
Maximization of the Minicircle DNA Vaccine Production Expressing SARS-CoV-2 RBD

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
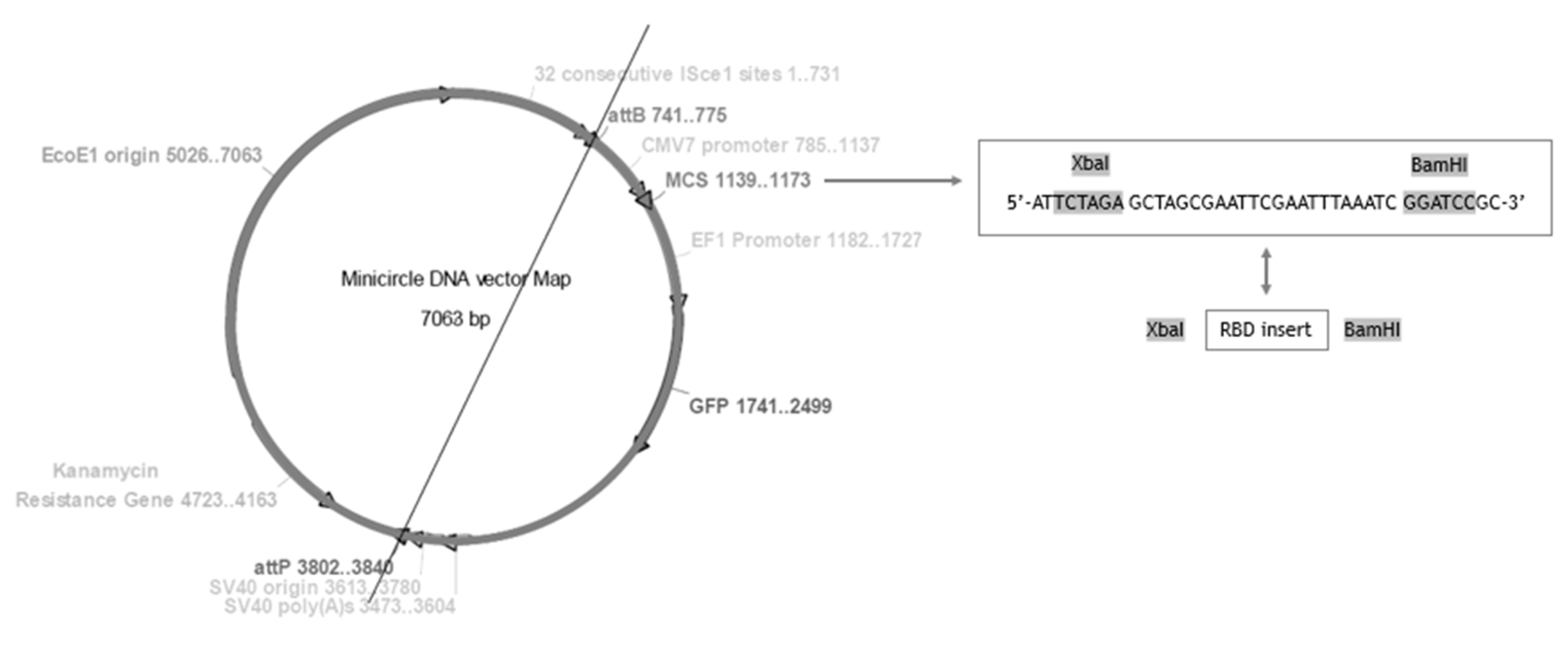
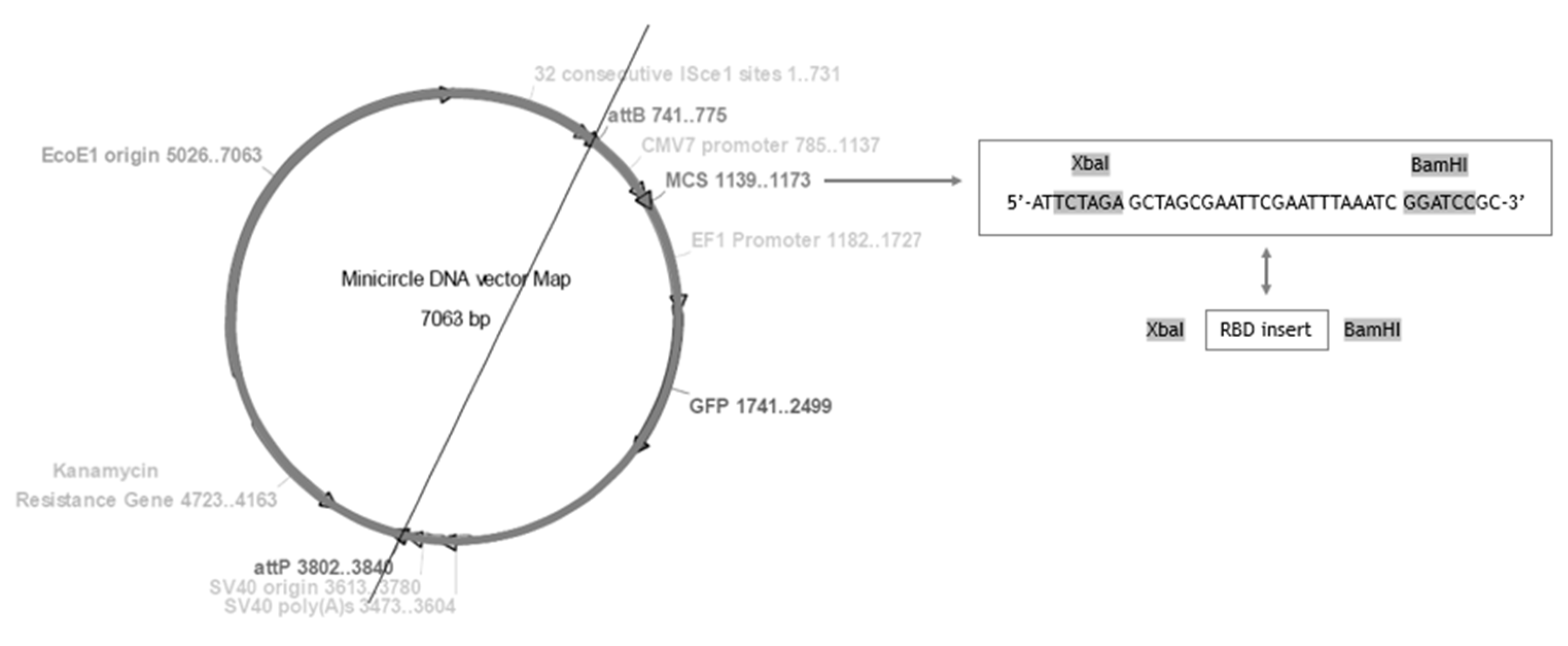
2.2. Construction of mcDNA-RBD Vector
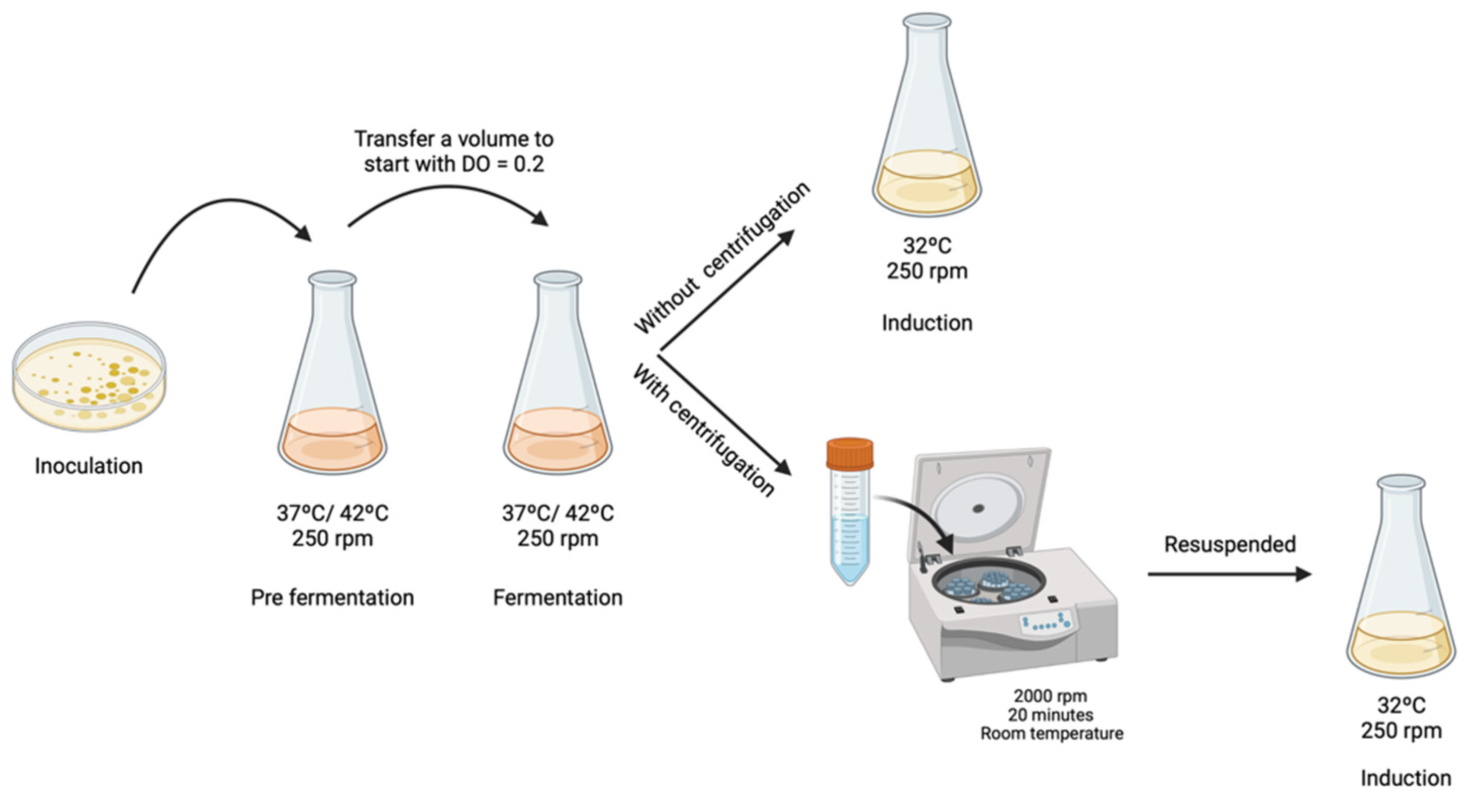
2.3. Fermentation and Induction Conditions in an Orbital Shaker
2.4. Design of Experiments (DoE)
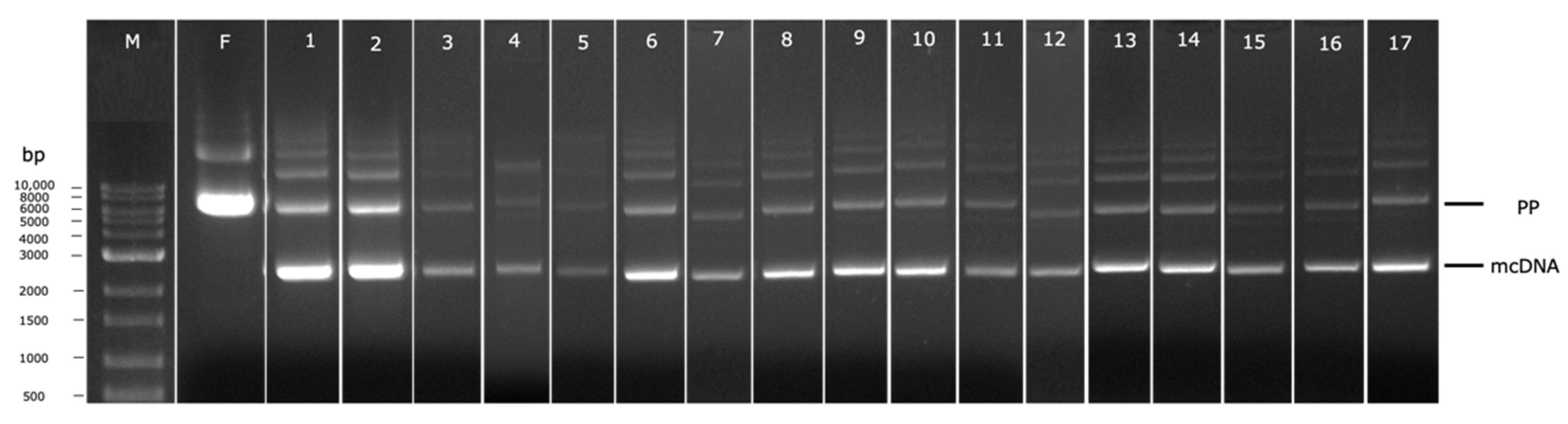
2.5. Agarose Gel Electrophoresis
2.6. Output Determination
2.7. Analytical Chromatography
2.8. Fermentation and Induction Conditions in Mini-Bioreactor
2.9. L-Arabinose Determination by HPLC Analysis
3. Results and Discussion
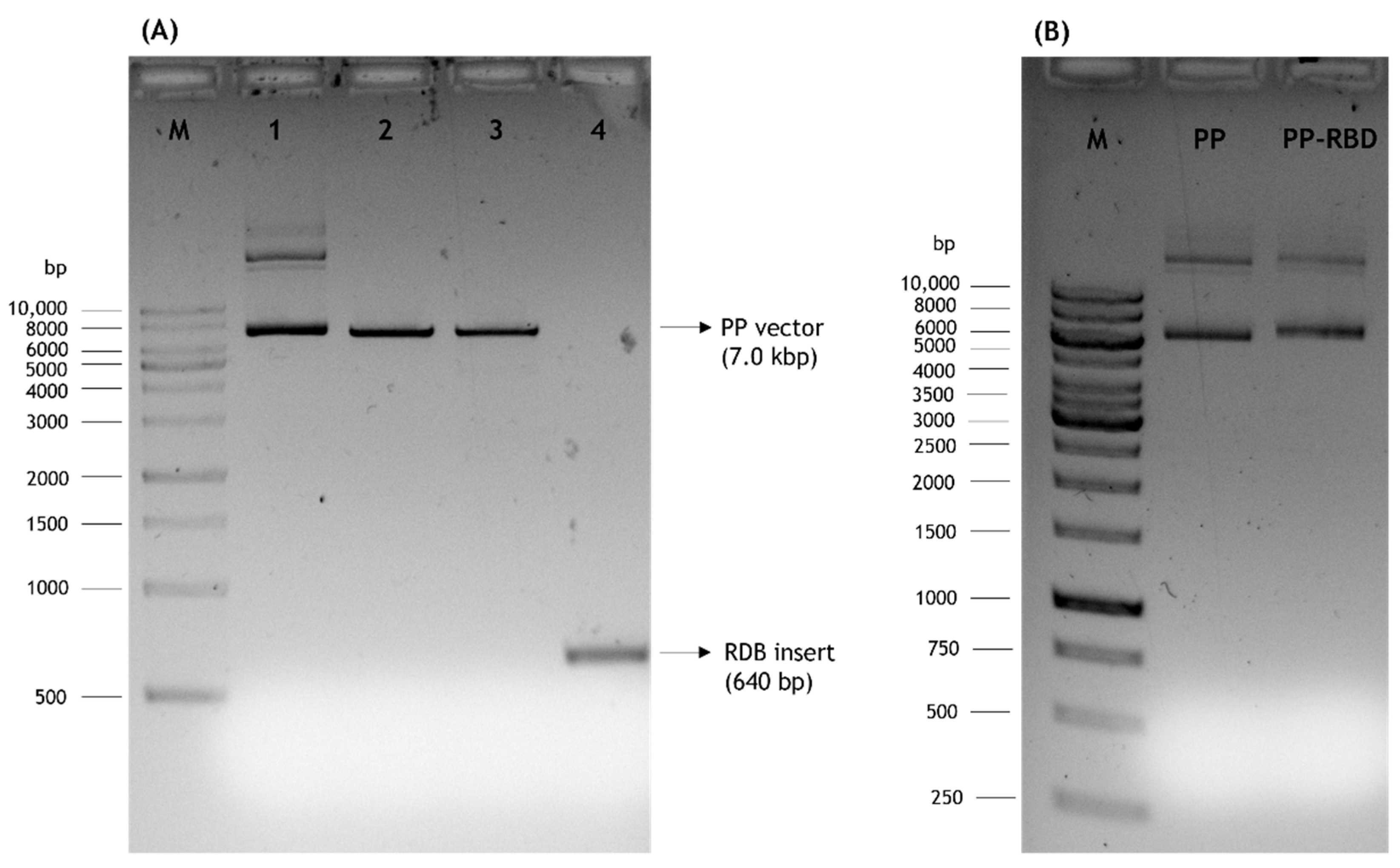
3.1. Cloning of RBD Gene Sequence into PP Vector
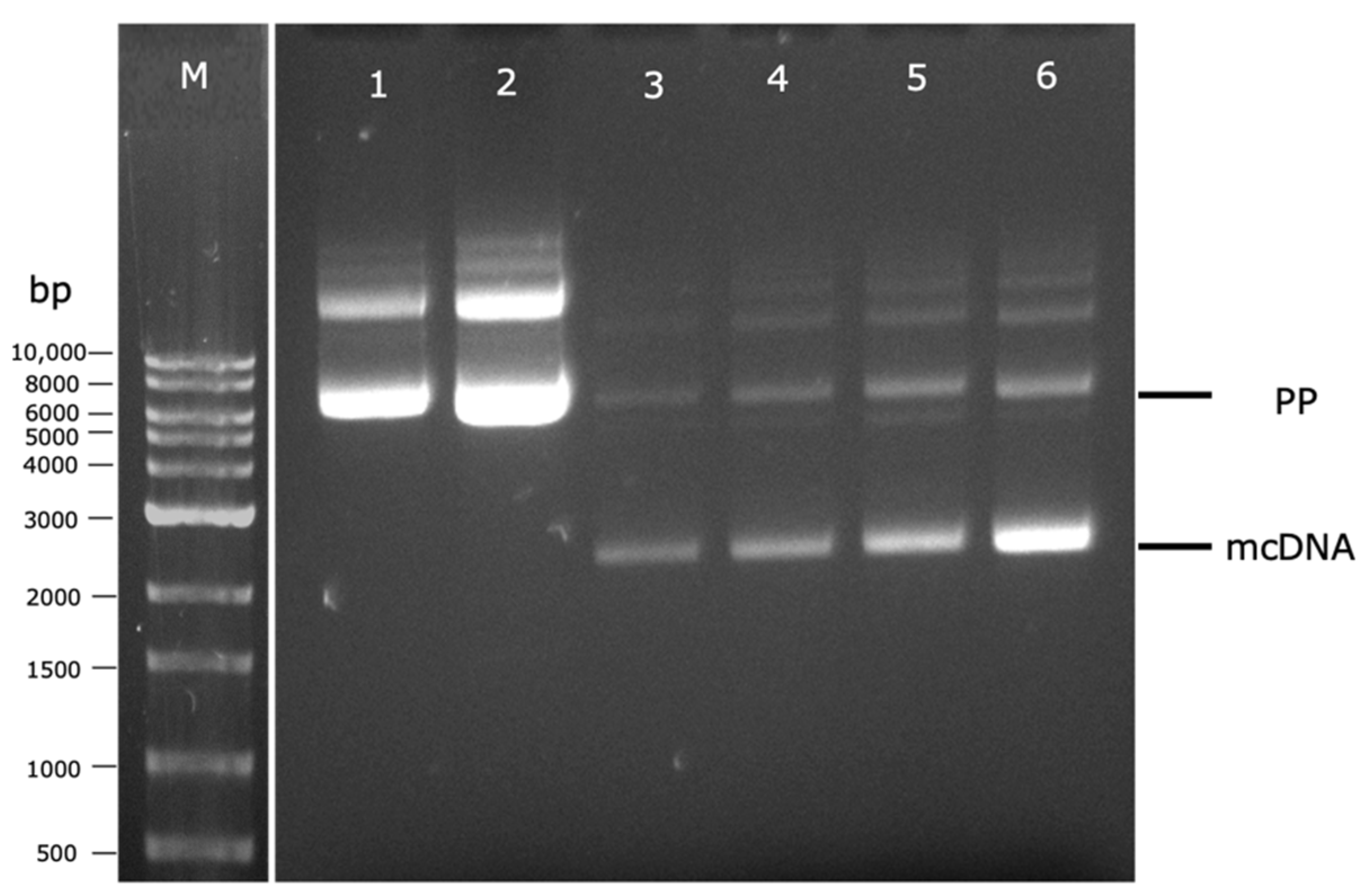
3.2. PP Amplification and Recombination into mcDNA
3.3. DoE Inputs for PP Recombination into mcDNA
3.4. Model Generation and Statistical Analysis
3.5. Input Effects on % of Recombined mcDNA
3.6. Output Optimization and Model Validation
3.7. Determination of PP and mcDNA Concentration
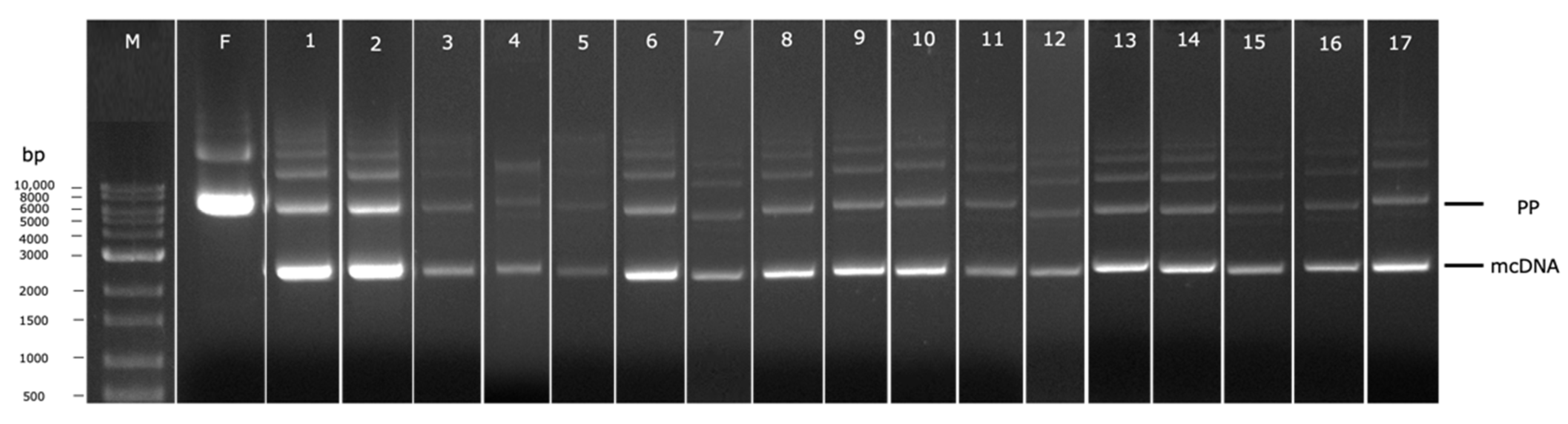
3.8. Scale up of the mcDNA Biosynthesis in Mini-Bioreactor
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antony, P.; Vijayan, R. Role of SARS-CoV-2 and ACE2 variations in COVID-19. Biomed. J. 2021, 44, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Takasawa, K.; Asada, K.; Shinkai, N.; Bolatkan, A.; Yamada, M.; Takahashi, S.; Machino, H.; Kobayashi, K.; Komatsu, M.; et al. Epigenetic Mechanisms Underlying COVID-19 Pathogenesis. Biomedicines 2021, 9, 1142. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Hassine, I.H. Covid-19 vaccines and variants of concern: A review. Rev. Med. Virol. 2021, 2021, e2313. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kumar, S.A.; Jhan, Y.Y.; Bishop, C.J. Engineering DNA vaccines against infectious diseases. Acta Biomater. 2018, 80, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Eusébio, D.; Neves, A.R.; Costa, D.; Biswas, S.; Alves, G.; Cui, Z.; Sousa, Â. Methods to improve the immunogenicity of plasmid DNA vaccines. Drug Discov. Today 2021, 26, 2575–2592. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Queiroz, J.A.; Sousa, F.; Sousa, Â. Minicircle DNA: The Future for DNA-Based Vectors? Trends Biotechnol. 2020, 38, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, G.; Zhang, Y.; Liu, X.; Tien, P. A novel minicircle vector based system for inhibting the replication and gene expression of Enterovirus 71 and Coxsackievirus A16. Antivir. Res. 2012, 96, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Simcikova, M.; Prather, K.L.J.; Prazeres, D.M.F.; Monteiro, G.A. On the dual effect of glucose during production of pBAD/AraC-based minicircles. Vaccine 2014, 32, 2843–2846. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.H.; Guo, X.Y.; Chen, Y.; He, C.Y.; Chen, Z.Y. Increasing the minicircle DNA purity using an enhanced triplex DNA technology to eliminate DNA contaminants. Mol. Ther.-Methods Clin. Dev. 2015, 1, 14062. [Google Scholar] [CrossRef]
- Chang, C.W.; Christensen, L.V.; Lee, M.; Kim, S.W. Efficient expression of vascular endothelial growth factor using minicircle DNA for angiogenic gene therapy. J. Control. Release 2008, 125, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, V.M.; Maia, C.J.; Queiroz, J.A.; Pichon, C.; Correia, I.J.; Sousa, F. Improved minicircle DNA biosynthesis for gene therapy applications. Hum. Gene Ther. Methods 2014, 25, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, V.; Melo-Diogo, D.; Costa, E.; Moreira, A.; Queiroz, J.; Pichon, C.; Correia, I.; Sousa, F. Minicircle DNA vectors for gene therapy: Advances and applications. Expert Opin. Biol. Ther. 2015, 15, 353–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenler, S.; Wiklander, O.P.B.; Badal-Tejedor, M.; Turunen, J.; Nordin, J.; Hallengärd, D.; Wahren, B.; Andaloussi, S.E.L.; Rutland, M.W.; Edvard Smith, C.I.; et al. Micro-minicircle Gene Therapy: Implications of Size on Fermentation, Complexation, Shearing Resistance, and Expression. Mol. Ther.-Nucleic Acids 2014, 3, e140. [Google Scholar] [CrossRef] [Green Version]
- Kay, M.A.; He, C.Y.; Chen, Z.Y. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010, 28, 1287–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; WikstrÄom, C. Design of Experiments, Principles and Applications, 3rd ed.; Umetrics AB: Umeå, Sweden, 2008. [Google Scholar]
- Myers, J.L.; Well, A.D.; Lorch, R.F. Research Design and Statistical Analysis, 3rd ed.; Routledge: Abingdon, UK, 2010. [Google Scholar]
- Liu, H.; Galbraith, S.C.; Ricart, B.; Stanton, C.; Smith-Goettler, B.; Verdi, L.; Connor, T.O.; Lee, S.; Yoon, S. Optimization of critical quality attributes in continuous twin-screw wet granulation via design space validated with pilot scale experimental data. Int. J. Pharm. 2017, 525, 249–263. [Google Scholar] [CrossRef]
- Hocharoen, L.; Noppiboon, S.; Kitsubun, P. Toward QbD Process Understanding on DNA Vaccine Purification Using Design of Experiment. Front. Bioeng. Biotechnol. 2021, 9, 657201. [Google Scholar] [CrossRef] [PubMed]
- Sousa, Â.; Almeida, A.M.; Faria, R.; Konate, K.; Boisguerin, P.; Queiroz, J.A.; Costa, D. Optimization of peptide-plasmid DNA vectors formulation for gene delivery in cancer therapy exploring design of experiments. Colloids Surf. B Biointerfaces 2019, 183, 110417. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.; Almeida, A.M.; Černigoj, U.; Sousa, F.; Queiroz, J.A. Histamine monolith versatility to purify supercoiled plasmid deoxyribonucleic acid from Escherichia coli lysate. J. Chromatogr. A 2014, 1355, 125–133. [Google Scholar] [CrossRef]
- Pedro, A.Q.; Oppolzer, D.; Bonifácio, M.J.; Maia, C.J.; Queiroz, J.A.; Passarinha, L.A. Evaluation of Mut(S) and Mut+ Pichia pastoris strains for membrane-bound catechol-O-methyltransferase biosynthesis. Appl. Biochem. Biotechnol. 2015, 175, 3840–3855. [Google Scholar] [CrossRef] [PubMed]
- Eusébio, D.; Almeida, A.M.; Alves, J.M.; Maia, C.J.; Queiroz, J.A.; Sousa, F.; Sousa, Â. The Performance of Minicircle DNA Versus Parental Plasmid in p53 Gene Delivery into HPV-18-Infected Cervical Cancer Cells. Nucleic Acid Ther. 2021, 31, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.M.; Muesing, M.A.; Polisky, B. Temperature-sensitive copy number mutants of ColE1 are located in an untranslated region of the plasmid genome. Proc. Natl. Acad. Sci. USA 1982, 79, 3570–3574. [Google Scholar] [CrossRef] [Green Version]
- Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Inducible Escherichia coli fermentation for increased plasmid DNA production. Biotechnol. Appl. Biochem. 2006, 45, 155–166. [Google Scholar]
- Ata-abadi, N.S.; Rezaei, N.; Dormiani, K.; Nasr-esfahani, M.H. Production of Minicircle DNA Vectors Using Site-Specific Recombinases. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1642, pp. 325–339. [Google Scholar]
- Chen, Z.Y.; He, C.Y.; Kay, M.A. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum. Gene Ther. 2005, 16, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, D.C. Design and Analysis of Experiments, 10th ed.; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Ali, R.B.; Ouada, S.B.; Leboulanger, C.; Sayadi, S.; Ouada, H.B. Bisphenol A removal by the Chlorophyta Picocystis sp.: Optimization and kinetic study. Int. J. Phytoremediat. 2021, 23, 818–828. [Google Scholar]
- Galindo, J.; Barrón, B.L.; Lara, A.R. Improved production of large plasmid DNA by enzyme-controlled glucose release. Ann. Microbiol. 2016, 66, 1337–1342. [Google Scholar] [CrossRef]
- Cruz, M.; Ramírez, E.A.; Sigala, J.C.; Utrilla, J.; Lara, A.R. Plasmid DNA production in proteome-reduced Escherichia coli. Microorganisms 2020, 8, 1444. [Google Scholar] [CrossRef]
- Islas-Lugo, F.; Vega-Estrada, J.; Alvis, C.A.; Ortega-López, J.; Montes-Horcasitas, M.C. Developing strategies to increase plasmid DNA production in Escherichia coli DH5α using batch culture. J. Biotechnol. 2016, 233, 66–73. [Google Scholar] [CrossRef]
- Gonçalves, G.A.L.; Prather, K.L.J.; Monteiro, G.A.; Carnes, A.E.; Prazeres, D.M.F. Plasmid DNA production with Escherichia coli GALG20, a pgi-gene knockout strain: Fermentation strategies and impact on downstream processing. J. Biotechnol. 2014, 186, 119–127. [Google Scholar] [CrossRef]
- Borja, G.M.; Mora, E.M.; Barrón, B.; Gosset, G.; Ramírez, O.T.; Lara, A.R. Engineering Escherichia coli to increase plasmid DNA production in high cell-density cultivations in batch mode. Microb. Cell Fact. 2012, 11, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, D.V.K.; Ramu, C.T.; Rao, J.V.; Narasu, M.L.; Bhujanga Rao, A.K.S. Impact of dissolved oxygen concentration on some key parameters and production of rhG-CSF in batch fermentation. J. Ind. Microbiol. Biotechnol. 2008, 35, 991–1000. [Google Scholar]
- Passarinha, L.A.; Diogo, M.M.; Queiroz, J.A.; Monteiro, G.A.; Fonseca, L.P.; Prazeres, D.M. Production of ColE1 type plasmid by Escherichia coli DH5α cultured under nonselective conditions. J. Microbiol. Biotechnol. 2006, 16, 20–24. [Google Scholar]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kennedy, R.D.; Ward, J.M.; Keshavarz-Moore, E. Effects of fermentation strategy on the characteristics of plasmid DNA production. Biotechnol. Appl. Biochem. 2003, 37, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Šimčíková, M.; Alves, C.P.A.; Brito, L.; Prather, K.L.J.; Prazeres, D.M.F.; Monteiro, G.A. Improvement of DNA minicircle production by optimization of the secondary structure of the 5′-UTR of ParA resolvase. Appl. Microbiol. Biotechnol. 2016, 100, 6725–6737. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay Number | Temperature (°C) | Antibiotic Concentration (µg/mL) | Induction Time (h) | mcDNA Recombined (%) |
|---|---|---|---|---|
| 1 | 30 | 50 | 1 | 81.43 |
| 2 | 30 | 0 | 1 | 92.75 |
| 3 | 30 | 25 | 3 | 58.73 |
| 4 | 30 | 50 | 5 | 26.98 |
| 5 | 30 | 0 | 5 | 47.85 |
| 6 | 34 | 25 | 1 | 57.78 |
| 7 | 34 | 0 | 3 | 22.60 |
| 8 | 34 | 25 | 3 | 42.01 |
| 9 | 34 | 25 | 3 | 41.24 |
| 10 | 34 | 25 | 3 | 40.95 |
| 11 | 34 | 50 | 3 | 28.55 |
| 12 | 34 | 25 | 5 | 23.40 |
| 13 | 38 | 50 | 1 | 43.40 |
| 14 | 38 | 0 | 1 | 38.84 |
| 15 | 38 | 25 | 3 | 21.56 |
| 16 | 38 | 50 | 5 | 16.51 |
| 17 | 38 | 0 | 5 | 18.68 |
| Output | R2 | Adjusted R2 | Predicted R2 | Adequate Precision |
|---|---|---|---|---|
| % of mcDNA | 0.9972 | 0.9937 | 0.9762 | 60.885 |
| Source | Sum of Squares | dF | Mean Square | F Value | p Value |
|---|---|---|---|---|---|
| Model | 6526.52 | 9 | 725.17 | 280.41 | <0.0001 |
| Temperature (A) | 2520.16 | 1 | 2520.16 | 974.50 | <0.0001 |
| [Antibiotic] (B) | 129.17 | 1 | 129.17 | 49.95 | 0.002 |
| Induction time (C) | 3268.14 | 1 | 3268.14 | 1263.73 | <0.0001 |
| AB | 149.47 | 1 | 149.47 | 57.80 | 0.0001 |
| AC | 341.91 | 1 | 341.91 | 132.21 | <0.0001 |
| BC | 33.13 | 1 | 33.13 | 12.81 | 0.0090 |
| A2 | 50.20 | 1 | 50.20 | 19.41 | 0.0031 |
| B2 | 0.88 | 1 | 0.88 | 0.34 | 0.5776 |
| C2 | 0.14 | 1 | 0.14 | 0.053 | 0.8244 |
| Residual | 18.10 | 1 | 2.59 | − | − |
| Lack of fit | 17.50 | 5 | 3.50 | 11.66 | 0.0808 |
| Output | Predicted Mean | SE Mean | 95% CI Low | 95% CI High | SE Predicted | 95% PI Low | 95% PI High |
|---|---|---|---|---|---|---|---|
| % of recombined mcDNA | 92.1131 | 1.43 | 88.72 | 95.50 | 2.15 | 87.02 | 97.21 |
| Time (h) | Initial Concentration of L-Arabinose (%) | |
|---|---|---|
| 0.010 | 1.000 | |
| 2 | 0.006 ± 0.0012 | 0.868 ± 0.0150 |
| 4 | 0.005 ± 0.0025 | 0.791 ± 0.0112 |
| 6 | 0.003 ± 0.0003 | 0.774 ± 0.0209 |
| 8 | 0.003 ± 0.0002 | 0.729 ± 0.0183 |
| 10 | 0.002 ± 0.0012 | 0.707 ± 0.0106 |
| Nucleic Acid | Orbital Shaker | Increment | Bioreactor |
|---|---|---|---|
| PP | 23.07 mg/L | 65.46 | 1.51 g/L |
| mcDNA | 16.48 mg/L | 69.97 | 1.15 g/L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventura, C.; Eusébio, D.; Gonçalves, A.M.; Barroca-Ferreira, J.; Costa, D.; Cui, Z.; Passarinha, L.A.; Sousa, Â. Maximization of the Minicircle DNA Vaccine Production Expressing SARS-CoV-2 RBD. Biomedicines 2022, 10, 990. https://doi.org/10.3390/biomedicines10050990
Ventura C, Eusébio D, Gonçalves AM, Barroca-Ferreira J, Costa D, Cui Z, Passarinha LA, Sousa Â. Maximization of the Minicircle DNA Vaccine Production Expressing SARS-CoV-2 RBD. Biomedicines. 2022; 10(5):990. https://doi.org/10.3390/biomedicines10050990
Chicago/Turabian StyleVentura, Cathy, Dalinda Eusébio, Ana M. Gonçalves, Jorge Barroca-Ferreira, Diana Costa, Zhengrong Cui, Luís A. Passarinha, and Ângela Sousa. 2022. "Maximization of the Minicircle DNA Vaccine Production Expressing SARS-CoV-2 RBD" Biomedicines 10, no. 5: 990. https://doi.org/10.3390/biomedicines10050990
APA StyleVentura, C., Eusébio, D., Gonçalves, A. M., Barroca-Ferreira, J., Costa, D., Cui, Z., Passarinha, L. A., & Sousa, Â. (2022). Maximization of the Minicircle DNA Vaccine Production Expressing SARS-CoV-2 RBD. Biomedicines, 10(5), 990. https://doi.org/10.3390/biomedicines10050990