
Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Protein Extraction and Western Blot Analysis
2.4. Protein Identification by Peptide Mass Fingerprinting (PMF)
2.5. Immunoprecipitation
2.6. Cells Proliferation Measurement by MTT Reagents and DAPI Staining
2.7. Recombinant GST Proteins Purification and Site-Directed Mutagenesis
2.8. Transfection of Plasmid DNA and si-RNA
2.9. Cytosolic and Nuclear Fractions Isolation
2.10. Immunostaining of Cells
2.11. Monolayer Wound Healing Assay
2.12. Chromatin Immunoprecipitation Sequencing (ChIP-Seq) and ChIP-PCR
2.13. RNA Isolation and Quantitative Reverse Transcriptase (RT-qPCR) to Measure Target Genes’ mRNA Level
2.14. Analysis of Survival Probability of Human Liver and Lung Cancer Patients
2.15. Preparation of Rat Liver by Insulin Treatment
2.16. Xenograft Experiment
2.17. Statistical Analysis
3. Results
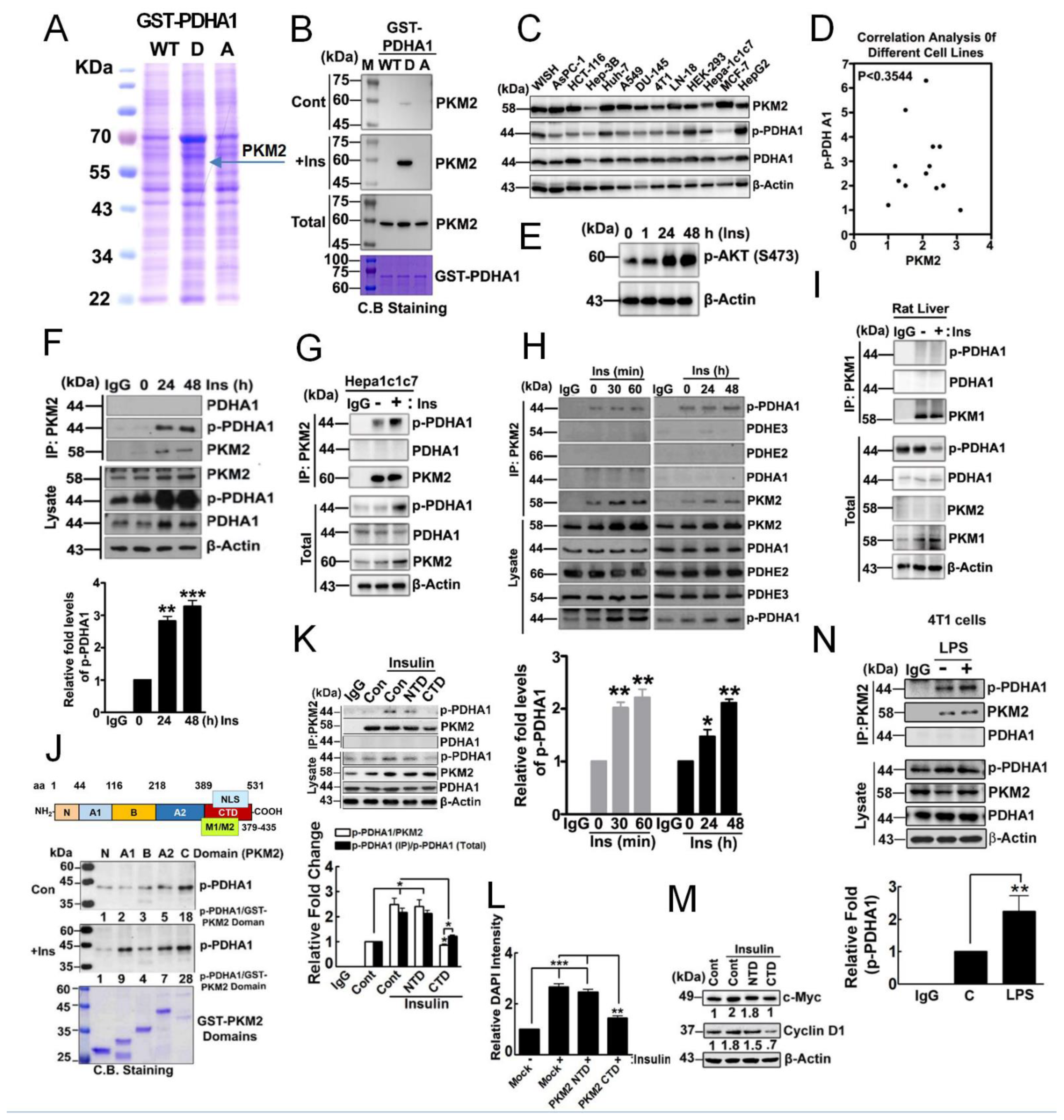
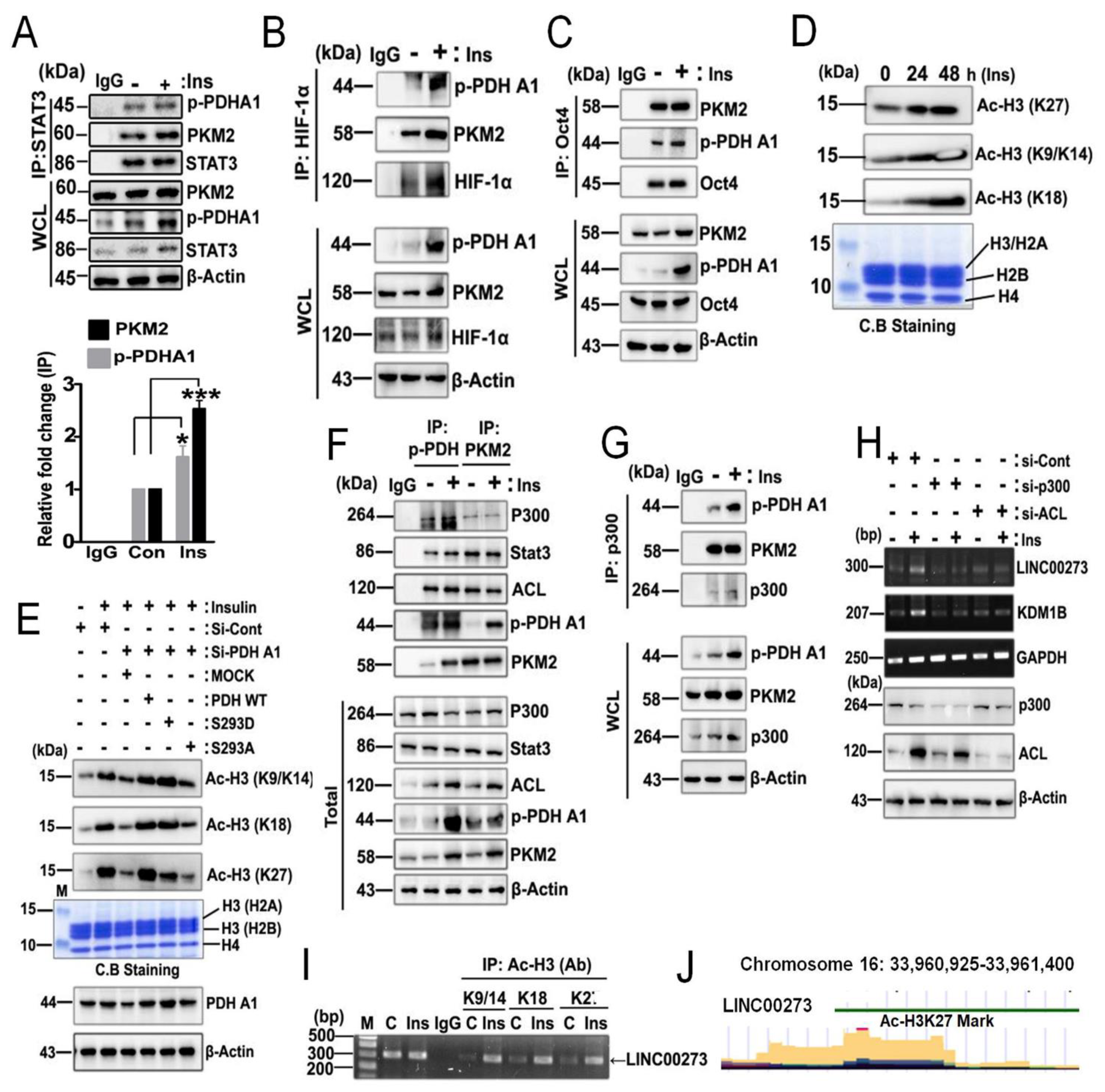
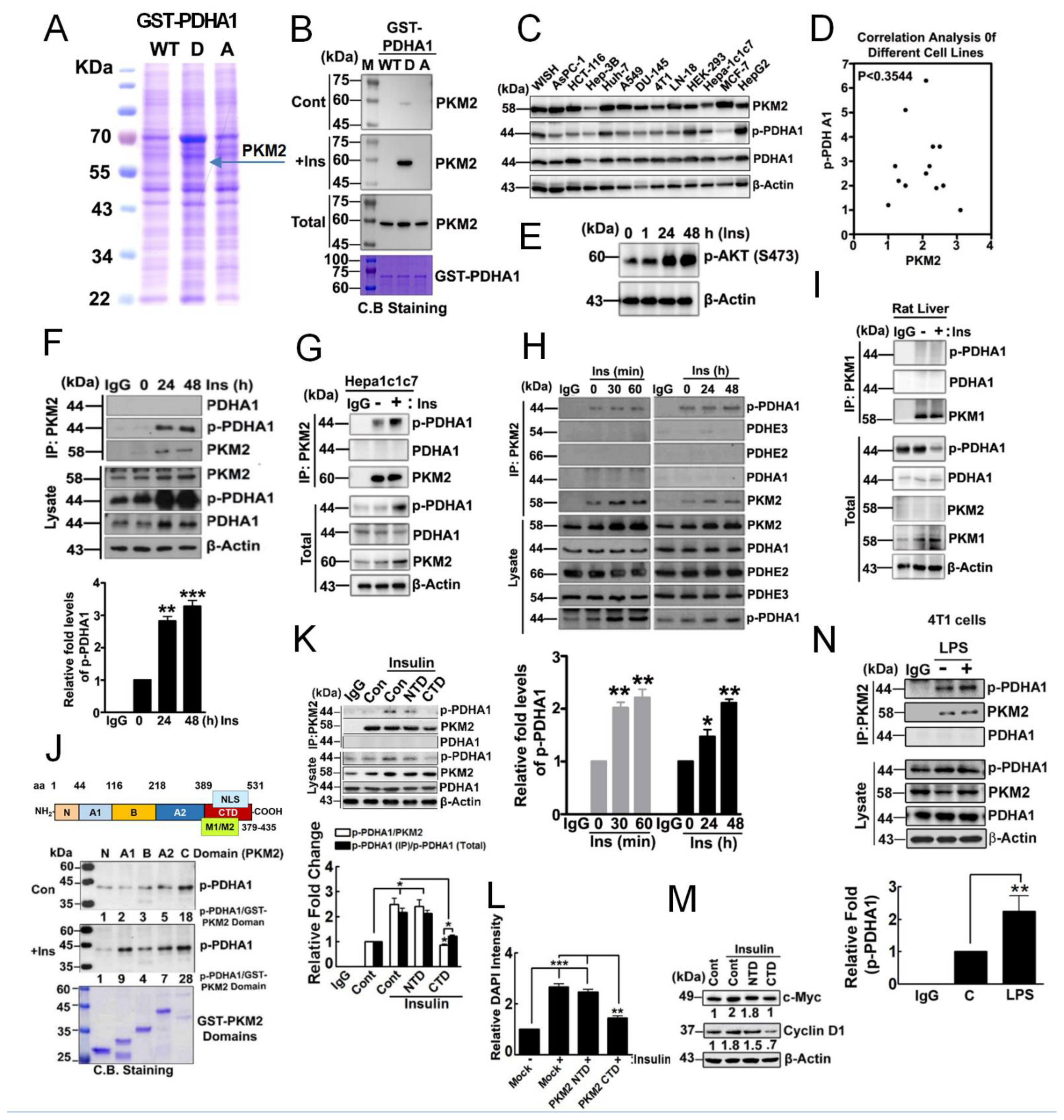
3.1. p-PDHA1 Interacts with PKM2
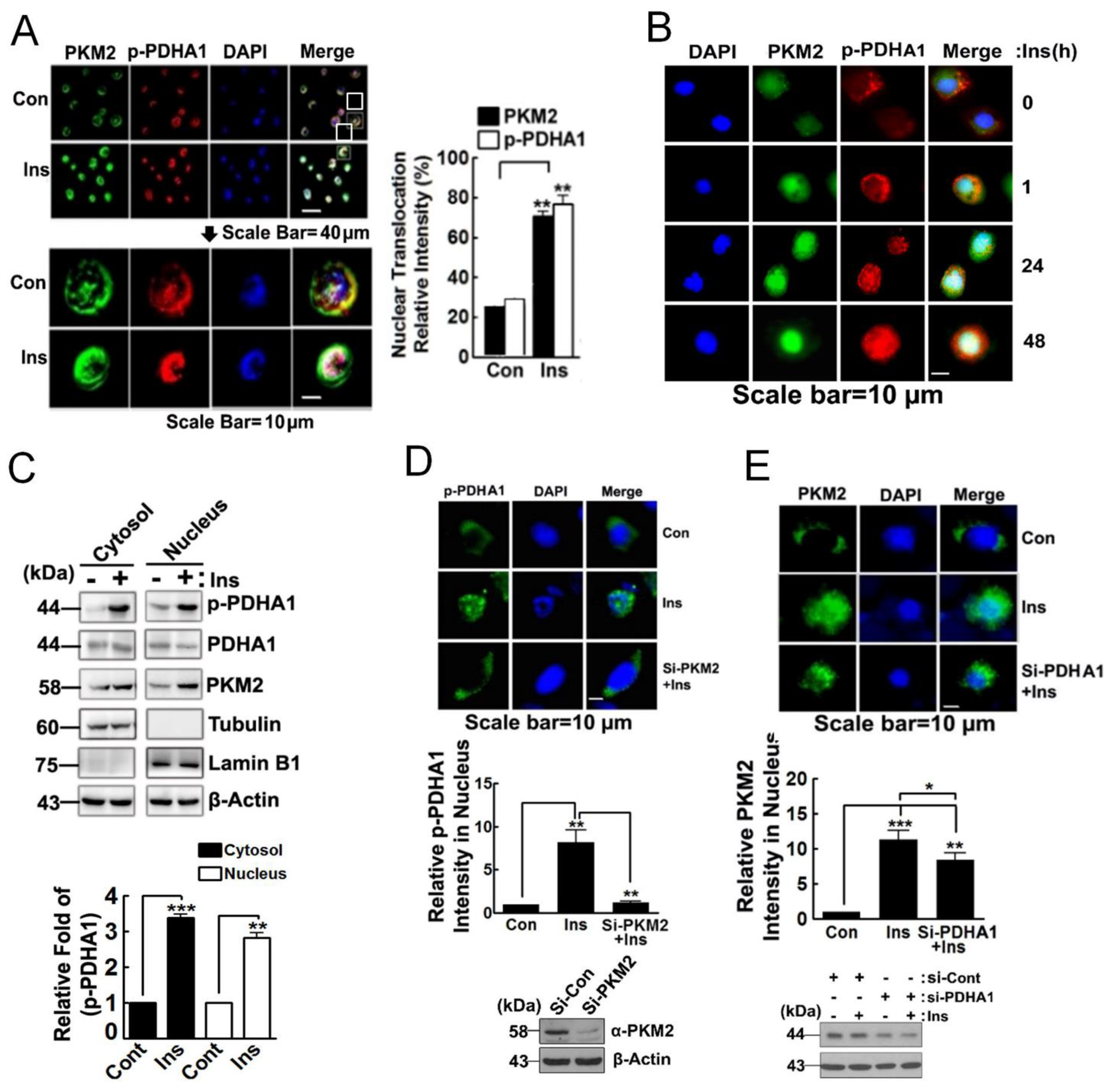
3.2. PKM2 Delivers p-PDHA1 to the Nucleus in Response to Insulin
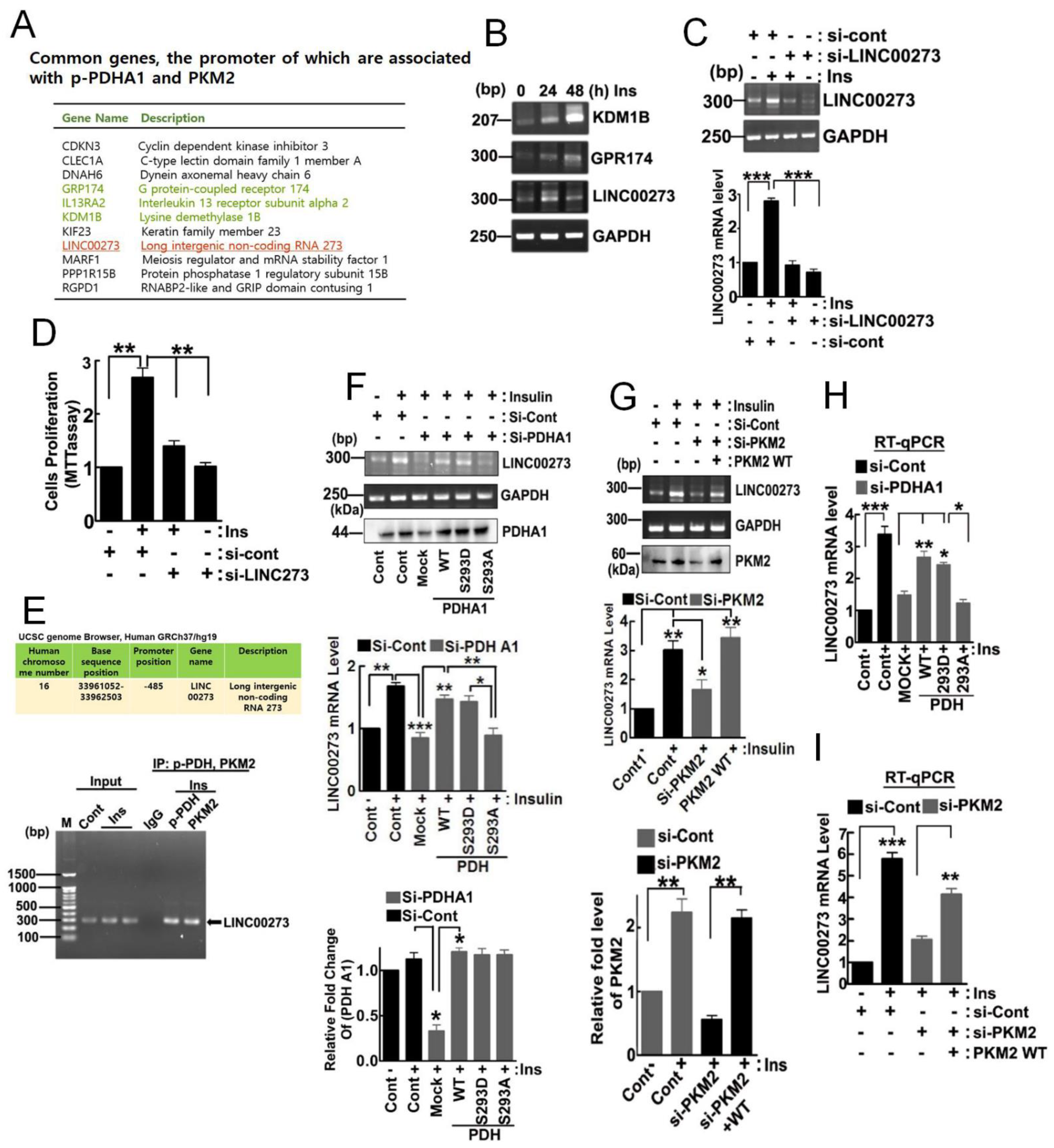
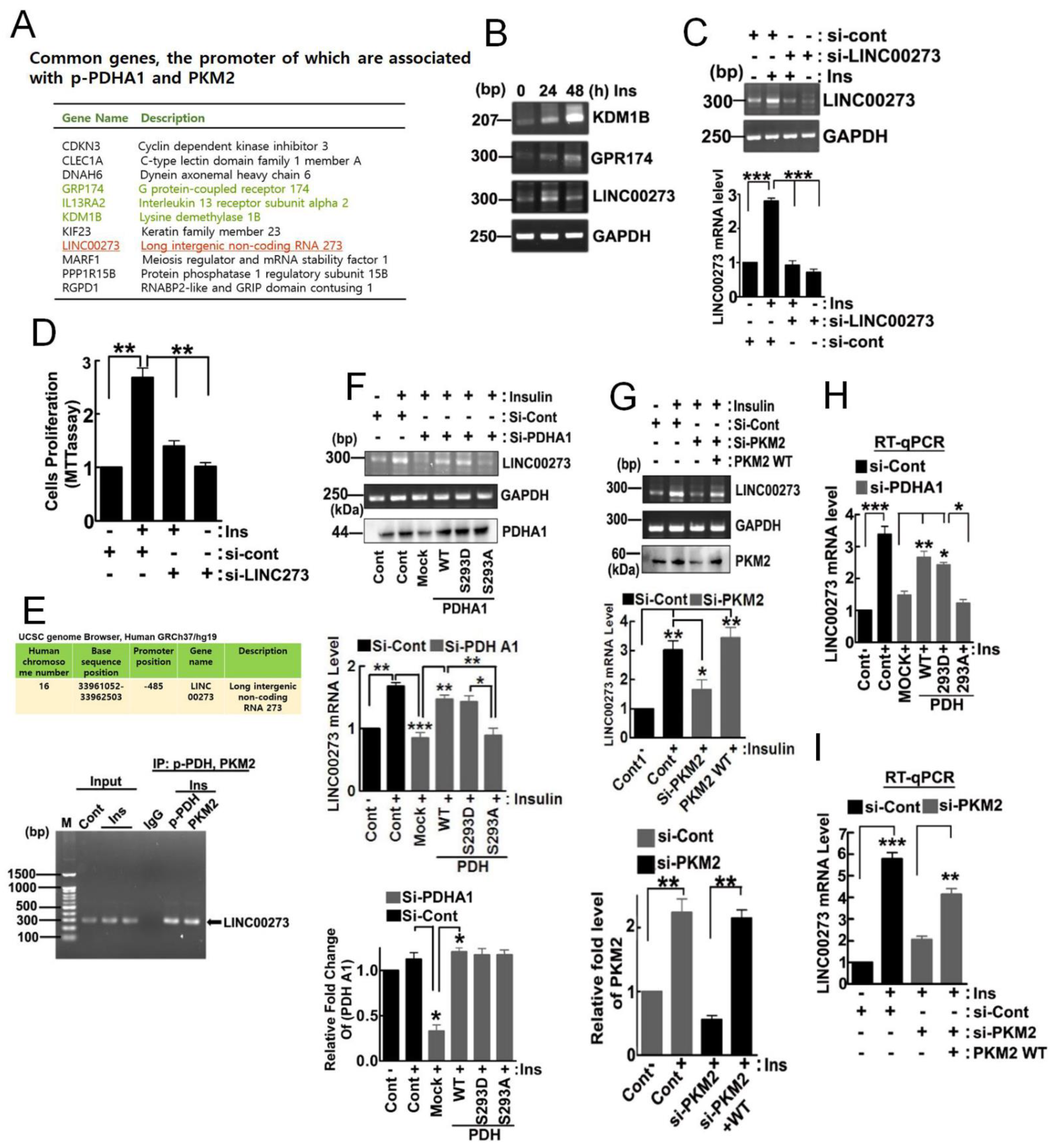
3.3. Target Genes of 1-PDHA1 and PKM2
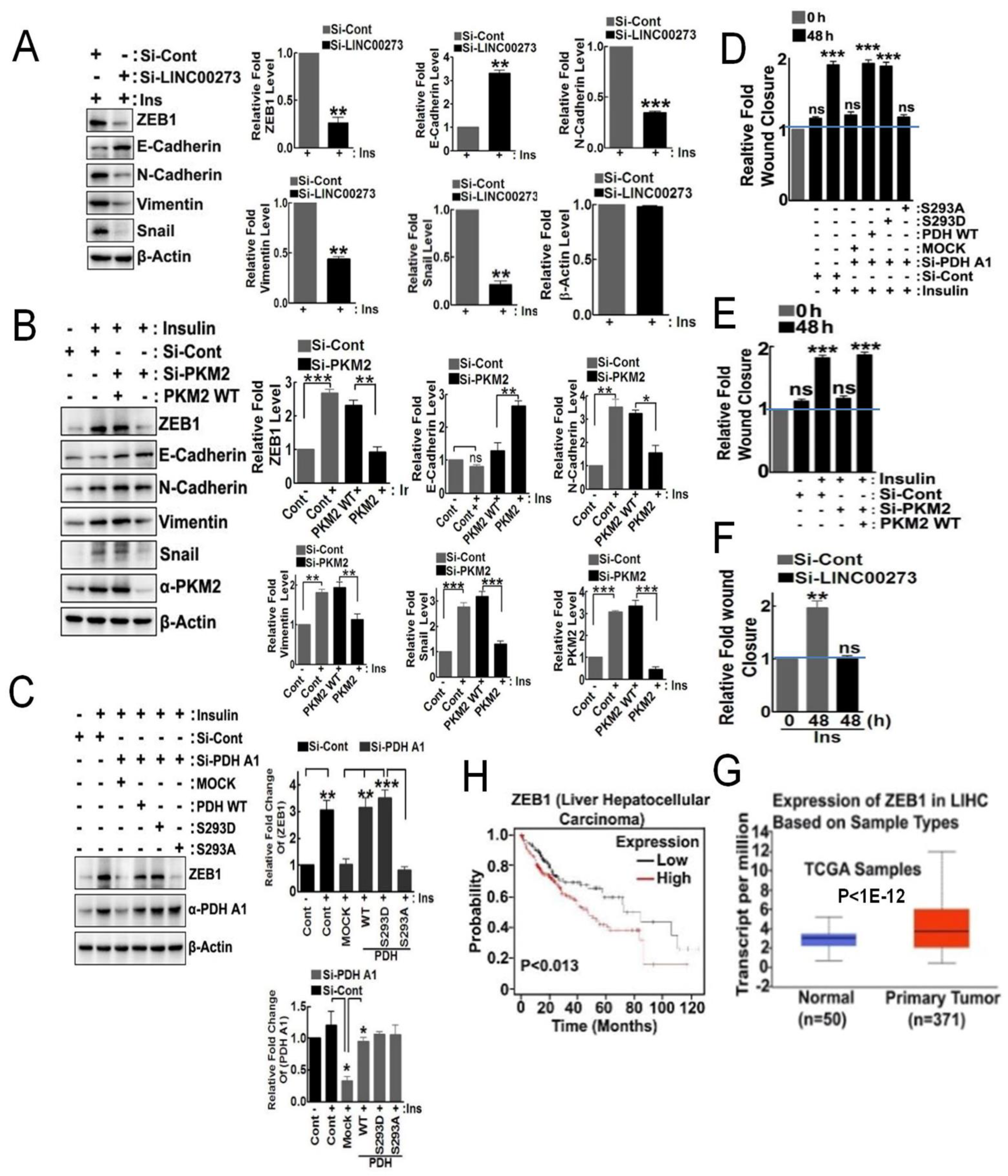
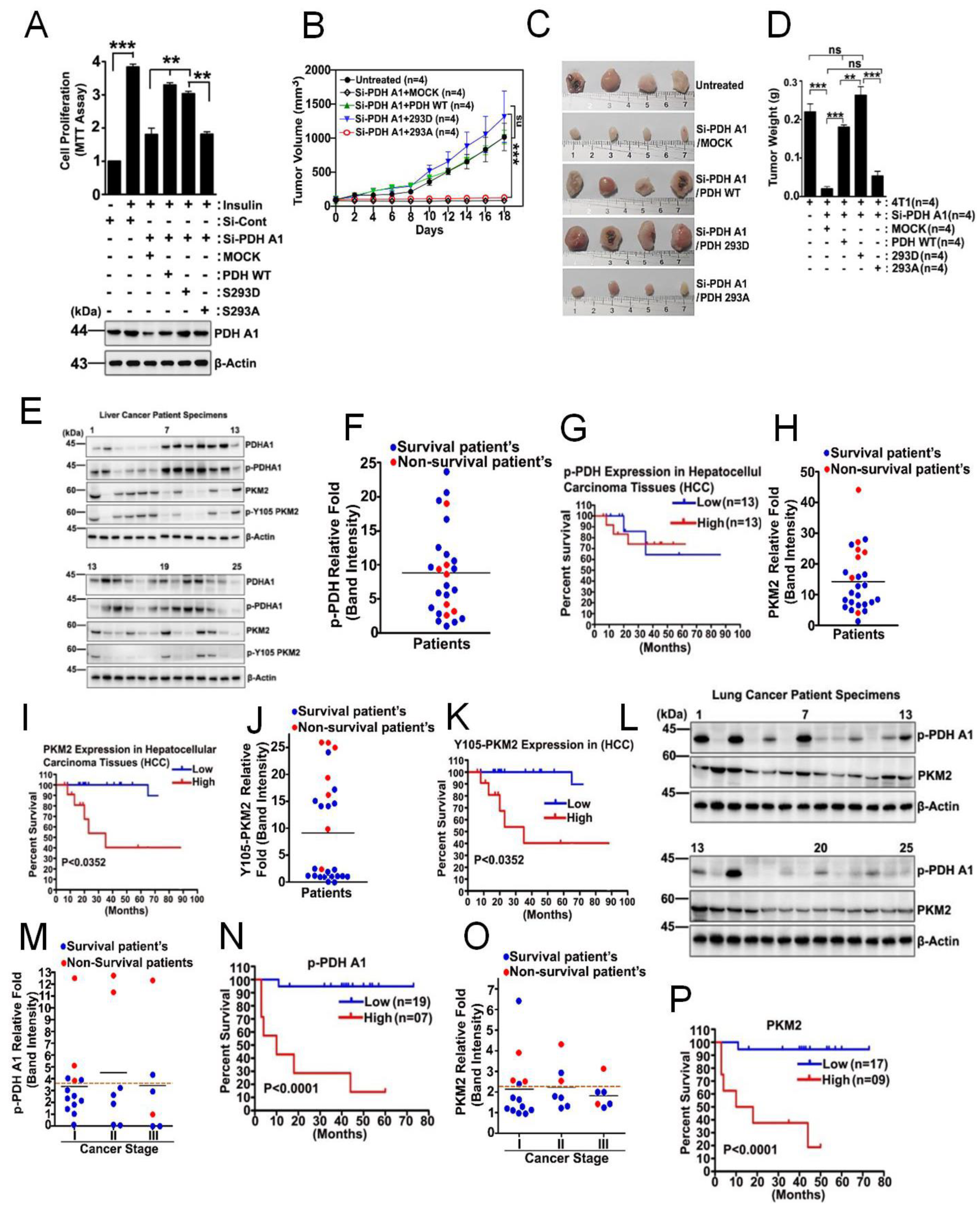
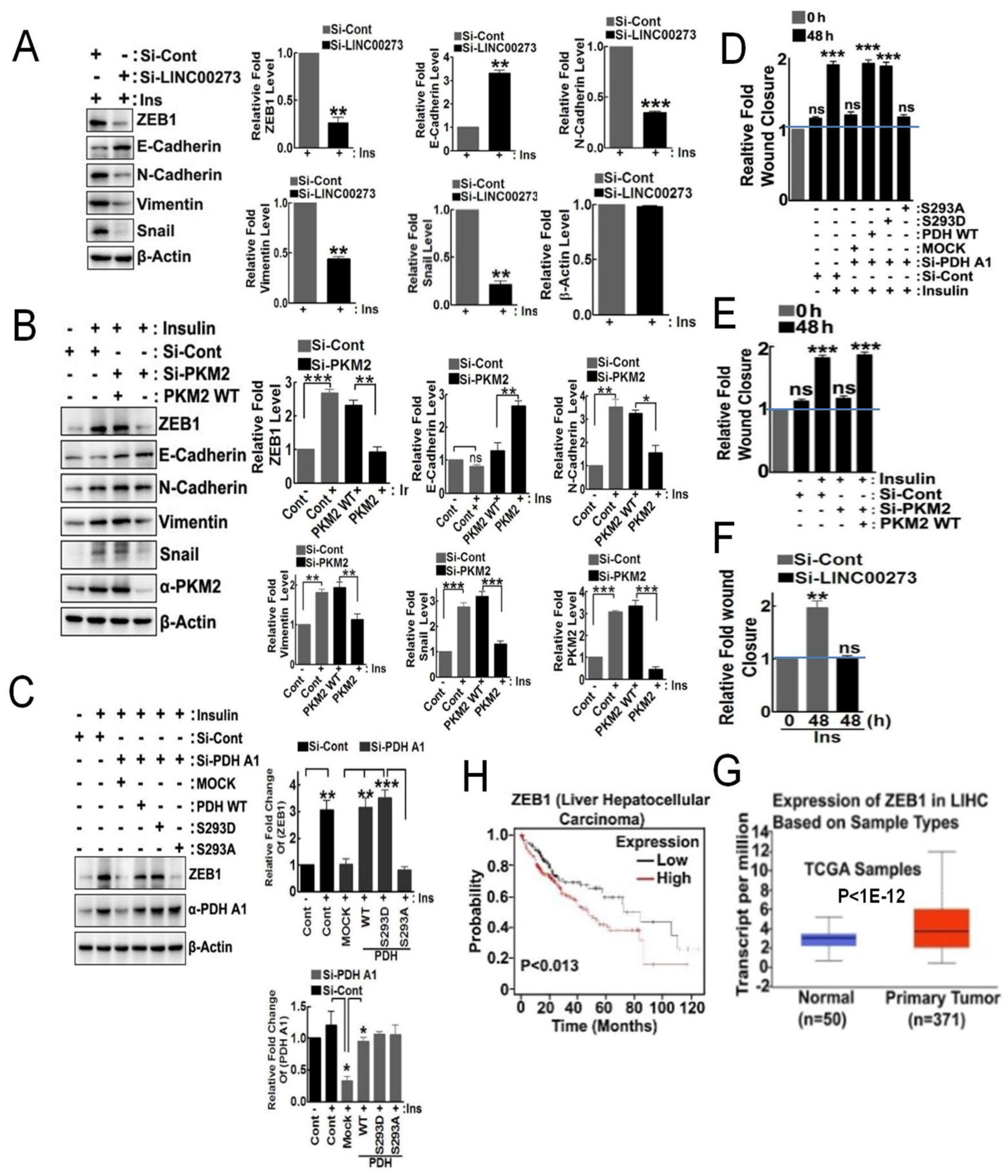
3.4. p-PDHA1 and PKM2 Induces Cell Migration through Expression of LINC00273 and ZEB1
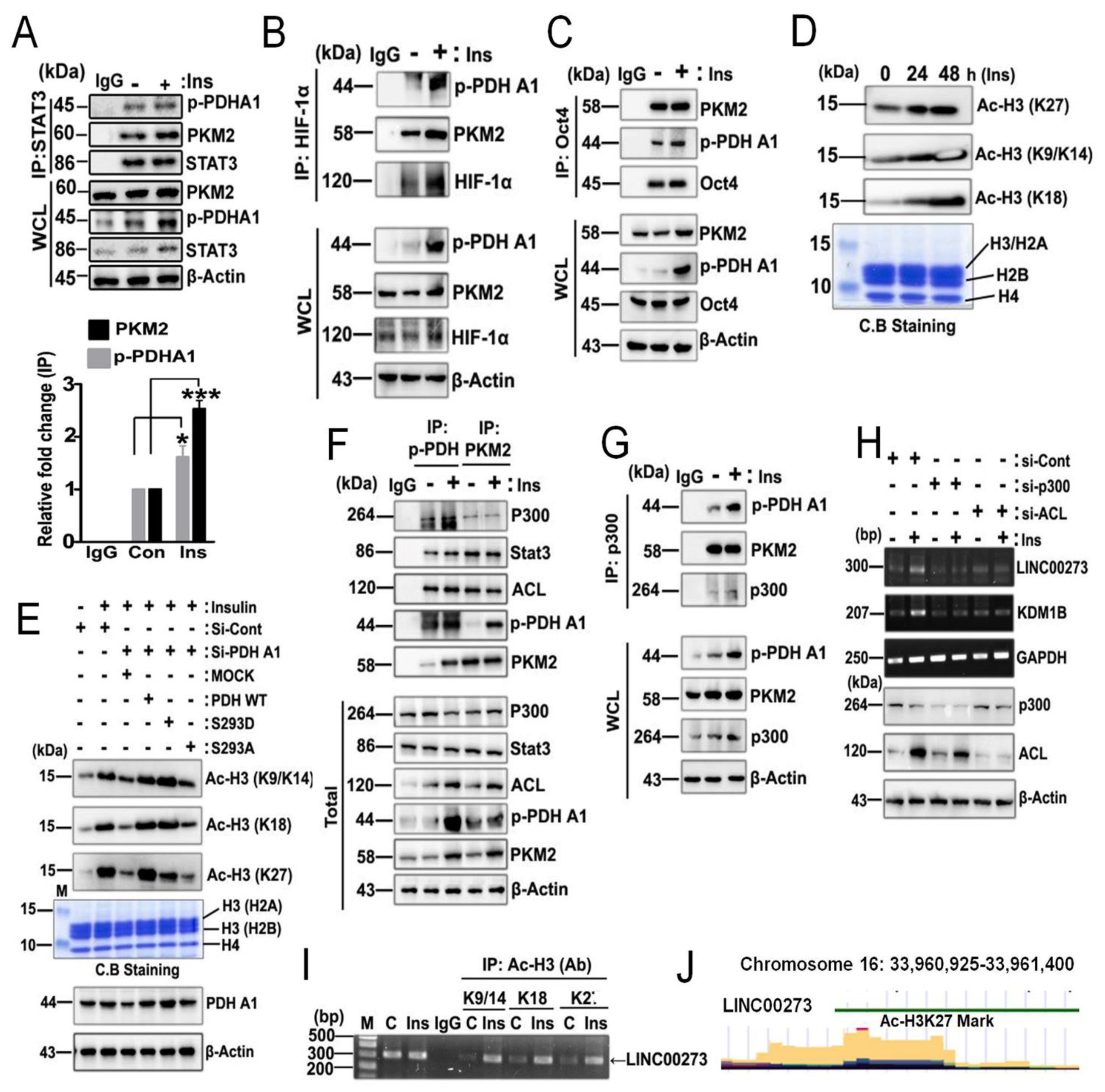
3.5. p-PDHA1/PKM2 Complex Increases Histone Acetylation in Response to Insulin
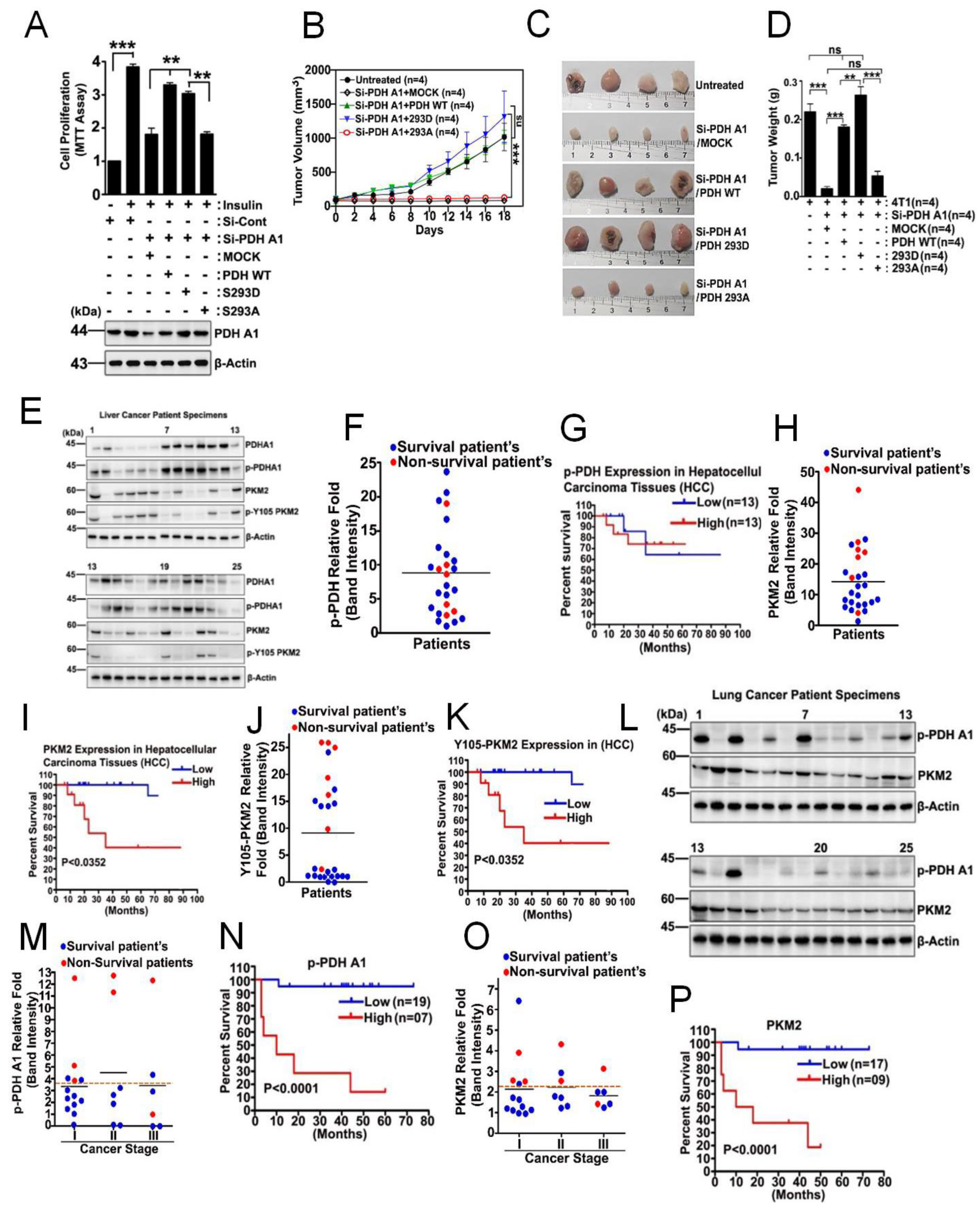
3.6. p-PDHA1 Is Relevant to Tumorigenesis in Liver
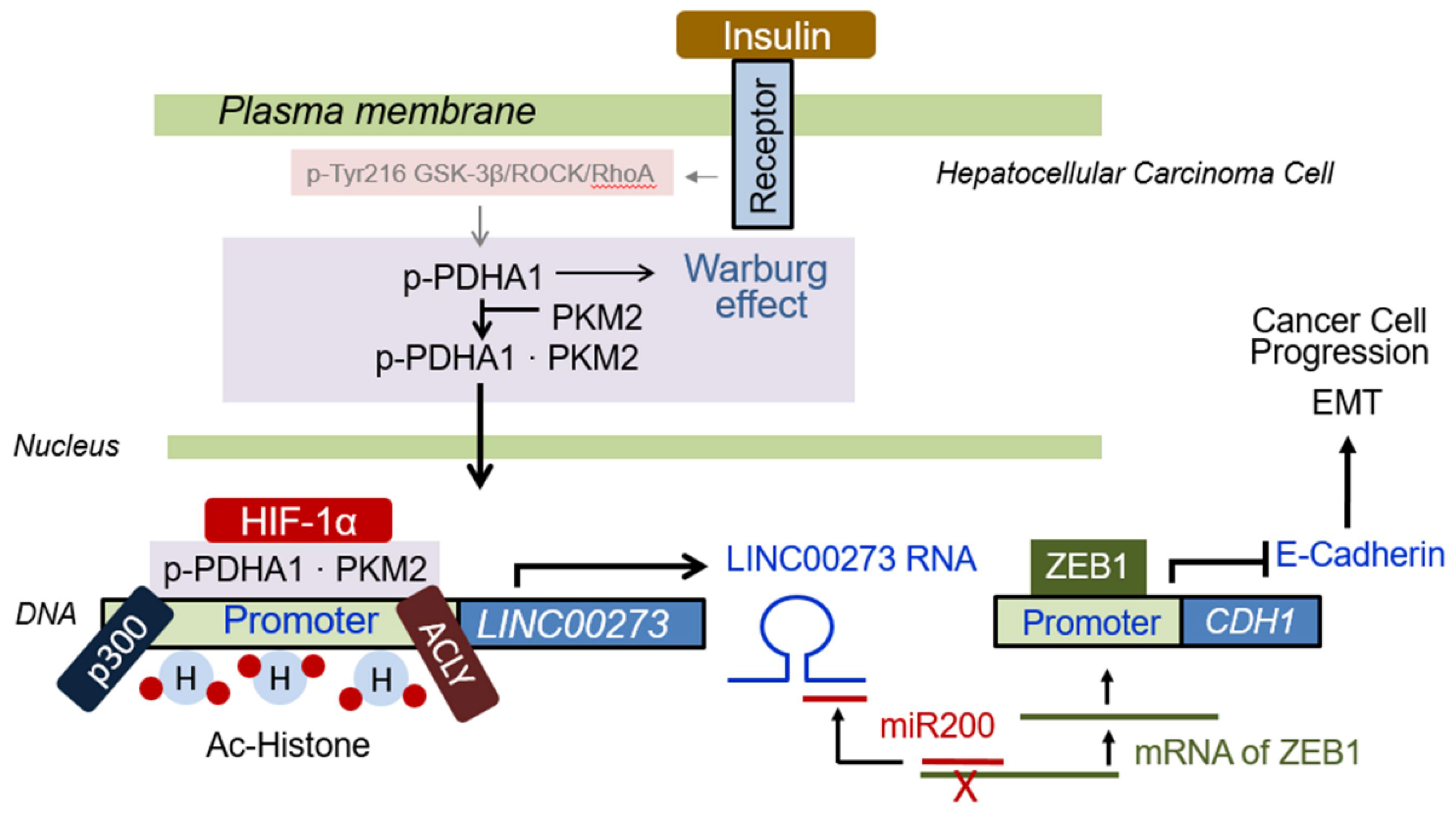
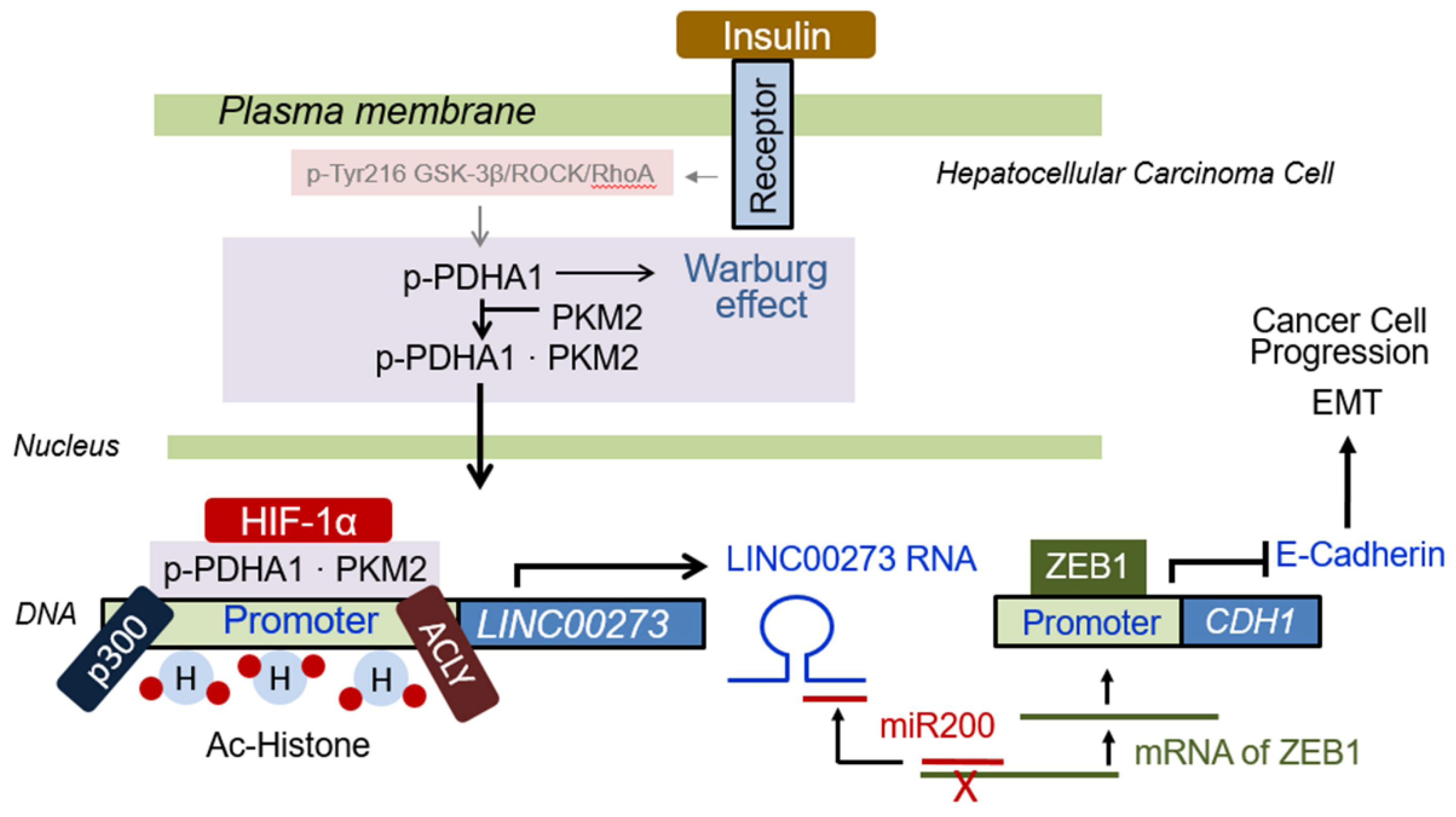
3.7. Proposed Schematic Illustration for Role of p-PDHA1/PKM2 Complex in Cancer Cells
4. Discussion
4.1. Functions of p-PDHA1/PKM2 Complex in the Nucleus
4.2. LINC00273, a Target Gene That Is Regulated by p-PDHA1 and PKM2
4.3. Elevated Levels of p-PDHA1 and PKM2 in Hepatocellular Carcinoma Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACL | ATP citrate lyase |
| EMT | Epithelial–mesenchymal transition |
| HAT | Histone acetyltransferase |
| PDH | Pyruvate dehydrogenase |
| PDHA1 | PDH E1α |
| PDC | Pyruvate dehydrogenase complex |
| GS | Glycogen synthase |
| GSK-3β | Glycogen synthase kinase-3β |
| ROCK | Rho-dependent coiled coil kinase |
| PDHK | Pyruvate dehydrogenase kinase |
| PKM2 | Pyruvate kinase M2 |
| NTD | N-terminal domain |
| CTD | C-terminal domain |
| LPS | Lipopolysaccharide |
| LDH | Lactate dehydrogenase |
| PEP | Phosphoenol pyruvate |
| LINC RNA | Long intergenic protein non-coding RNA |
References
- Clayton, P.E.; Banerjee, I.; Murray, P.G.; Renehan, A.G. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat. Rev. Endocrinol. 2011, 7, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.A.; Saltiel, A.R. Spatial compartmentalization of signal transduction in insulin action. Bioessays 2001, 23, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Noel, A.; Barrier, L.; Rinaldi, F.; Hubert, C.; Fauconneau, B.; Ingrand, S. Lithium chloride and staurosporine potentiate the accumulation of phosphorylated glycogen synthase kinase 3beta/Tyr216, resulting in glycogen synthase kinase 3beta activation in SH-SY5Y human neuroblastoma cell lines. J. Neurosci. Res. 2011, 89, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Jeon, J.H.; Min, B.K.; Ha, C.M.; Thoudam, T.; Park, B.Y.; Lee, I.K. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef]
- Li, Z.; Yang, P.; Li, Z. The multifaceted regulation and functions of PKM2 in tumor progression. Biochim. Biophys. Acta 2014, 1846, 285–296. [Google Scholar] [CrossRef]
- Lee, Y.B.; Min, J.K.; Kim, J.G.; Cap, K.C.; Islam, R.; Hossain, A.J.; Dogsom, O.; Hamza, A.; Mahmud, S.; Choi, D.R.; et al. Multiple functions of pyruvate kinase M2 in various cell types. J. Cell Physiol. 2021, 237, 128–148. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell. Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Kim, J.G.; Park, Y.; Cho, J.Y.; Cap, K.C.; Kho, A.R.; Chung, W.S.; Suh, S.W.; Park, J.B. Insulin induces phosphorylation of pyruvate dehydrogenase through RhoA activation pathway in HepG2 cells. FASEB J. 2019, 33, 2072–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.G.; Kwon, H.J.; Wu, G.; Park, Y.; Lee, J.Y.; Kim, J.; Kim, S.C.; Choe, M.; Kang, S.G.; Seo, G.Y.; et al. RhoA GTPase oxidation stimulates cell proliferation via nuclear factor-kappaB activation. Free Radic. Biol. Med. 2017, 103, 57–68. [Google Scholar] [CrossRef]
- Kim, J.G.; Mahmud, S.; Min, J.K.; Lee, Y.B.; Kim, H.; Kang, D.C.; Park, H.S.; Seong, J.; Park, J.B. RhoA GTPase phosphorylated at tyrosine 42 by src kinase binds to beta-catenin and contributes transcriptional regulation of vimentin upon Wnt3A. Redox. Biol. 2021, 40, 101842. [Google Scholar] [CrossRef]
- Fernandez, J.; Gharahdaghi, F.; Mische, S.M. Routine identification of proteins from sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels or polyvinyl difluoride membranes using matrix assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS). Electrophoresis 1998, 19, 1036–1045. [Google Scholar] [CrossRef]
- Kim, J.G.; Choi, K.C.; Hong, C.W.; Park, H.S.; Choi, E.K.; Kim, Y.S.; Park, J.B. Tyr42 phosphorylation of RhoA GTPase promotes tumorigenesis through nuclear factor (NF)-kappaB. Free Radic. Biol. Med. 2017, 112, 69–83. [Google Scholar] [CrossRef]
- Cap, K.C.; Jung, Y.J.; Choi, B.Y.; Hyeon, S.J.; Kim, J.G.; Min, J.K.; Islam, R.; Hossain, A.J.; Chung, W.S.; Suh, S.W.; et al. Distinct dual roles of p-Tyr42 RhoA GTPase in tau phosphorylation and ATP citrate lyase activation upon different Abeta concentrations. Redox. Biol. 2020, 32, 101446. [Google Scholar] [CrossRef]
- Jana, S.; Jana, J.; Patra, K.; Mondal, S.; Bhat, J.; Sarkar, A.; Sengupta, P.; Biswas, A.; Mukherjee, M.; Tripathi, S.P.; et al. LINCRNA00273 promotes cancer metastasis and its G-Quadruplex promoter can serve as a novel target to inhibit cancer invasiveness. Oncotarget 2017, 8, 110234–110256. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Rahaman, A.; Biswas, I.; Mukherjee, G.; Chatterjee, S.; Bhattacharjee, S.; Mandal, D.P. TGFbeta mediated LINC00273 upregulation sponges mir200a-3p and promotes invasion and metastasis by activating ZEB1. J. Cell Physiol. 2020, 235, 7159–7172. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.; Xu, Y.P.; Zhao, D.; Li, F.L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.T.; Guan, K.L.; et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell 2013, 52, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Chien, M.H.; Liu, H.Y.; Chang, Y.C.; Chen, C.K.; Lee, W.J.; Kuo, T.C.; Hsiao, M.; Hua, K.T.; Cheng, T.Y. Nuclear translocation of PKM2/AMPK complex sustains cancer stem cell populations under glucose restriction stress. Cancer Lett. 2018, 421, 28–40. [Google Scholar] [CrossRef]
- Morfouace, M.; Lalier, L.; Oliver, L.; Cheray, M.; Pecqueur, C.; Cartron, P.F.; Vallette, F.M. Control of glioma cell death and differentiation by PKM2-Oct4 interaction. Cell Death Dis. 2014, 5, e1036. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [Green Version]
- Freedman, S.J.; Sun, Z.Y.; Poy, F.; Kung, A.L.; Livingston, D.M.; Wagner, G.; Eck, M.J. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 5367–5372. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Banerjee, P.; Liu, X.; Yu, J.; Gibbons, D.L.; Wu, P.; Scott, K.L.; Diao, L.; Zheng, X.; Wang, J.; et al. The epithelial-to-mesenchymal transition activator ZEB1 initiates a prometastatic competing endogenous RNA network. J. Clin. Investig. 2018, 128, 1267–1282. [Google Scholar] [CrossRef] [Green Version]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef]
- Grooteclaes, M.L.; Frisch, S.M. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 2000, 19, 3823–3828. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, M.J.; Li, C.M.; Xu, Y.; An, J.; Huang, Y.; Cyster, J.G. The lysophosphatidylserine receptor GPR174 constrains regulatory T cell development and function. J. Exp. Med. 2015, 212, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Verdegaal, E.M.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011, 24, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Vasilatos, S.N.; Qin, Y.; Katz, T.A.; Cao, C.; Wu, H.; Tasdemir, N.; Levine, K.M.; Oesterreich, S.; Davidson, N.E.; et al. Functional characterization of lysine-specific demethylase 2 (LSD2/KDM1B) in breast cancer progression. Oncotarget 2017, 8, 81737–81753. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, X.; Yin, Y.; Zheng, J.T.; Jiang, C.F.; Wang, J.; Shen, H.; Li, C.Y.; Wang, M.; Liu, L.Z.; et al. Insulin regulates glucose consumption and lactate production through reactive oxygen species and pyruvate kinase M2. Oxid Med. Cell Longev. 2014, 2014, 504953. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Hunter, T. Metabolic Kinases Moonlighting as Protein Kinases. Trends Biochem. Sci. 2018, 43, 301–310. [Google Scholar] [CrossRef]
- Xu, D.; Shao, F.; Bian, X.; Meng, Y.; Liang, T.; Lu, Z. The Evolving Landscape of Noncanonical Functions of Metabolic Enzymes in Cancer and Other Pathologies. Cell Metab. 2021, 33, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, X.; Gao, P. Diabetes Mellitus and Risk of Hepatocellular Carcinoma. Biomed. Res. Int. 2017, 2017, 5202684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donadon, V.; Balbi, M.; Casarin, P.; Vario, A.; Alberti, A. Association between hepatocellular carcinoma and type 2 diabetes mellitus in Italy: Potential role of insulin. World J. Gastroenterol. 2008, 14, 5695–5700. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, A.J.; Islam, R.; Kim, J.-G.; Dogsom, O.; Cap, K.C.; Park, J.-B. Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation. Biomedicines 2022, 10, 1256. https://doi.org/10.3390/biomedicines10061256
Hossain AJ, Islam R, Kim J-G, Dogsom O, Cap KC, Park J-B. Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation. Biomedicines. 2022; 10(6):1256. https://doi.org/10.3390/biomedicines10061256
Chicago/Turabian StyleHossain, Abu Jubayer, Rokibul Islam, Jae-Gyu Kim, Oyungerel Dogsom, Kim Cuong Cap, and Jae-Bong Park. 2022. "Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation" Biomedicines 10, no. 6: 1256. https://doi.org/10.3390/biomedicines10061256
APA StyleHossain, A. J., Islam, R., Kim, J.-G., Dogsom, O., Cap, K. C., & Park, J.-B. (2022). Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation. Biomedicines, 10(6), 1256. https://doi.org/10.3390/biomedicines10061256