
Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- ATR, along with ataxia-telangiectasia mutated kinase (ATM), plays a significant role in activating the DNA response. ATR under physiological conditions is known to protect against replication stress and to safeguard genomic stability during replication by preventing the breakage or ‘collapse’ of stalled replication forks [17]. ATR promotes the recruitment of ATRIP, the regulatory partner of ATR, thereby allowing the ATR–ATRIP complex to recognize the replication protein A bound ssDNA at DNA damage sites or stressed replication forks [18]. The ATR-dependent DNA damage repair is known to follow the HR pathway [19]. ATR initiates the S and G2 cell cycle checkpoint through CHK1 phosphorylation [15], which in turn mediates CDC25A-C phosphorylation, leading to blocking of CDK1 and CDK2 (thus preventing cell cycle progression) [20]. Inhibition of these pathways causes a dramatic increase in replication initiation, or, rather, a lack of cell cycle arrest with an eventual mitotic catastrophe due to the lack of DNA repair. Furthermore, ATR-CHK1 activation has been associated with the expression of PD-L1 expression on irradiated tumor cells in a type I IFN signaling-dependent manner [21,22].
- DNA-PKcs, which belongs to the phosphoinositide 3-kinase (PI3K)-related protein kinase (PIKK) family, just like ATR and ATM, plays a central role in regulating c-NHEJ. c-NHEJ can repair DSBs of varying complexity, such as those with incompatible ends or damaged bases [23]. The DSB end-recognition is detected by the Ku70/80 heterodimer (Ku), which serves as a scaffold for the assembly of the components of the c-NHEJ machinery, including DNA-PKcs, the XRCC4-DNA ligase IV complex, and XRCC4-like factor (XLF) [24]. The role of DNA-PK in regulating the cell cycle is still not completely understood. However, DNA-PK demonstrates cross-talk between ATM/ATR and plays a role in checkpoint recovery, albeit in a cellular context [25,26]. DNA-PK inhibitors have also been documented to substantially enhance PD-L1 expression in irradiated cancer cells via cGAS/STING pathway activation in a p53-deficient background [27].
2. Materials and Methods
2.1. Cell Lines
2.2. Small Molecule Inhibitors
2.3. Ionizing Radiation (IR) Source and Setup
2.4. Western Blot
2.5. Neutral Comet Assay
2.6. Short Term Single and Combined Drug Response Analyses
2.7. Clonogenic Survival Assay
2.8. Whole Exome Sequencing
2.9. Statistical Data Acquisition
3. Results
3.1. Short-Term Application of DNA-PKi (AZD7648) and ATRi (Ceralasertib) Leads to Cell Death and Radiosensitization
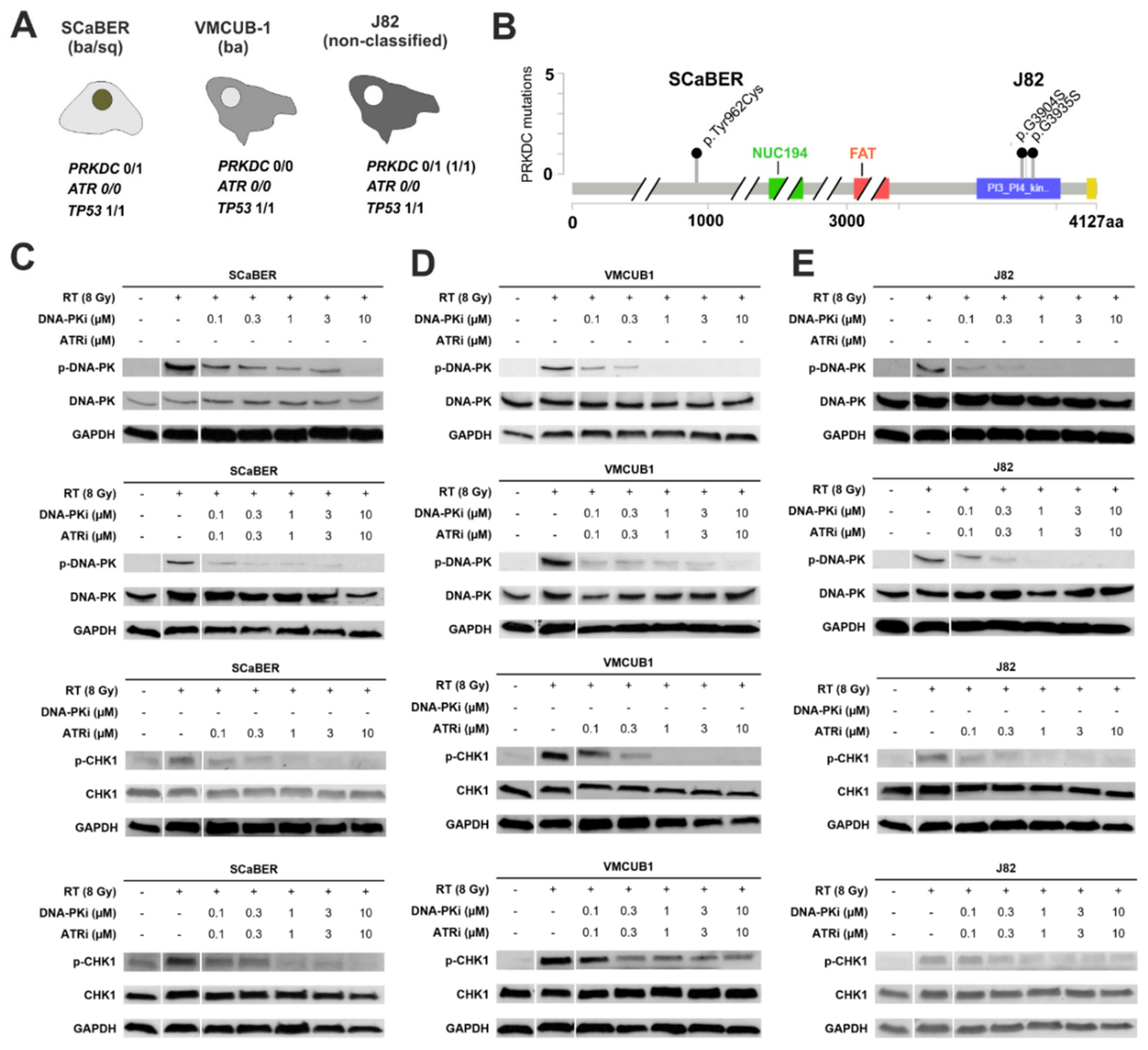
3.2. Pharmacological Inhibition of DNA-PK and ATR Effectively Blocks Downstream Signaling Involved in DDR
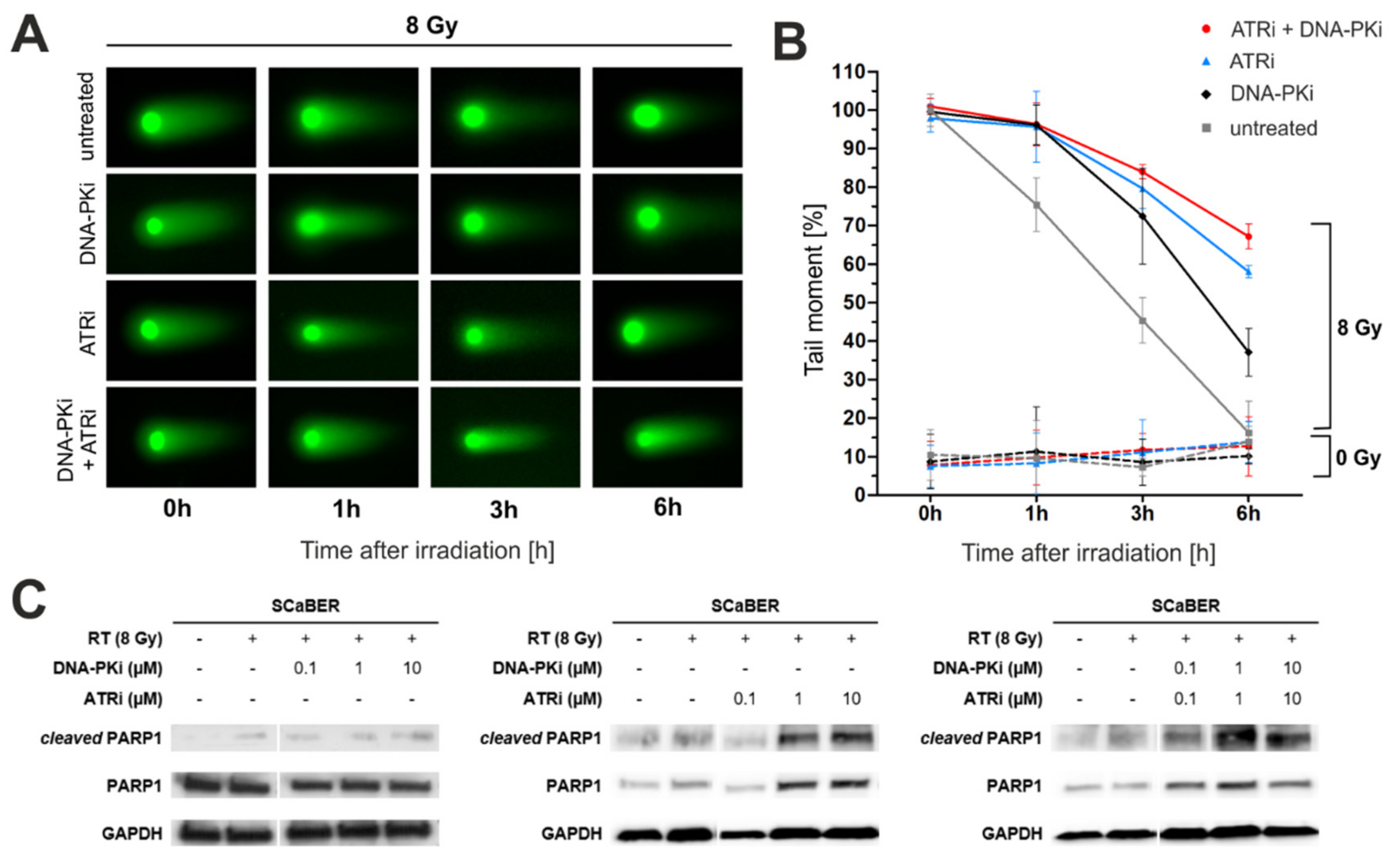
3.3. Pharmacological Inhibition of DNA-PK and ATR Impedes DNA Damage Response after Exposure to Ionizing Radiation
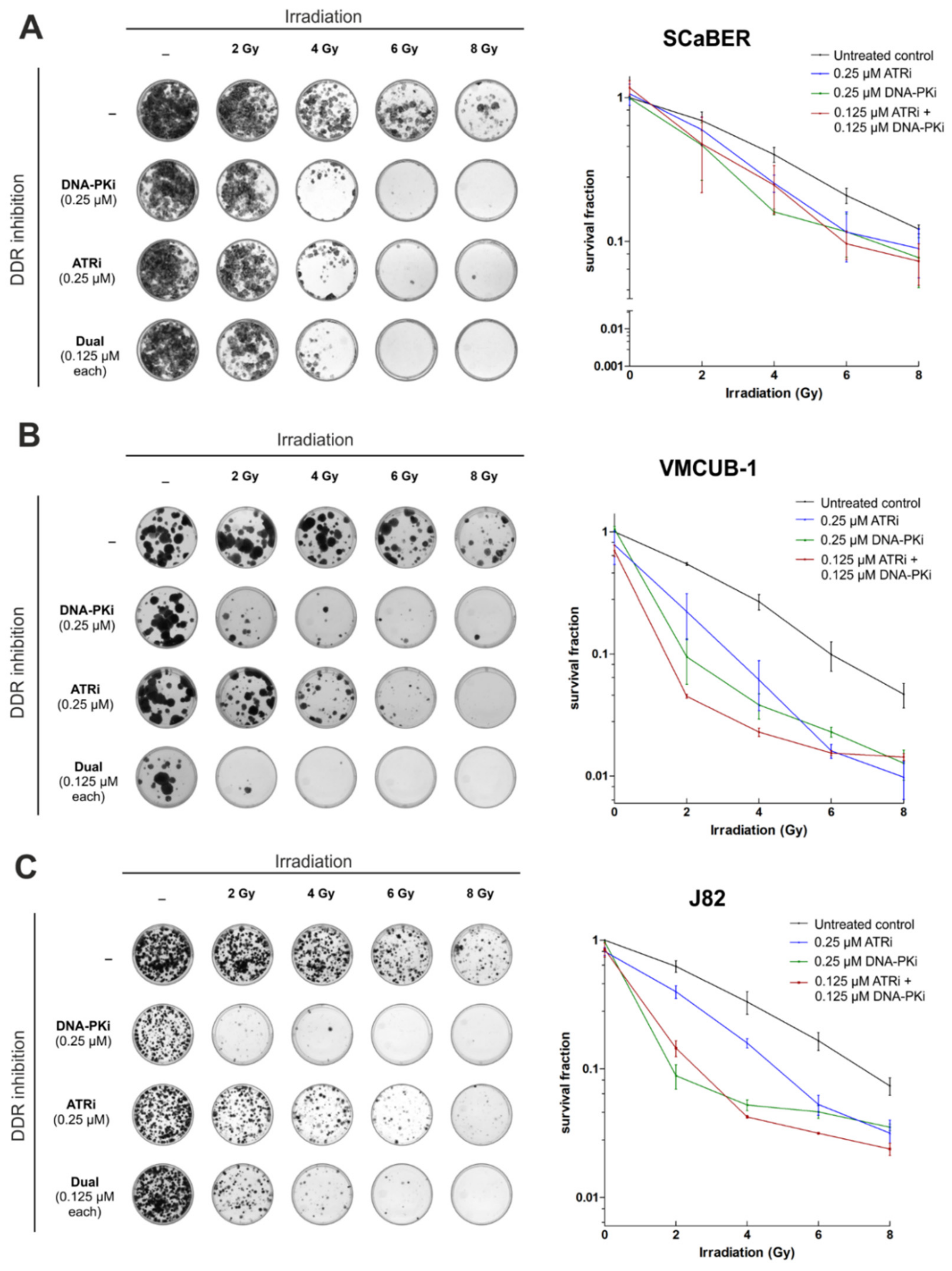
3.4. AZD7648 and Ceralasertib at Nano-Molar Concentrations Drastically Reduce Clonogenic Survival after Radiation Exposure
3.5. Combination of AZD4678 and Ceralasertib Shows Potential Synergism Depending on Cell Line and Drug-/IR-Dose
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messing, E.M.; Tangen, C.M.; Lerner, S.P.; Sahasrabudhe, D.M.; Koppie, T.M.; Wood, D.P., Jr.; Mack, P.C.; Svatek, R.S.; Evans, C.P.; Hafez, K.S.; et al. Effect of Intravesical Instillation of Gemcitabine vs. Saline Immediately following Resection of Suspected Low-Grade Non-Muscle-Invasive Bladder Cancer on Tumor Recurrence: SWOG S0337 Randomized Clinical Trial. JAMA 2018, 319, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Brandau, S.; Suttmann, H. Thirty years of BCG immunotherapy for non-muscle invasive bladder cancer: A success story with room for improvement. Biomed. Pharmacother. 2007, 61, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Niegisch, G.; Gerullis, H.; Lin, S.W.; Pavlova, J.; Gondos, A.; Rudolph, A.; Haas, G.; Hennies, N.; Kramer, M.W. A Real-World Data Study to Evaluate Treatment Patterns, Clinical Characteristics and Survival Outcomes for First- and Second-Line Treatment in Locally Advanced and Metastatic Urothelial Cancer Patients in Germany. J. Cancer 2018, 9, 1337–1348. [Google Scholar] [CrossRef]
- Von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef]
- James, N.D.; Hussain, S.A.; Hall, E.; Jenkins, P.; Tremlett, J.; Rawlings, C.; Crundwell, M.; Sizer, B.; Sreenivasan, T.; Hendron, C.; et al. Radiotherapy with or without chemotherapy in muscle-invasive bladder cancer. N. Engl. J. Med. 2012, 366, 1477–1488. [Google Scholar] [CrossRef] [Green Version]
- Dumont, F.; Altmeyer, A.; Bischoff, P. Radiosensitising agents for the radiotherapy of cancer: Novel molecularly targeted approaches. Expert Opin. Ther. Pat. 2009, 19, 775–799. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin. 2020, 70, 404–423. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [Green Version]
- Mamo, T.; Mladek, A.C.; Shogren, K.L.; Gustafson, C.; Gupta, S.K.; Riester, S.M.; Maran, A.; Galindo, M.; van Wijnen, A.J.; Sarkaria, J.N.; et al. Inhibiting DNA-PK CS radiosensitizes human osteosarcoma cells. Biochem. Biophys. Res. Commun. 2017, 486, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G(2)-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [Green Version]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Powell, S.N.; Iliakis, G.; Wang, Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004, 64, 7139–7143. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, M.; Martinez-Pastor, B.; Fernandez-Capetillo, O. ATR activation in response to ionizing radiation: Still ATM territory. Cell Div. 2006, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Tubbs, A.; Zhang, C.; Tang, M.; Sridharan, S.; Wang, C.; Jiang, D.; Su, D.; Zhang, H.; Chen, Z.; et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 2020, 39, e104036. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spagnolo, L.; Rivera-Calzada, A.; Pearl, L.H.; Llorca, O. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell 2006, 22, 511–519. [Google Scholar] [CrossRef]
- Li, F.; Mladenov, E.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Shift in G 1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation. Cells 2021, 11, 63. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G 2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Carr, M.I.; Chiu, L.Y.; Guo, Y.; Xu, C.; Lazorchak, A.S.; Yu, H.; Qin, G.; Qi, J.; Marelli, B.; Lan, Y.; et al. DNA-PK Inhibitor Peposertib Amplifies Radiation-Induced Inflammatory Micronucleation and Enhances TGFβ/PD-L1 Targeted Cancer Immunotherapy. Mol. Cancer Res. 2022, 20, 568–582. [Google Scholar] [CrossRef]
- Rose, M.; Maurer, A.; Wirtz, J.; Bleilevens, A.; Waldmann, T.; Wenz, M.; Eyll, M.; Geelvink, M.; Gereitzig, M.; Rüchel, N.; et al. EGFR activity addiction facilitates anti-ERBB based combination treatment of squamous bladder cancer. Oncogene 2020, 39, 6856–6870. [Google Scholar] [CrossRef]
- Heide, T.; Maurer, A.; Eipel, M.; Knoll, K.; Geelvink, M.; Veeck, J.; Knuechel, R.; van Essen, J.; Stoehr, R.; Hartmann, A.; et al. Multiregion human bladder cancer sequencing reveals tumour evolution, bladder cancer phenotypes and implications for targeted therapy. J. Pathol. 2019, 248, 230–242. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Stone, H.B.; Bernhard, E.J.; Coleman, C.N.; Deye, J.; Capala, J.; Mitchell, J.B.; Brown, J.M. Preclinical Data on Efficacy of 10 Drug-Radiation Combinations: Evaluations, Concerns, and Recommendations. Transl. Oncol. 2016, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, F.W.; Finlay, M.R.V.; Ting, A.K.T.; Beattie, D.; Lamont, G.M.; Fallan, C.; Wrigley, G.L.; Schimpl, M.; Howard, M.R.; Williamson, B.; et al. The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2 H-pyran-4-yl)-7,9-dihydro-8 H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor. J. Med. Chem. 2020, 63, 3461–3471. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Qvarnström, O.F.; Simonsson, M.; Eriksson, V.; Turesson, I.; Carlsson, J. γH2AX and cleaved PARP-1 as apoptotic markers in irradiated breast cancer BT474 cellular spheroids. Int. J. Oncol. 2009, 35, 41–47. [Google Scholar] [CrossRef]
- Lobo, N.; Kulkarni, M.; Hughes, S.; Nair, R.; Khan, M.S.; Thurairaja, R. Urologic Complications Following Pelvic Radiotherapy. Urology 2018, 122, 1–9. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Nakamura, K.; Karmokar, A.; Farrington, P.M.; James, N.H.; Ramos-Montoya, A.; Bickerton, S.J.; Hughes, G.D.; Illidge, T.M.; Cadogan, E.B.; Davies, B.R.; et al. Inhibition of DNA-PK with AZD7648 Sensitizes Tumor Cells to Radiotherapy and Induces Type I IFN-Dependent Durable Tumor Control. Clin. Cancer Res. 2021, 27, 4353–4366. [Google Scholar] [CrossRef]
- Hong, C.R.; Buckley, C.D.; Wong, W.W.; Anekal, P.V.; Dickson, B.D.; Bogle, G.; Hicks, K.O.; Hay, M.P.; Wilson, W.R. Radiosensitisation of SCCVII tumours and normal tissues in mice by the DNA-dependent protein kinase inhibitor AZD7648. Radiother. Oncol. 2022, 166, 162–170. [Google Scholar] [CrossRef]
- Karukonda, P.; Odhiambo, D.; Mowery, Y.M. Pharmacologic inhibition of ataxia telangiectasia and Rad3-related (ATR) in the treatment of head and neck squamous cell carcinoma. Mol. Carcinog. 2022, 61, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Wang, F.; Li, K.; Li, S.; Zhao, C.; Fan, C.; Wang, J. Significance of TP53 mutation in bladder cancer disease progression and drug selection. PeerJ 2019, 7, e8261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouw, K.W. DNA Repair Pathway Alterations in Bladder Cancer. Cancers 2017, 9, 28. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Cato Guldberg and Peter Waage, the history of the Law of Mass Action, and its relevance to clinical pharmacology. Br. J. Clin. Pharmacol. 2016, 81, 52–55. [Google Scholar] [CrossRef] [Green Version]
- Minato, A.; Fujimoto, N.; Kubo, T. Squamous Differentiation Predicts Poor Response to Cisplatin-Based Chemotherapy and Unfavorable Prognosis in Urothelial Carcinoma of the Urinary Bladder. Clin. Genitourin. Cancer 2017, 15, e1063–e1067. [Google Scholar] [CrossRef]
- Mijnes, J.; Veeck, J.; Gaisa, N.T.; Burghardt, E.; de Ruijter, T.C.; Gostek, S.; Dahl, E.; Pfister, D.; Schmid, S.C.; Knüchel, R.; et al. Promoter methylation of DNA damage repair (DDR) genes in human tumor entities: RBBP8/CtIP is almost exclusively methylated in bladder cancer. Clin. Epigenet. 2018, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, D.; Morra, F.; Giannella, R.; Visconti, R.; Cerrato, A.; Celetti, A. New combinatorial strategies to improve the PARP inhibitors efficacy in the urothelial bladder Cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 91. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chughtai, A.A.; Pannhausen, J.; Dinger, P.; Wirtz, J.; Knüchel, R.; Gaisa, N.T.; Eble, M.J.; Rose, M. Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR. Biomedicines 2022, 10, 1277. https://doi.org/10.3390/biomedicines10061277
Chughtai AA, Pannhausen J, Dinger P, Wirtz J, Knüchel R, Gaisa NT, Eble MJ, Rose M. Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR. Biomedicines. 2022; 10(6):1277. https://doi.org/10.3390/biomedicines10061277
Chicago/Turabian StyleChughtai, Ahmed Ali, Julia Pannhausen, Pia Dinger, Julia Wirtz, Ruth Knüchel, Nadine T. Gaisa, Michael J. Eble, and Michael Rose. 2022. "Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR" Biomedicines 10, no. 6: 1277. https://doi.org/10.3390/biomedicines10061277
APA StyleChughtai, A. A., Pannhausen, J., Dinger, P., Wirtz, J., Knüchel, R., Gaisa, N. T., Eble, M. J., & Rose, M. (2022). Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR. Biomedicines, 10(6), 1277. https://doi.org/10.3390/biomedicines10061277