. Pathol. 2018, 246, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.; Gay, C.M.; Byers, L.A. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Transl. Lung Cancer Res. 2018, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; He, S.-B.; Yao, Y.-Z.; Qu, J.-G.; Xie, R.; Ma, Y.-Q.; Zong, M.-H.; Chen, J.-X. Tre2 (USP6NL) promotes colorectal cancer cell proliferation via Wnt/β-catenin pathway. Cancer Cell Int. 2019, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Liu, H.; Liu, Y.; Liu, T.; Wang, H.; Qiao, F.; Song, L.; Zhang, L. USP6NL mediated by LINC00689/miR-142-3p promotes the development of triple-negative breast cancer. BMC Cancer 2020, 20, 998. [Google Scholar] [CrossRef]
- Park, H.-B.; Kim, J.-W.; Baek, K.-H. Regulation of Wnt Signaling through Ubiquitination and Deubiquitination in Cancers. Int. J. Mol. Sci. 2020, 21, 3904. [Google Scholar] [CrossRef]
- Shahoumi, L.A.; Khodadadi, H.; Bensreti, H.; Baban, B.; Yeudall, W.A. EPS8 phosphorylation by Src modulates its oncogenic functions. Br. J. Cancer 2020, 123, 1078–1088. [Google Scholar] [CrossRef]
- Avanzato, D.; Pupo, E.; Ducano, N.; Isella, C.; Bertalot, G.; Luise, C.; Pece, S.; Bruna, A.; Rueda, O.M.; Caldas, C.; et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res. 2018, 78, 3432–3444. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, K.C.; Llongueras, J.P.; Capilla-González, V.; Prasad, H.; Hack, A.; Smith, C.; Guerrero-Cázares, H.; Quiñones-Hinojosa, A.; Rao, R. A leak pathway for luminal protons in endosomes drives oncogenic signalling in glioblastoma. Nat. Commun. 2015, 6, 6289. [Google Scholar] [CrossRef] [Green Version]
- Ujifuku, K.; Mitsutake, N.; Takakura, S.; Matsuse, M.; Saenko, V.; Suzuki, K.; Hayashi, K.; Matsuo, T.; Kamada, K.; Nagata, I.J.C.l. MiR-195, miR-455-3p and miR-10a∗ are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010, 296, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Stevens, M.F.G.; Laughton, C.A.; Madhusudan, S.; Bradshaw, T.D. Acquired Resistance to Temozolomide in Glioma Cell Lines: Molecular Mechanisms and Potential Translational Applications. Oncology 2010, 78, 103–114. [Google Scholar] [CrossRef]
- Detre, S.; Saclani, J.G.; Dowsett, M.A. “quickscore” method for immunohistochemical semiquantitation: Validation for oestrogen receptor in breast carcinomas. J. Clin. Pathol. 1995, 48, 876–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Gines, C.; Gil-Benso, R.; Ferrer-Luna, R.; Benito, R.; Serna, E.; Gonzalez-Darder, J.; Quilis, V.; Monleon, D.; Celda, B.; Cerdá-Nicolas, M. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod. Pathol. 2010, 23, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, S.; Roos, W.P.; Kaina, B. Rad51 and BRCA2—New Molecular Targets for Sensitizing Glioma Cells to Alkylating Anticancer Drugs. PLoS ONE 2011, 6, e27183. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Long, Y.H.; Wang, S.Q.; Li, Y.F.; Zhang, J.H. Phosphorylation of H2A.X(T)(yr39) positively regulates DNA damage response and is linked to cancer progression. FEBS J. 2016, 283, 4462–4473. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Burke, P.; Schooler, K.; Wiley, H.S. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol. Biol. Cell 2001, 12, 1897–1910. [Google Scholar] [CrossRef] [Green Version]
- Acconcia, F.; Sigismund, S.; Polo, S. Ubiquitin in trafficking: The network at work. Exp. Cell Res. 2009, 315, 1610–1618. [Google Scholar] [CrossRef]
- Yun, S.-I.; Kim, H.H.; Yoon, J.H.; Park, W.S.; Hahn, M.-J.; Kim, H.C.; Chung, C.H.; Kim, K.K. Ubiquitin specific protease 4 positively regulates the WNT/β-catenin signaling in colorectal cancer. Mol. Oncol. 2015, 9, 1834–1851. [Google Scholar] [CrossRef]
- Ward, J.A.; McLellan, L.; Stockley, M.; Gibson, K.R.; Whitlock, G.A.; Knights, C.; Harrigan, J.A.; Jacq, X.; Tate, E.W. Quantitative chemical proteomic profiling of ubiquitin specific proteases in intact cancer cells. ACS Chem. Biol. 2016, 11, 3268–3272. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Hong, A.; Park, H.I.; Shin, W.H.; Yoo, L.; Jeon, S.J.; Chung, K.C. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J. Cell. Physiol. 2017, 232, 3664–3676. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Novellasdemunt, L.; Foglizzo, V.; Cuadrado, L.; Antas, P.; Kucharska, A.; Encheva, V.; Snijders, A.P.; Li, V.S. USP7 is a tumor-specific WNT activator for APC-mutated colorectal cancer by mediating β-catenin deubiquitination. Cell Rep. 2017, 21, 612–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.R.; Johnston, P.G.; Longley, D.B. Anti-apoptotic mechanisms of drug resistance in cancer. Curr. Cancer Drug Targets 2009, 9, 307–319. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr Protoc Pharm. 2013, 61, 14.25.1–14.25.14. [Google Scholar] [CrossRef] [Green Version]
- Yeom, S.Y.; Nam, D.H.; Park, C. RRAD promotes EGFR-mediated STAT3 activation and induces temozolomide resistance of malignant glioblastoma. Mol. Cancer Ther. 2014, 13, 3049–3061. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.Q.; Xu, X.H.; Liu, F.; Li, C.Y.; Li, Y.J.; Li, X.R.; Jiang, G.Y.; Hu, F.; Liu, D.; Pan, F. The binding of PD-L1 and Akt facilitates glioma cell invasion upon starvation via Akt/Autophagy/F-Actin signaling. Front. Oncol. 2019, 9, 1347. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Zhang, J.; Ren, X. PD-L1 regulates tumorigenesis and autophagy of ovarian cancer by activating mTORC signaling. Biosci. Rep. 2019, 39, BSR20191041. [Google Scholar] [CrossRef] [Green Version]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.J.C.d.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Hasan-Olive, M.M.; Zhu, H.; Denisova, O.; Grudic, A.; Latif, M.A.; Saed, H.; Varughese, J.K.; Røsland, G.V.; Yang, N. Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. Int. J. Cancer 2019, 144, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P. Untangling autophagy measurements: All fluxed up. Circ. Res. 2015, 116, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, I.-C.; Su, Y.-K.; Chuang, H.-Y.; Yadav, V.K.; Setiawan, S.A.; Fong, I.-H.; Yeh, C.-T.; Huang, H.-C.; Lin, C.-M. Ubiquitin-Specific Protease 6 n-Terminal-like Protein (USP6NL) and the Epidermal Growth Factor Receptor (EGFR) Signaling Axis Regulates Ubiquitin-Mediated DNA Repair and Temozolomide-Resistance in Glioblastoma. Biomedicines 2022, 10, 1531. https://doi.org/10.3390/biomedicines10071531
Su I-C, Su Y-K, Chuang H-Y, Yadav VK, Setiawan SA, Fong I-H, Yeh C-T, Huang H-C, Lin C-M. Ubiquitin-Specific Protease 6 n-Terminal-like Protein (USP6NL) and the Epidermal Growth Factor Receptor (EGFR) Signaling Axis Regulates Ubiquitin-Mediated DNA Repair and Temozolomide-Resistance in Glioblastoma. Biomedicines. 2022; 10(7):1531. https://doi.org/10.3390/biomedicines10071531
Chicago/Turabian StyleSu, I-Chang, Yu-Kai Su, Hao-Yu Chuang, Vijesh Kumar Yadav, Syahru Agung Setiawan, Iat-Hang Fong, Chi-Tai Yeh, Hui-Chuan Huang, and Chien-Min Lin. 2022. "Ubiquitin-Specific Protease 6 n-Terminal-like Protein (USP6NL) and the Epidermal Growth Factor Receptor (EGFR) Signaling Axis Regulates Ubiquitin-Mediated DNA Repair and Temozolomide-Resistance in Glioblastoma" Biomedicines 10, no. 7: 1531. https://doi.org/10.3390/biomedicines10071531
APA StyleSu, I.-C., Su, Y.-K., Chuang, H.-Y., Yadav, V. K., Setiawan, S. A., Fong, I.-H., Yeh, C.-T., Huang, H.-C., & Lin, C.-M. (2022). Ubiquitin-Specific Protease 6 n-Terminal-like Protein (USP6NL) and the Epidermal Growth Factor Receptor (EGFR) Signaling Axis Regulates Ubiquitin-Mediated DNA Repair and Temozolomide-Resistance in Glioblastoma. Biomedicines, 10(7), 1531. https://doi.org/10.3390/biomedicines10071531