
Apoptosis and (in) Pain—Potential Clinical Implications

 , , and
, , and
Abstract
1. Introduction
2. Definition and Mechanisms of Pain—A Brief Review
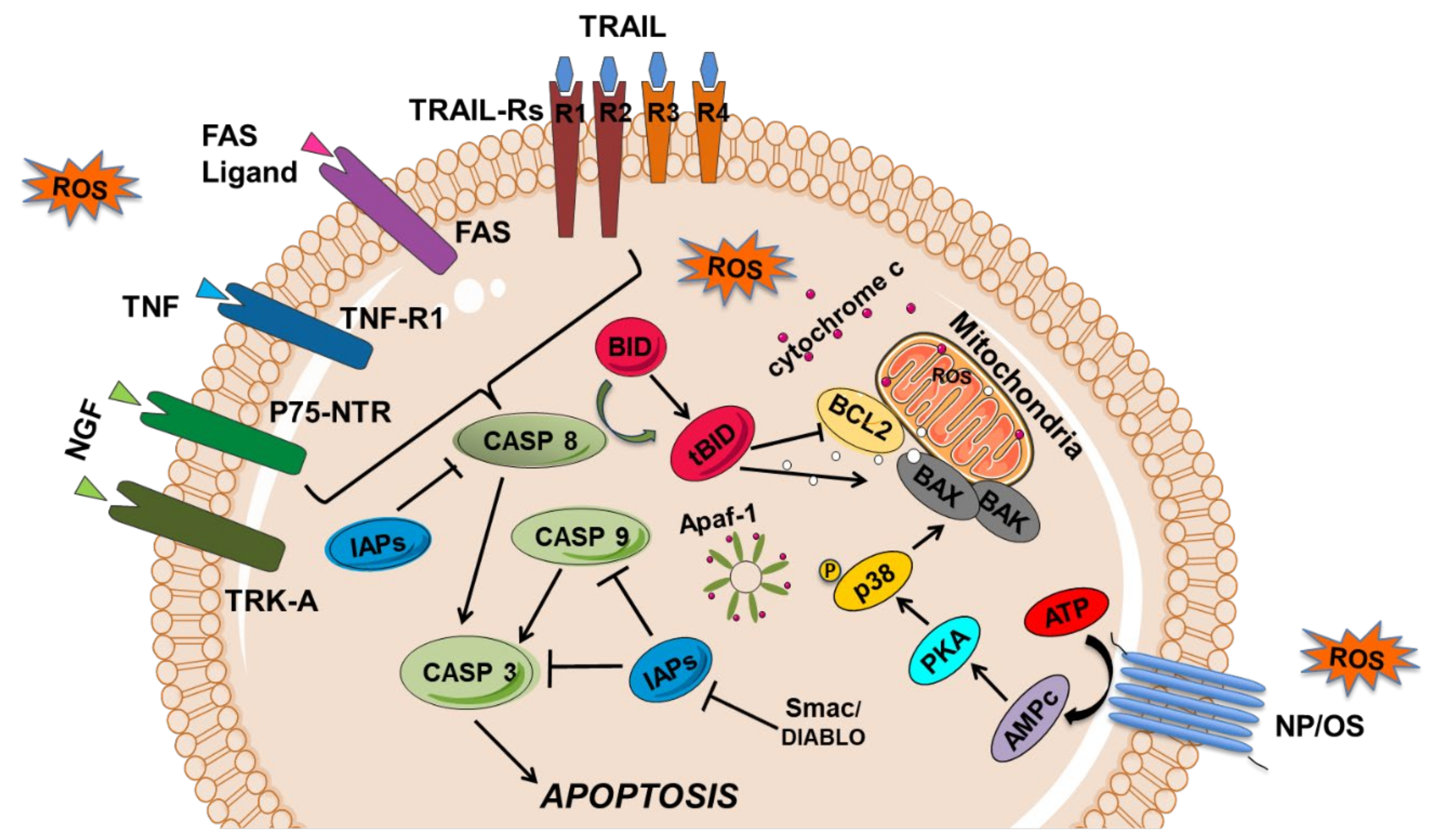
3. Apoptosis Pathways as Mediators of Pain Formation
3.1. Apoptosis Cell Signaling Pathways
3.1.1. The Extrinsic or Membrane Pathway of Apoptosis
3.1.2. The Intrinsic or Mitochondrial Apoptotic Pathway
3.2. Ubiquitin Proteasome Pathway and Apoptosis
3.3. Endoplasmic Reticulum Stress Signalling and Apoptosis
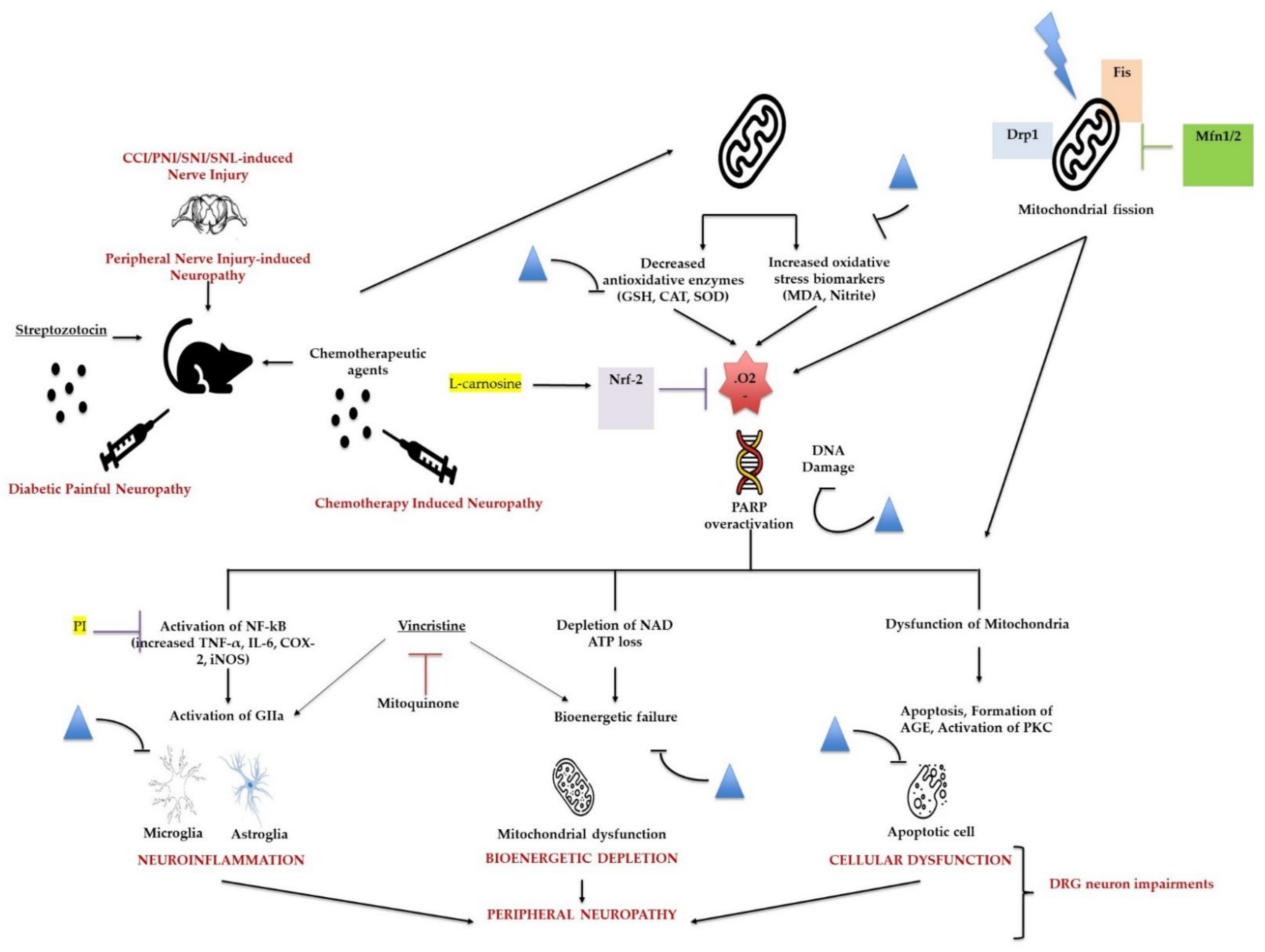
3.4. Oxidative Stress
3.5. Inflammation
3.6. The p38MAPK Pathway and Apoptosis
4. Apoptosis and Clinical Implications
4.1. Apoptosis and Neuropathic Pain
4.2. Biomarkers and Circulating Mediators in Pain
4.3. Pain Therapy through Modulating Apoptosis Activities
5. Conclusions and New Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corasaniti, M.T.; Amantea, D.; Russo, R.; Bagetta, G. The crucial role of neuronal plasticity in pain and cell death. Cell Death Differ. 2006, 13, 534–536. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.; Treede, R.-D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [PubMed]
- van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef]
- Teixeira-Santos, L.; Albino-Teixeira, A.; Pinho, D. Neuroinflammation, oxidative stress and their interplay in neuropathic pain: Focus on specialized pro-resolving mediators and NADPH oxidase inhibitors as potential therapeutic strategies. Pharmacol. Res. 2020, 162, 105280. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Sui, B.; Xu, T.-Q.; Liu, J.-W.; Wei, W.; Zheng, C.-X.; Guo, B.-L.; Wang, Y.-Y.; Yang, Y.-L. Understanding the role of mitochondria in the pathogenesis of chronic pain. Postgrad. Med. J. 2013, 89, 709–714. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 16, 17002. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic pain: From mechanisms to treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Ji, R.-R.; Nackley, A.; Huh, B.Y.; Terrando, N.; Maixner, D.W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef]
- Carrasco, C.; Naziroǧlu, M.; Rodríguez, A.B.; Pariente, J.A. Neuropathic pain: Delving into the oxidative origin and the possible implication of transient receptor potential channels. Front. Physiol. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Reichling, D.B.; Levine, J.D. Pain and death: Neurodegenerative disease mechanisms in the nociceptor. Ann. Neurol. 2011, 69, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, H.; Della Penna, K.; Benz, R.; Xu, J.; Gerhold, D.; Holder, D.; Koblan, K. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience 2002, 114, 529–546. [Google Scholar] [CrossRef]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised international association for the study of pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef]
- Nobelstiftelsen. Nobel Lectures in Physiology or Medicine; Elsevier for the Nobel Foundation: Amsterdam, The Netherlands; London, UK, 1965; pp. 1922–1941. [Google Scholar]
- Valenzuela-Moguillansky, C.; Reyes-Reyes, A.; Gaete, M.I. Exteroceptive and interoceptive body-self awareness in fibromyalgia patients. Front. Hum. Neurosci. 2017, 11, 117. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef]
- McEntire, D.M.; Kirkpatrick, D.R.; Dueck, N.P.; Kerfeld, M.J.; Smith, T.A.; Nelson, T.J.; Reisbig, M.D.; Agrawal, D.K. Pain transduction: A pharmacologic perspective. Expert Rev. Clin. Pharmacol. 2016, 9, 1069–1080. [Google Scholar] [CrossRef]
- Garcia-Larrea, L.; Peyron, R. Pain matrices and neuropathic pain matrices: A review. Pain 2013, 154, S29–S43. [Google Scholar] [CrossRef]
- Orr, P.M.; Shank, B.C.; Black, A.C. The role of pain classification systems in pain management. Crit. Care Nurs. Clin. N. Am. 2017, 29, 407–418. [Google Scholar] [CrossRef]
- Adams, J.D. Editorial: Apoptosis is critical to pain control. Open J. Apoptosis 2013, 2, 23–24. [Google Scholar] [CrossRef]
- Ogawa, N.; Kurokawa, T.; Mori, Y. Sensing of redox status by TRP channels. Cell Calcium 2016, 60, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Braidy, N. Thermo-sensitive TRP channels: Novel targets for treating chemotherapy-induced peripheral pain. Front. Physiol. 2017, 8, 1040. [Google Scholar] [CrossRef] [PubMed]
- Jardín, I.; López, J.J.; Diez, R.; Sánchez-Collado, J.; Cantonero, C.; Albarrán, L.; Woodard, G.E.; Redondo, P.C.; Salido, G.M.; Smani, T.; et al. TRPs in pain sensation. Front. Physiol. 2017, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Yowtak, J.; Wang, J.; Kim, H.Y.; Lu, Y.; Chung, K.; Chung, J.M. Effect of antioxidant treatment on spinal GABA neurons in a neuropathic pain model in the mouse. Pain 2013, 154, 2469–2476. [Google Scholar] [CrossRef]
- Mai, L.; Huang, F.; Zhu, X.; He, H.; Fan, W. Role of nerve growth factor in orofacial pain. J. Pain Res. 2020, 13, 1875–1882. [Google Scholar] [CrossRef]
- Braidy, N.; Smani, T.; Naziroglu, M. Editorial: Involvements of TRP channels, oxidative stress and apoptosis in neurodegenerative diseases. Front. Physiol. 2021, 12, 649230. [Google Scholar] [CrossRef]
- Duitama, M.; Vargas-López, V.; Casas, Z.; Albarracin, S.L.; Sutachan, J.-J.; Torres, Y.P. TRP channels role in pain associated with neurodegenerative diseases. Front. Neurosci. 2020, 14, 782. [Google Scholar] [CrossRef]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 Contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef]
- Carniglia, L.; Ramírez, D.; Durand, D.; Saba, J.; Turati, J.; Caruso, C.; Scimonelli, T.N.; Lasaga, M. Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediat. Inflamm. 2017, 2017, 5048616. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.R.; Wen, Y.-R.; Decosterd, I.; Ji, R.-R. Do glial cells control pain? Neuron Glia Biol. 2007, 3, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Areti, A.; Yerra, V.G.; Naidu, V.; Kumar, A. Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Sun, B.; Liu, Z.; Yao, X.; Wang, H.; Shen, X.; Jiang, H.; Chen, J. Advanced oxidative protein products cause pain hypersensitivity in rats by inducing dorsal root ganglion neurons apoptosis via NADPH Oxidase 4/c-Jun N-terminal kinase pathways. Front. Mol. Neurosci. 2017, 10, 195. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-F.; Lu, K.-T.; Hsu, J.-L.; Lee, C.-H.; Cheng, M.-Y.; Ro, L.-S. The role of autophagy and apoptosis in neuropathic pain formation. Int. J. Mol. Sci. 2022, 23, 2685. [Google Scholar] [CrossRef]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef]
- Duke, R.C.; Ojcius, D.M.; Young, J.D.-E. Cell suicide in health and disease. Sci. Am. 1996, 275, 80–87. [Google Scholar] [CrossRef]
- Thompson, C.B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Sekiguchi, Y.; Konno, S.-I.; Kobayashi, H.; Homma, Y.; Kikuchi, S.-I. Comparison of neuropathic pain and neuronal apoptosis following nerve root or spinal nerve compression. Eur. Spine J. 2009, 18, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, T.; Ni, H.; Liu, Q.; An, K.; Tao, J.; Chen, Y.; Xu, L.S.; Zhu, C.; Yao, M.; et al. Endoplasmic reticulum stress promoting caspase signaling pathway-dependent apoptosis contributes to bone cancer pain in the spinal dorsal horn. Mol. Pain 2019, 15, 1744806919876150. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E.; Kischkel, F.C.; Hellbardt, S.; Chinnaiyan, A.M.; Krammer, P.H.; Dixit, V.M. CD95 (APO-1/Faz)—associating signalling proteins. Cell Death Differ. 1996, 3, 161–170. [Google Scholar] [PubMed]
- Lewin, G.R.; Nykjaer, A. Pro-neurotrophins, sortilin, and nociception. Eur. J. Neurosci. 2014, 39, 363–374. [Google Scholar] [CrossRef]
- Deng, Y.; Yang, L.; Xie, Q.; Yang, F.; Li, G.; Zhang, G.; Li, S.; Wu, Z.; Wang, J.; Kang, X. Protein kinase a is involved in neuropathic pain by activating the p38MAPK pathway to mediate spinal cord cell apoptosis. Mediat. Inflamm. 2020, 2020, 6420425-17. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Liu, X.-H.; Kwon, D.; Schielke, G.P.; Yang, G.-Y.; Silverstein, F.S.; Barks, J.D.E. Mice deficient in interleukin-1 converting enzyme are resistant to neonatal hypoxic-ischemic brain damage. J. Cereb. Blood Flow Metab. 1999, 19, 1099–1108. [Google Scholar] [CrossRef]
- Sarmento-Ribeiro, A.B.; Dourado, M.; Paiva, A.; Freitas, A.; Silva, T.; Regateiro, F.; Oliveira, C.R. Apoptosis deregulation influences chemoresistance to azaguanine in human leukemic cell lines. Cancer Investig. 2012, 30, 331–342. [Google Scholar] [CrossRef]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef]
- Martins, L.M.; Earnshaw, W.C. Apoptosis: Alive and kicking in 1997. Trends Cell Biol. 1997, 7, 111–114. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Green, D.R. Apoptotic Pathways. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Li, B.; Dou, Q.P. Bax degradation by the ubiquitin/proteasome-dependent pathway: Involvement in tumor survival and progression. Proc. Natl. Acad. Sci. USA 2000, 97, 3850–3855. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Deng, Y.; Zhou, J. Role of the ubiquitin system in chronic pain. Front. Mol. Neurosci. 2021, 14, 674914. [Google Scholar] [CrossRef] [PubMed]
- Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Ubiquitin-mediated degradation of the proapoptotic active form of bid. J. Biol. Chem. 2000, 275, 21648–21652. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Wang, J.; Yang, X.; Hsu, H.-C.; Mountz, J.D. Regulation of apoptosis proteins in cancer cells by ubiquitin. Oncogene 2004, 23, 2009–2015. [Google Scholar] [CrossRef]
- Qiu, J.H.; Asai, A.; Chi, S.; Saito, N.; Hamada, H.; Kirino, T. Proteasome inhibitors induce cytochrome c–caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J. Neurosci. 2000, 20, 259–265. [Google Scholar] [CrossRef]
- George, D.E.; Tepe, J.J. Advances in proteasome enhancement by small molecules. Biomolecules 2021, 11, 1789. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Q.; Cheng, H.; Yan, G.; Xing, C. Upregulation of ubiquitin conjugating enzyme E2B (Ube2b) ameliorates neuropathic pain by regulating Kcna2 (potassium voltage-gated channel subfamily A member 2) in primary afferent neurons. Bioengineered 2021, 12, 7470–7480. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.S.; Berg, S.; Alkass, K.; Druid, H.; Hart, D.A.; Svensson, C.I.; Kosek, E. NF-κB-Associated pain-related neuropeptide expression in patients with degenerative disc disease. Int. J. Mol. Sci. 2019, 20, 658. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.E.; Eskew, O.; Wong, T.; Tchivileva, I.E.; Oladosu, F.A.; O’Buckley, S.C.; Nackley, A.G. Nuclear factor-kappa B regulates pain and COMT expression in a rodent model of inflammation. Brain Behav. Immun. 2015, 50, 196–202. [Google Scholar] [CrossRef]
- Niederberger, E.; Geisslinger, G. The IKK-NF-κB pathway: A source for novel molecular drug targets in pain therapy? FASEB J. 2008, 22, 3432–3442. [Google Scholar] [CrossRef]
- Fakhri, S.; Abbaszadeh, F.; Jorjani, M. On the therapeutic targets and pharmacological treatments for pain relief following spinal cord injury: A mechanistic review. Biomed. Pharmacother. 2021, 139, 111563. [Google Scholar] [CrossRef]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol. 2012, 86, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J. The contribution of mitochondria to sensory processing and pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 119–146. [Google Scholar] [CrossRef]
- Bennett, G.J.; Doyle, T.; Salvemini, D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat. Rev. Neurol. 2014, 10, 326–336. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Lim, T.; Rone, M.B.; Lee, S.; Antel, J.P.; Zhang, J. Mitochondrial and bioenergetic dysfunction in trauma-induced painful peripheral neuropathy. Mol. Pain 2015, 11, 58. [Google Scholar] [CrossRef]
- English, K.; Barton, M.C. HDAC6: A key link between mitochondria and development of peripheral neuropathy. Front. Mol. Neurosci. 2021, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Todorova, V.; Blokland, A. Mitochondria and synaptic plasticity in the mature and aging nervous system. Curr. Neuropharmacol. 2016, 15, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Bittar, A.; Jun, J.; La, J.-H.; Wang, J.; Leem, J.; Chung, J.M. Reactive oxygen species affect spinal cell type-specific synaptic plasticity in a model of neuropathic pain. Pain 2017, 158, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Wu, F.; Sun, X.; Xiang, Z.; Yang, L.; Huang, S.; Lu, Z.; Sun, Y.; Yu, W.-F. Intrathecal infusion of hydrogen-rich normal saline attenuates neuropathic pain via inhibition of activation of spinal astrocytes and microglia in rats. PLoS ONE 2014, 9, e97436. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Wang, H.; Han, B.; Zhang, L.; Guo, J. Tempol attenuates neuropathic pain by inhibiting nitric oxide production. Anal. Cell. Pathol. 2019, 2019, 8253850. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Kim, H.K.; Chung, J.M.; Chung, K. Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain 2007, 131, 262–271. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, I.; Chun, S.W. Reactive oxygen species donors increase the responsiveness of dorsal horn neurons and induce mechanical hyperalgesia in rats. Neural Plast. 2015, 2015, 293423. [Google Scholar] [CrossRef]
- Muscoli, C.; Dagostino, C.; Ilari, S.; Lauro, F.; Gliozzi, M.; Bardhi, E.; Palma, E.; Mollace, V.; Salvemini, D. Posttranslational nitration of tyrosine residues modulates glutamate transmission and contributes to N-Methyl-D-aspartate-Mediated thermal hyperalgesia. Mediat. Inflamm. 2013, 2013, 950947. [Google Scholar] [CrossRef]
- Son, Y.-K.; Cheong, N.-H.; Kim, H.-T.; Chung, D.; Kang, G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 1–6. [Google Scholar] [CrossRef]
- Shapovalov, G.; Lehen’Kyi, V.; Skryma, R.; Prevarskaya, N. TRP channels in cell survival and cell death in normal and transformed cells. Cell Calcium 2011, 50, 295–302. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, R.; Lazcano, P.; Ferrari, F.; Pinto-Pardo, N.; González-Billault, C.; Utreras, E. TNF-α increases production of reactive oxygen species through Cdk5 activation in nociceptive neurons. Front. Physiol. 2018, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Averitt, D.L.; Maier, C.; Basu, A. The effects of nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2) activation in preclinical models of peripheral neuropathic pain. Antioxidants 2022, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Staurengo-Ferrari, L.; Badaro-Garcia, S.; Hohmann, M.S.N.; Manchope, M.F.; Zaninelli, T.; Casagrande, R.; Verri, W. Contribution of Nrf2 modulation to the mechanism of action of analgesic and anti-inflammatory drugs in pre-clinical and clinical stages. Front. Pharmacol. 2019, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Haanen, C.; Vermes, I. Apoptosis and inflammation. Mediat. Inflamm. 1995, 4, 5–15. [Google Scholar] [CrossRef]
- Wyllie, A.; Kerr, J.; Currie, A. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [CrossRef]
- Wallach, D.; Kovalenko, A. Keeping inflammation at bay. eLife 2014, 3, e02583. [Google Scholar] [CrossRef]
- Haslett, C.; Savill, J.S.; Whyte, M.K.B.; Stern, M.; Dransfield, I.; Meagher, L.C.; Nagata, S. Granulocyte apoptosis and the control of inflammation. Philos. Trans. R. Soc. B: Biol. Sci. 1994, 345, 327–333. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K. Switching from apoptosis to pyroptosis: Gasdermin-elicited inflammation and antitumor immunity. Int. J. Mol. Sci. 2021, 22, 426. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.P.; Dransfield, I.; Rossi, A.G. Phagocytosis of apoptotic cells. Methods 2008, 44, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.E.; Yuan, J.; Horvitz, H.R. Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 1991, 7, 663–698. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.J. Apoptosis. Immunol. Today 1993, 14, 126–130. [Google Scholar] [CrossRef]
- Arienti, S.; Barth, N.; Dorward, D.A.; Rossi, A.G.; Dransfield, I. Regulation of apoptotic cell clearance during resolution of inflammation. Front. Pharmacol. 2019, 10, 891. [Google Scholar] [CrossRef]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef]
- Martin, S.; Reutelingsperger, C.E.M.; McGahon, A.J.; Rader, J.; Van Schie, R.C.A.A.; LaFace, D.M.; Green, D. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef]
- Huynh, M.-L.N.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef]
- Segawa, K.; Nagata, S. an apoptotic ‘eat me’ signal: Phosphatidylserine exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef]
- Ravichandran, K.S.; Lorenz, U. Engulfment of apoptotic cells: Signals for a good meal. Nat. Rev. Immunol. 2007, 7, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Maruyama, T.; Urade, Y.; Nagata, S. Immunosuppression via adenosine receptor activation by adenosine monophosphate released from apoptotic cells. eLife 2014, 3, e02172. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Sarang, Z.; Kiss, B.; Garabuczi, É.; Köröskényi, K. Anti-inflammatory mechanisms triggered by apoptotic cells during their clearance. Front. Immunol. 2017, 8, 909. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.; Ravichandran, K. Clearing the Dead: Apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748. [Google Scholar] [CrossRef] [PubMed]
- Bournazou, I.; Pound, J.D.; Duffin, R.; Bournazos, S.; Melville, L.A.; Brown, S.B.; Rossi, A.G.; Gregory, C.D. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J. Clin. Investig. 2008, 119, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Riwaldt, S.; Corydon, T.J.; Pantalone, D.; Sahana, J.; Wise, P.; Wehland, M.; Krüger, M.; Melnik, D.; Kopp, S.; Infanger, M.; et al. Role of apoptosis in wound healing and apoptosis alterations in microgravity. Front. Bioeng. Biotechnol. 2021, 9, 498. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, G.; Zhang, P.; Fan, J. Programmed cell death and its role in inflammation. Mil. Med. Res. 2015, 2, 12. [Google Scholar] [CrossRef]
- Bosurgi, L.; Hughes, L.D.; Rothlin, C.V.; Ghosh, S. Death begets a new beginning. Immunol. Rev. 2017, 280, 8–25. [Google Scholar] [CrossRef]
- Poon, I.; Lucas, C.; Rossi, A.G.; Ravichandran, K. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Pierini, R.; Juruj, C.; Perret, M.; Jones, C.L.; Mangeot, P.; Weiss, D.S.; Henry, T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012, 19, 1709–1721. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Serrano-del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef]
- Faouzi, S.; Burckhardt, B.E.; Hanson, J.C.; Campe, C.B.; Schrum, L.W.; Rippe, R.A.; Maher, J.J. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-κB-independent, caspase-3-dependent pathway. J. Biol. Chem. 2001, 276, 49077–49082. [Google Scholar] [CrossRef]
- Fujimoto, I.; Pan, J.; Takizawa, T.; Nakanishi, Y. Virus clearance through apoptosis-dependent phagocytosis of influenza a virus-infected cells by macrophages. J. Virol. 2000, 74, 3399–3403. [Google Scholar] [CrossRef]
- Simon, H.-U. Targeting apoptosis in the control of inflammation. Eur. Respir. J. 2003, 22, 20s–21s. [Google Scholar] [CrossRef]
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171. [Google Scholar] [CrossRef]
- Schoenberger, J.; Bauer, J.; Moosbauer, J.; Eilles, C.; Grimm, D. Innovative strategies in in vivo apoptosis imaging. Curr. Med. Chem. 2008, 15, 187–194. [Google Scholar] [CrossRef]
- Kasuya, Y.; Umezawa, H.; Hatano, M. Stress-activated protein kinases in spinal cord injury: Focus on roles of p38. Int. J. Mol. Sci. 2018, 19, 867. [Google Scholar] [CrossRef]
- Shin, J.; Yin, Y.; Park, H.; Park, S.; Triantafillu, U.L.; Kim, Y.; Kim, S.R.; Lee, S.Y.; Kim, D.K.; Hong, J.; et al. p38 siRNA-encapsulated PLGA nanoparticles alleviate neuropathic pain behavior in rats by inhibiting microglia activation. Nanomedicine 2018, 13, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-Q.; Liu, S.; He, D.-D.; Liu, Y.-P.; Song, X.-J. Activation of the cAMP-PKA signaling pathway in rat dorsal root ganglion and spinal cord contributes toward induction and maintenance of bone cancer pain. Behav. Pharmacol. 2014, 25, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Materazzi, S.; Fusi, C.; Benemei, S.; Pedretti, P.; Patacchini, R.; Nilius, B.; Prenen, J.; Creminon, C.; Geppetti, P.; Nassini, R. TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflügers Arch. Eur. J. Physiol. 2012, 463, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Kahya, M.C.; Nazıroğlu, M.; Övey, I.S. Modulation of diabetes-induced oxidative stress, apoptosis, and ca2+ entry through TRPM2 and TRPV1 channels in dorsal root ganglion and hippocampus of diabetic rats by melatonin and selenium. Mol. Neurobiol. 2016, 54, 2345–2360. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.; Patte-Mensah, C.; Eckert, A.; Mensah-Nyagan, A.G. Sciatic nerve injury induces apoptosis of dorsal root ganglion satellite glial cells and selectively modifies neurosteroidogenesis in sensory neurons. Glia 2009, 58, 169–180. [Google Scholar] [CrossRef]
- Wiberg, R.; Novikova, L.N.; Kingham, P. Evaluation of apoptotic pathways in dorsal root ganglion neurons following peripheral nerve injury. NeuroReport 2018, 29, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, L.D.C.; Ghelardini, C.; Calvani, M.; Nicolai, R.; Mosconi, L.; Toscano, A.; Pacini, A.; Bartolini, A. Neuroprotective effects of acetyl-L-carnitine on neuropathic pain and apoptosis: A role for the nicotinic receptor. J. Neurosci. Res. 2008, 87, 200–207. [Google Scholar] [CrossRef]
- Campana, W.M.; Myers, R.R. Exogenous erythropoietin protects against dorsal root ganglion apoptosis and pain following peripheral nerve injury. Eur. J. Neurosci. 2003, 18, 1497–1506. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, S.; Hu, L.; Gu, Y.-F.; Cai, Y.; Wu, D.; Liu, W.-T.; Jiang, C.-Y.; Kong, X.; Zhang, G.-Q. Inhibition of apoptosis signal-regulating kinase by paeoniflorin attenuates neuroinflammation and ameliorates neuropathic pain. J. Neuroinflammation 2019, 16, 83. [Google Scholar] [CrossRef]
- Pi, Z.; Lin, H.; Yang, J. Isoflurane reduces pain and inhibits apoptosis of myocardial cells through the phosphoinositide 3-kinase/protein kinase B signaling pathway in mice during cardiac surgery. Mol. Med. Rep. 2018, 17, 6497–6505. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Drummond, J.C.; Cole, D.J.; Kelly, P.J.; Spurlock, M.P.; Patel, P.M. Effect of isoflurane on neuronal apoptosis in rats subjected to focal cerebral ischemia. Anesthesia Analg. 2004, 98, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Dong, Y.; Wu, X.; Zhang, J.; McAuliffe, S.; Pan, C.; Zhang, Y.; Ichinose, F.; Yue, Y.; Xie, Z. The potential dual effects of anesthetic isoflurane on Aβ-induced apoptosis. Curr. Alzheimer Res. 2011, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.-P.; Zhang, H.; Wang, W.; Wei, Y.-Y.; Zhai, M.-Z.; Wang, W.; Xu, L.-X.; Li, Y.-Q. Inhibition of spinal astrocytic c-Jun N-terminal kinase (JNK) activation correlates with the analgesic effects of ketamine in neuropathic pain. J. Neuroinflammation 2011, 8, 6. [Google Scholar] [CrossRef]
- Tatsumi, E.; Yamanaka, H.; Kobayashi, K.; Yagi, H.; Sakagami, M.; Noguchi, K. RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. Glia 2014, 63, 216–228. [Google Scholar] [CrossRef]
- Yamazaki, M.; Sakuma, T.; Kato, K.; Furuya, T.; Koda, M. Granulocyte colony-stimulating factor reduced neuropathic pain associated with thoracic compression myelopathy: Report of two cases. J. Spinal Cord Med. 2013, 36, 40–43. [Google Scholar] [CrossRef][Green Version]
- Leng, Y.-F.; Gao, X.-M.; Wang, S.-X.; Xing, Y.-H. Effects of tetramethylpyrazine on neuronal apoptosis in the superficial dorsal horn in a rat model of neuropathic pain. Am. J. Chin. Med. 2012, 40, 1229–1239. [Google Scholar] [CrossRef]
- Ito, S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef]
- Ahmed, A.; Ahmed, M.; Li, J.; Gu, H.F.; Bakalkin, G.; Stark, A.; Harris, H.E. Proteasome inhibitor MG132 modulates inflammatory pain by central mechanisms in adjuvant arthritis. Int. J. Rheum. Dis. 2014, 20, 25–32. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective targeting of TNF receptors as a novel therapeutic approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Chen, X.-J.; Wang, L.; Song, X.-Y. Mitoquinone alleviates vincristine-induced neuropathic pain through inhibiting oxidative stress and apoptosis via the improvement of mitochondrial dysfunction. Biomed. Pharmacother. 2020, 125, 110003. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.K.; Levine, J.D. Caspase signalling in neuropathic and inflammatory pain in the rat. Eur. J. Neurosci. 2004, 20, 2896–2902. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Broom, D.C.; Youn, D.-H.; Mills, C.D.; Kohno, T.; Suter, M.R.; Moore, K.A.; Decosterd, I.; Coggeshall, R.E.; Woolf, C.J. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina ii of the dorsal horn after peripheral nerve injury. J. Neurosci. 2005, 25, 7317–7323. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, C.; Marchena, A.M.; Holguín-Arévalo, M.S.; Martín-Partido, G.; Rodríguez, A.B.; Paredes, S.D.; Pariente, J.A. Anti-inflammatory effects of melatonin in a rat model of caerulein-induced acute pancreatitis. Cell Biochem. Funct. 2013, 31, 585–590. [Google Scholar] [CrossRef]
- Carrasco, C.; Rodríguez, A.B.; Pariente, J.A. Effects of melatonin on the oxidative damage and pancreatic antioxidant defenses in cerulein-induced acute pancreatitis in rats. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 442–446. [Google Scholar] [CrossRef]
- Basu, P.; Basu, A. In vitro and in vivo effects of flavonoids on peripheral neuropathic pain. Molecules 2020, 25, 1171. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Liu, F.-W.; Ren, F.; Nie, Z. Neuroprotective effect corilagin in spinal cord injury rat model by inhibiting nuclear factor-kb, inflammation and apoptosis. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 41–48. [Google Scholar] [CrossRef]
- Urdzikova, L.M.; Kárová, K.; Ruzicka, J.; Kloudova, A.; Shannon, C.; Dubisova, J.; Murali, R.; Kubinová; Syková, E.; Jhanwar-Uniyal, M.; et al. The anti-inflammatory compound curcumin enhances locomotor and sensory recovery after spinal cord injury in rats by immunomodulation. Int. J. Mol. Sci. 2015, 17, 49. [Google Scholar] [CrossRef]
- Meng, F.X.; Hou, J.M.; Sun, T.S. Effect of oxidative stress induced by intracranial iron overload on central pain after spinal cord injury. J. Orthop. Surg. Res. 2017, 12, 24. [Google Scholar] [CrossRef]
- Renno, W.M.; Al-Khaledi, G.; Mousa, A.; Karam, S.M.; Abul, H.; Asfar, S. (−)-Epigallocatechin-3-gallate (EGCG) modulates neurological function when intravenously infused in acute and, chronically injured spinal cord of adult rats. Neuropharmacology 2014, 77, 100–119. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, L.; Li, N.; Mai, N.; Zhang, Y.; Pan, S. Geraniol promotes functional recovery and attenuates neuropathic pain in rats with spinal cord injury. Can. J. Physiol. Pharmacol. 2017, 95, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.C.; Lee, J.Y.; Lim, E.J.; Baik, H.H.; Oh, T.H.; Yune, T.Y. Inhibition of ROS-induced p38MAPK and ERK activation in microglia by acupuncture relieves neuropathic pain after spinal cord injury in rats. Exp. Neurol. 2012, 236, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Coronel, M.; Labombarda, F.; De Nicola, A.; González, S. Progesterone reduces the expression of spinal cyclooxygenase-2 and inducible nitric oxide synthase and prevents allodynia in a rat model of central neuropathic pain. Eur. J. Pain 2013, 18, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Crown, E.D.; Gwak, Y.S.; Ye, Z.; Johnson, K.M.; Hulsebosch, C.E. Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp. Neurol. 2008, 213, 257–267. [Google Scholar] [CrossRef]
- Pecze, L.; Viskolcz, B.; Oláh, Z. Molecular surgery concept from bench to bedside: A focus on TRPV1+ pain-sensing neurons. Front. Physiol. 2017, 8, 378. [Google Scholar] [CrossRef]
- Uslusoy, F.; Nazıroğlu, M.; Çiğ, B. Inhibition of the TRPM2 and TRPV1 channels through hypericum perforatum in sciatic nerve injury-induced rats demonstrates their key role in apoptosis and mitochondrial oxidative stress of sciatic nerve and dorsal root ganglion. Front. Physiol. 2017, 8, 335. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drugs | Administration and Time of the Experiment | Main Results | Model | Localization of Changes | References |
|---|---|---|---|---|---|
| Apocynin | Intrathecal lumbar injection; studied 42 days after injury | ↓ NP, mechanical sensitivity | Animal model of SCI—Rat | Behavioral testing only | [145] |
| Corilagin | IP injection for 4 weeks after injury | ↓ Oxidative stress, NF-ƘB, ↓ TNF-α, IL6 ↓ Caspase 3, ↓ Pain | Animal model of SCI—Rat | Spinal cord | [148] |
| Curcumin | IP injection for 28 days after injury | ↓ IL-2, NF-ƘB, TNF-α ↓ Pain | Animal model of SCI—Rat | Spinal cord | [149] |
| Deferoxamine | IP injection on the first day after injury, followed by once a week during 12 weeks | ↓ Oxidative stress ↓ Iron overload markers ↓ Pain | Animal model of SCI—Rat | Hind limb sensory cortex, hippocampus, thalamus | [150] |
| Epigallocatechin 3-gallate | IV infusion for 36 h after 4 h post-lesion (acute) and after 12 months (chronic) | ↓ Glial activity ↓ NP ↓ Lesion size area ↑ Number of spinal neurons | Animal model of SCI—Rat | Spinal cord | [151] |
| Geraniol | IV infusion 6h after lesion; evaluation at 4 weeks and 8 weeks | ↓TNF-α, ↓NMDAR1 ↓ Caspase 3, glial activation ↑ HO-1, Nrf2. ↓ NP | Animal model of SCI—Rat | Spinal cord | [152] |
| Acupuncture | Once a day on 31 to 33 d after lesion | ↓ Inflammation inhibitor, ROS scavenger, ↓ ERK inhibitor, ↓ p38MAPK inhibitor ↓ NP | Animal model of SCI—Rat | Spinal cord (L4–L5) | [153] |
| Progesterone | Daily sc injections (6 h, 1 day, 28 days after injury) | ↓ NF-ƘB, COX-2, iNOS ↓ NP | Animal model of SCI—Rat | Spinal cord (L4–L5) | [154] |
| SB203580 (P38MAPK antagonist) | 35 days Intraspinal injection in L4/L5 space | SCI ↑-P38MAPK ↓ P38MAPK, NP with antagonist | Animal model of SCI—Rat | Spinal cord (dorsal horn neurons) | [155] |
| Resiniferatoxin (molecular neurosurgery) | One time | ↓ TRPV1 ↓ NP, cancer pain | Clinical trial phase 2 | [156] | |
| Hypericum perforatum | Gastric gavage for 4 weeks | ↓ TRPV1, TRPM8, ROS ↓ NP | Animal model of CCI injury of the sciatic nerve—Rat | Sciatic nerve and DRG | [157] |
| VAS2870 (NADPH oxidase inhibitor) Roscovitine (CdK5 activity inhibitor) | 24 h 1 h | ↓NOX1, NOX2/NADPH oxidase complex, TNF-α ↓Inflammatory pain | Primary culture of mouse nociceptive neurons | Trigeminal ganglia, DRG, and HEK293 cells | [83] |
| Isoflurane (low concentration) | One time (6 h or 30 min) | ↑ PI3K/AKT activation ↑Cytosolic calcium levels | Cell line Mice primary neurons | Mice primary neurons H4 human neuroglioma cells | [134] |
| SP600125 (JNK inhibitor) | One time | AOPP ↑ JNK, and ↑ DRG neurons apoptosis by activating caspase 3 and PARP-1. ↓ by SP600125 | Cell culture Rat model | Rat DRG neurons | [34] |
| Paeoniflorin (ASK1 inhibitor) | One time | ↓ ASK1, glial cells, and neuroinflammation, ↓ ↓ ↓ TNF-α, IL-1 | Animal model of CCI injury of the sciatic nerve—Rat | Spinal dorsal horn | [131] |
| G-CSF | One time | ↑ MOR, autophagic activity, BCL-2 | Clinical trial | [137]] | |
| Tetramethylpyrazine | One time (3, 7 and 14 days post-surgery) | ↑ BCL-2 ↓ Caspase-3 expression ↓ NP | Animal model of CCI injury of the sciatic nerve—Rat | Superficial spinal dorsal horn (L4–L5)—laminae I and II | [138] |
| MG132 (Proteasome inhibitor) | One time | ↓ Activation of NF-κB ↓ Inflammatory pain | Rat model of rheumatoid arthritis and SCI | DRG | [140] |
| Mitoquinone | One time | ↓ Caspase-3 and BAX ↑ BCL-2 ↓Vincristine-induced neuropathic pain | Vincristine-induced neuropathic pain model in ICR mice Primary neuron culture of DRG | Lumbar (L4–L5) DRG Spinal cord (L4–L5) Sciatic nerve | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, H.; Sarmento-Ribeiro, A.B.; Andrade, J.P.; Dourado, M. Apoptosis and (in) Pain—Potential Clinical Implications. Biomedicines 2022, 10, 1255. https://doi.org/10.3390/biomedicines10061255
Ribeiro H, Sarmento-Ribeiro AB, Andrade JP, Dourado M. Apoptosis and (in) Pain—Potential Clinical Implications. Biomedicines. 2022; 10(6):1255. https://doi.org/10.3390/biomedicines10061255
Chicago/Turabian StyleRibeiro, Hugo, Ana Bela Sarmento-Ribeiro, José Paulo Andrade, and Marília Dourado. 2022. "Apoptosis and (in) Pain—Potential Clinical Implications" Biomedicines 10, no. 6: 1255. https://doi.org/10.3390/biomedicines10061255
APA StyleRibeiro, H., Sarmento-Ribeiro, A. B., Andrade, J. P., & Dourado, M. (2022). Apoptosis and (in) Pain—Potential Clinical Implications. Biomedicines, 10(6), 1255. https://doi.org/10.3390/biomedicines10061255