
Metformin Protects against Diabetic Cardiomyopathy: An Association between Desmin–Sarcomere Injury and the iNOS/mTOR/TIMP-1 Fibrosis Axis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Determination of Blood Levels of Glucose and HbA1c
2.4. Histological Examination
2.5. Immunohistochemistry of iNOS and Desmin
2.6. Transmission Electron Microscopy (TEM)
2.7. Western Blotting Analysis of mTOR and AMPK
2.8. Cardiac TIMP-1 Real-Time Polymerase Chain Reaction (qPCR)
2.9. Heart Rate (HR) and Electrocardiograph (ECG) Recordings
2.10. Statistical Analysis
3. Results
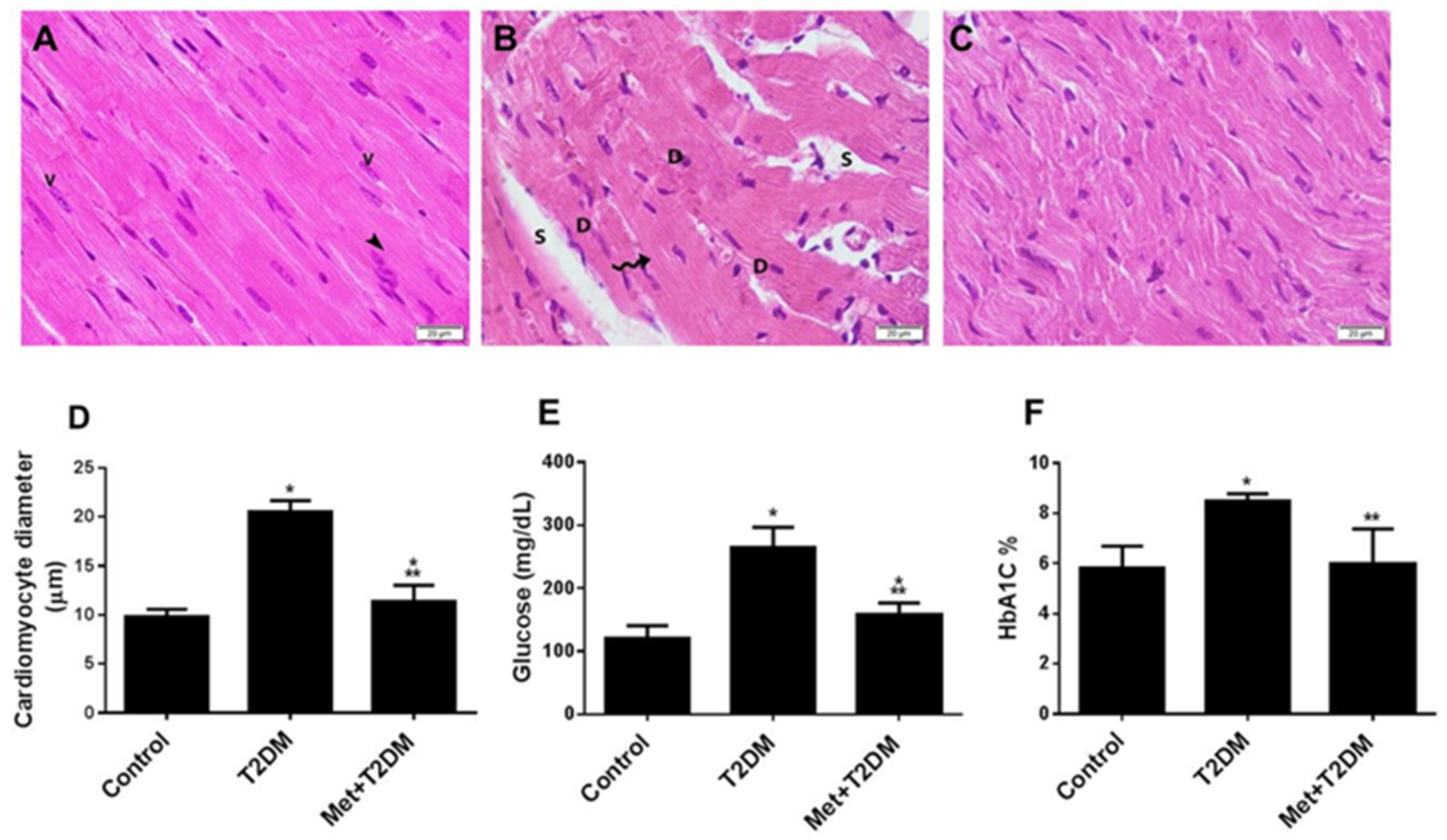
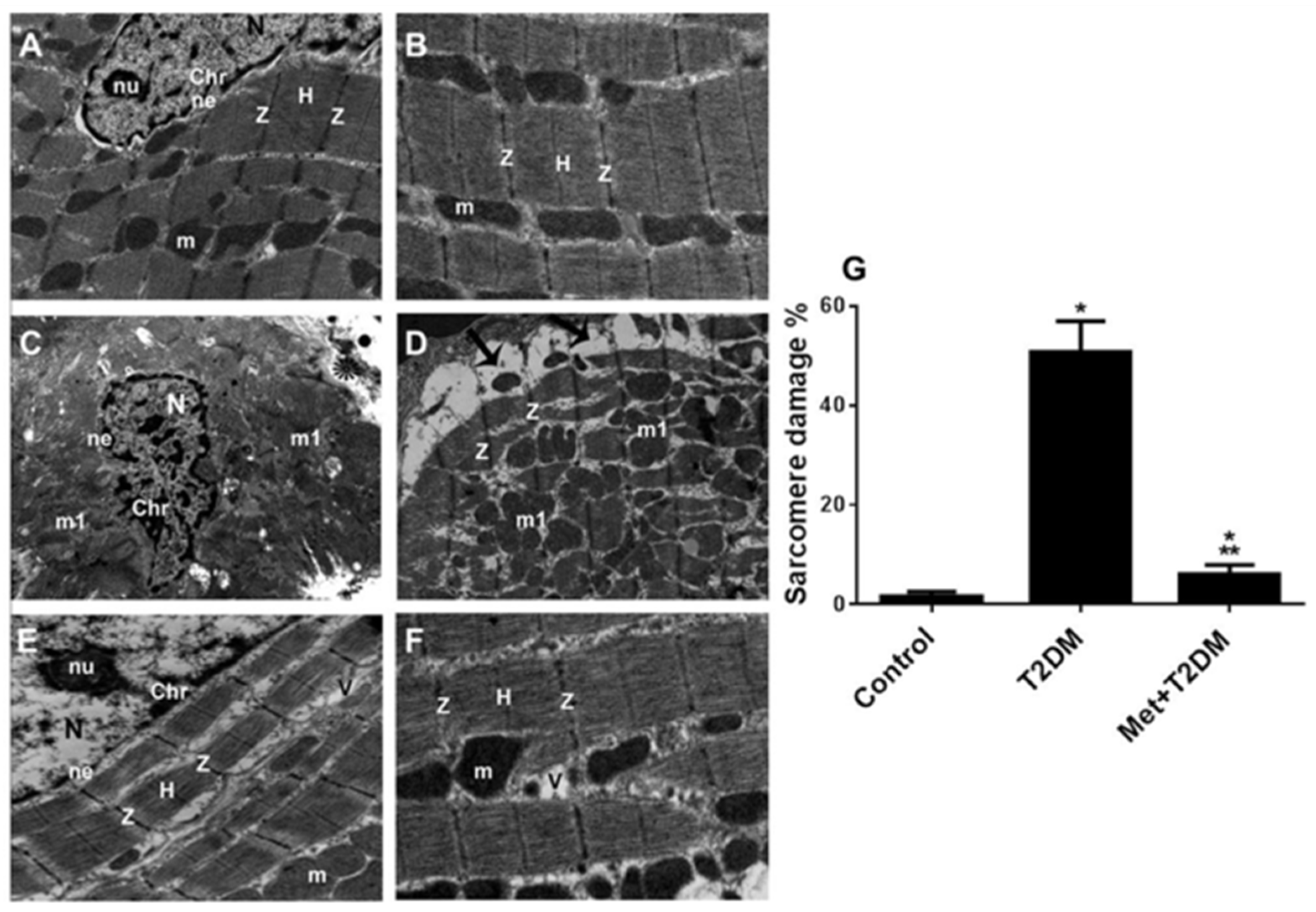
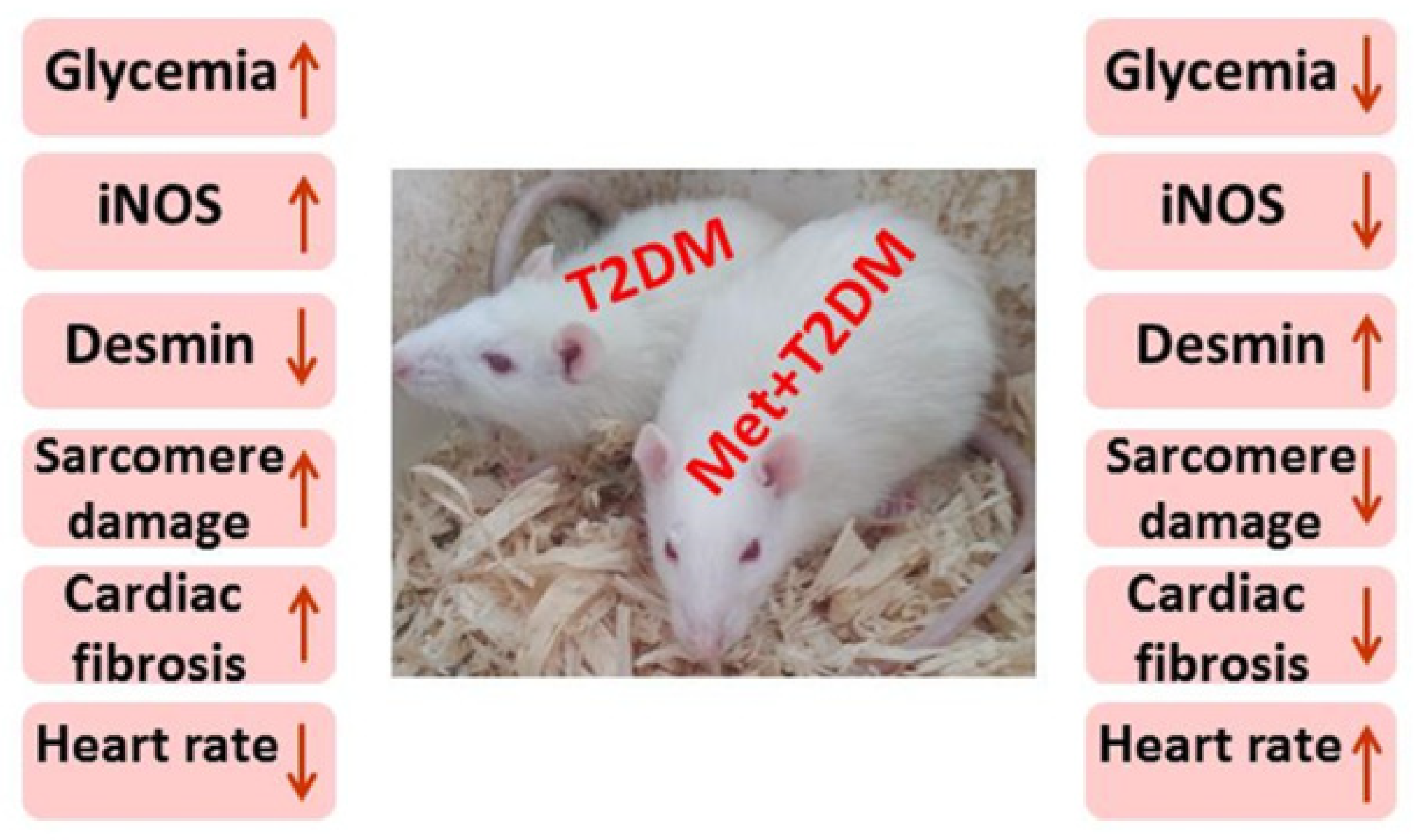
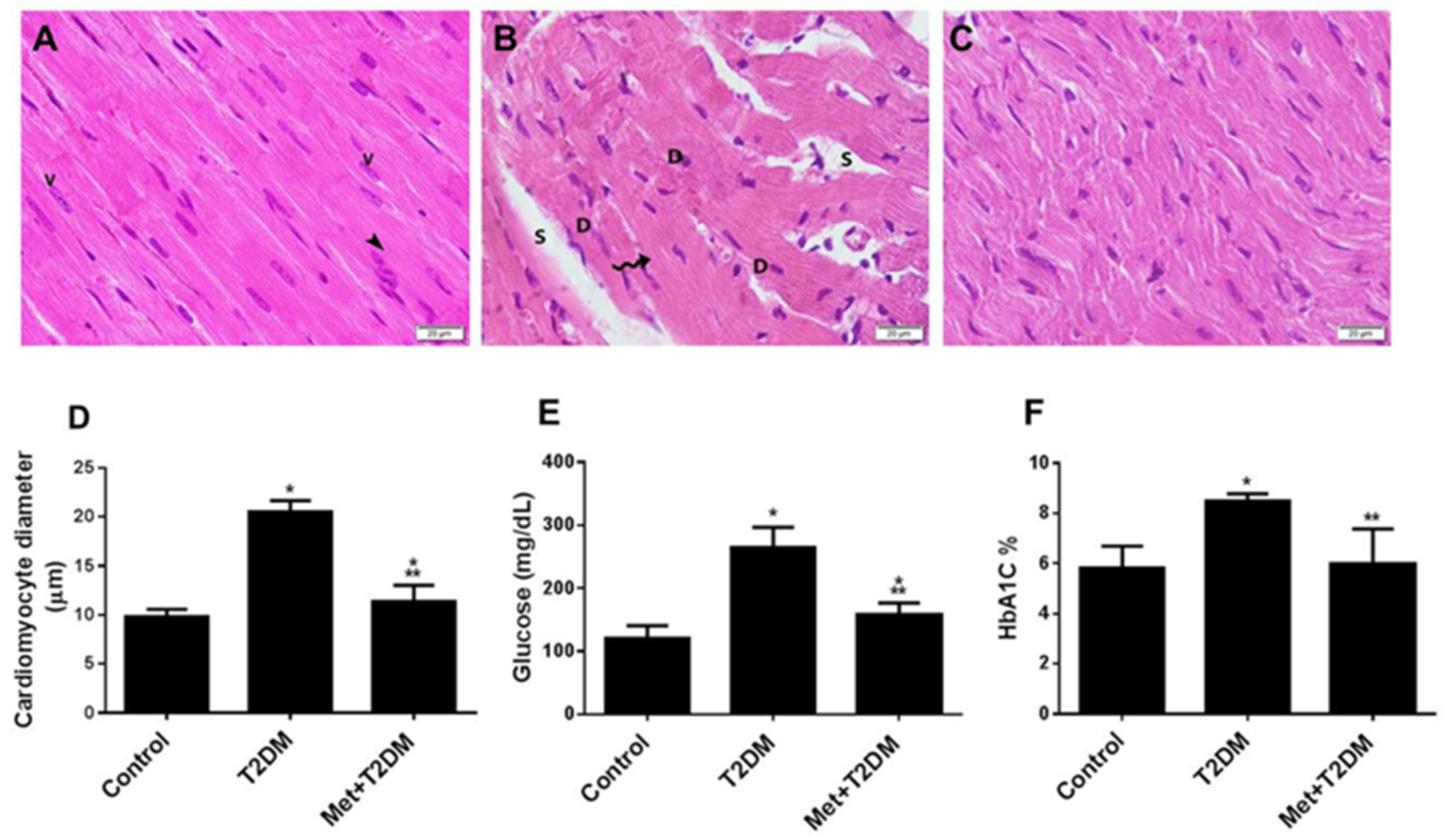
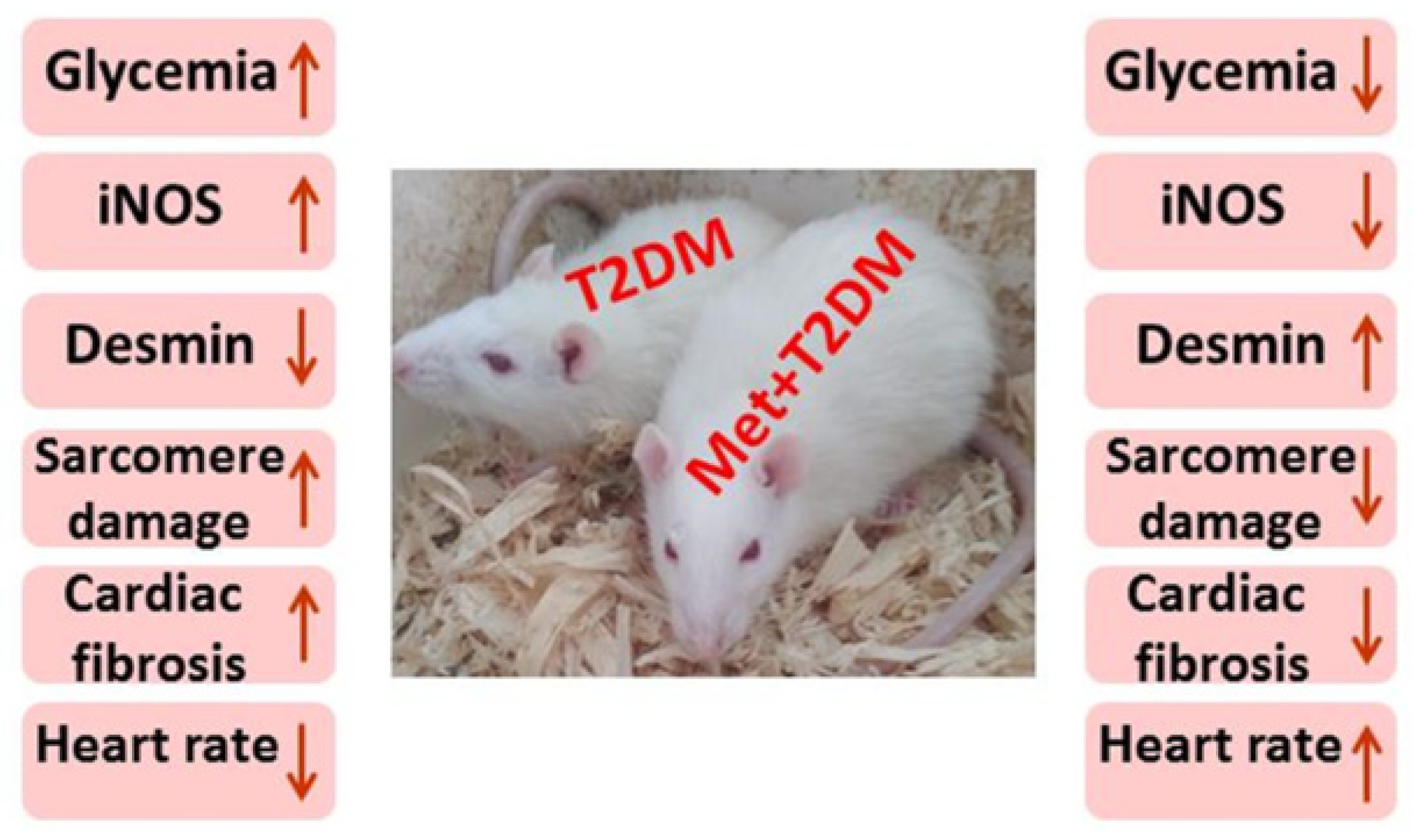
3.1. The Induction of Diabetic Cardiomyopathy Is Inhibited by Metformin
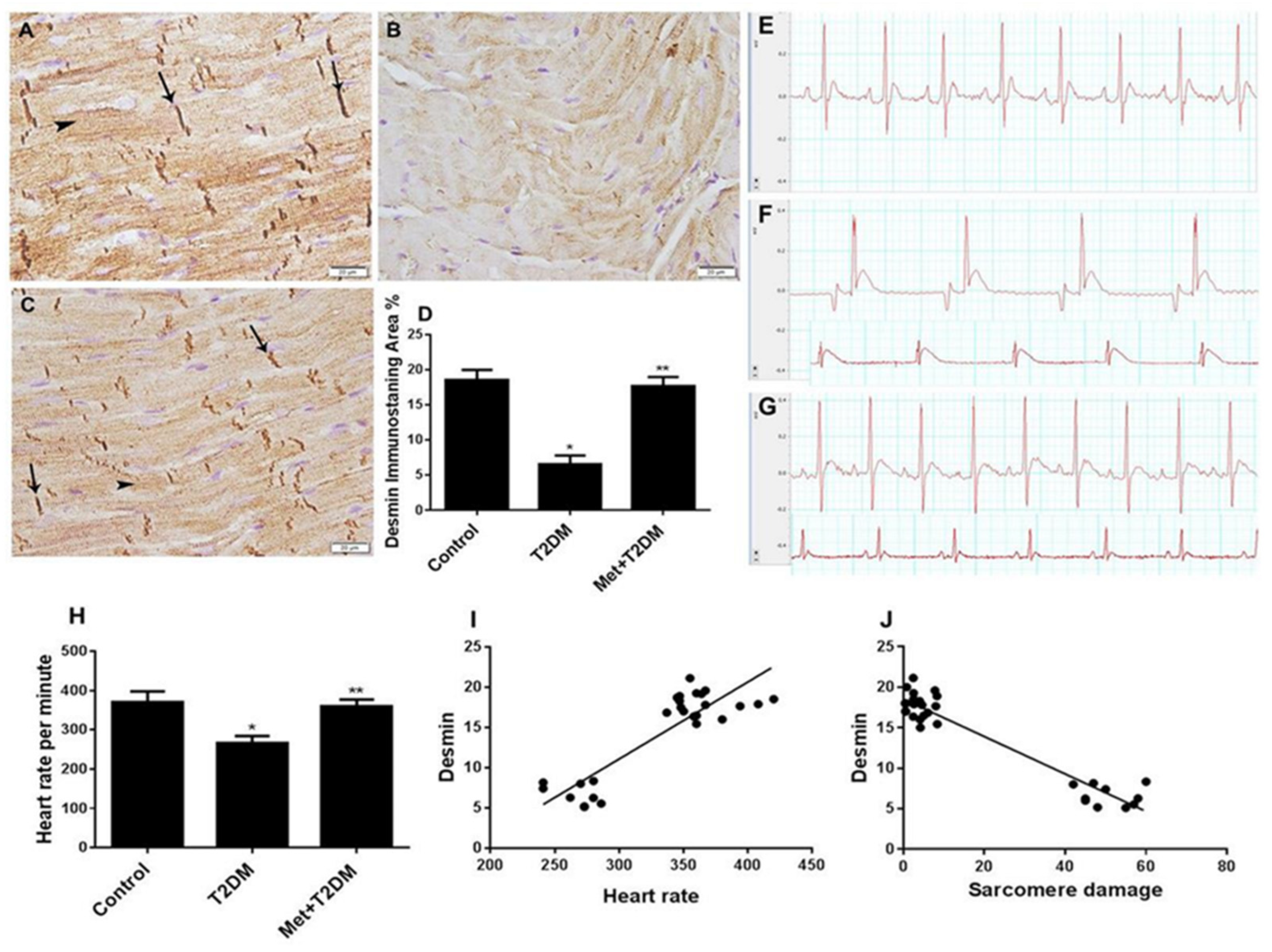
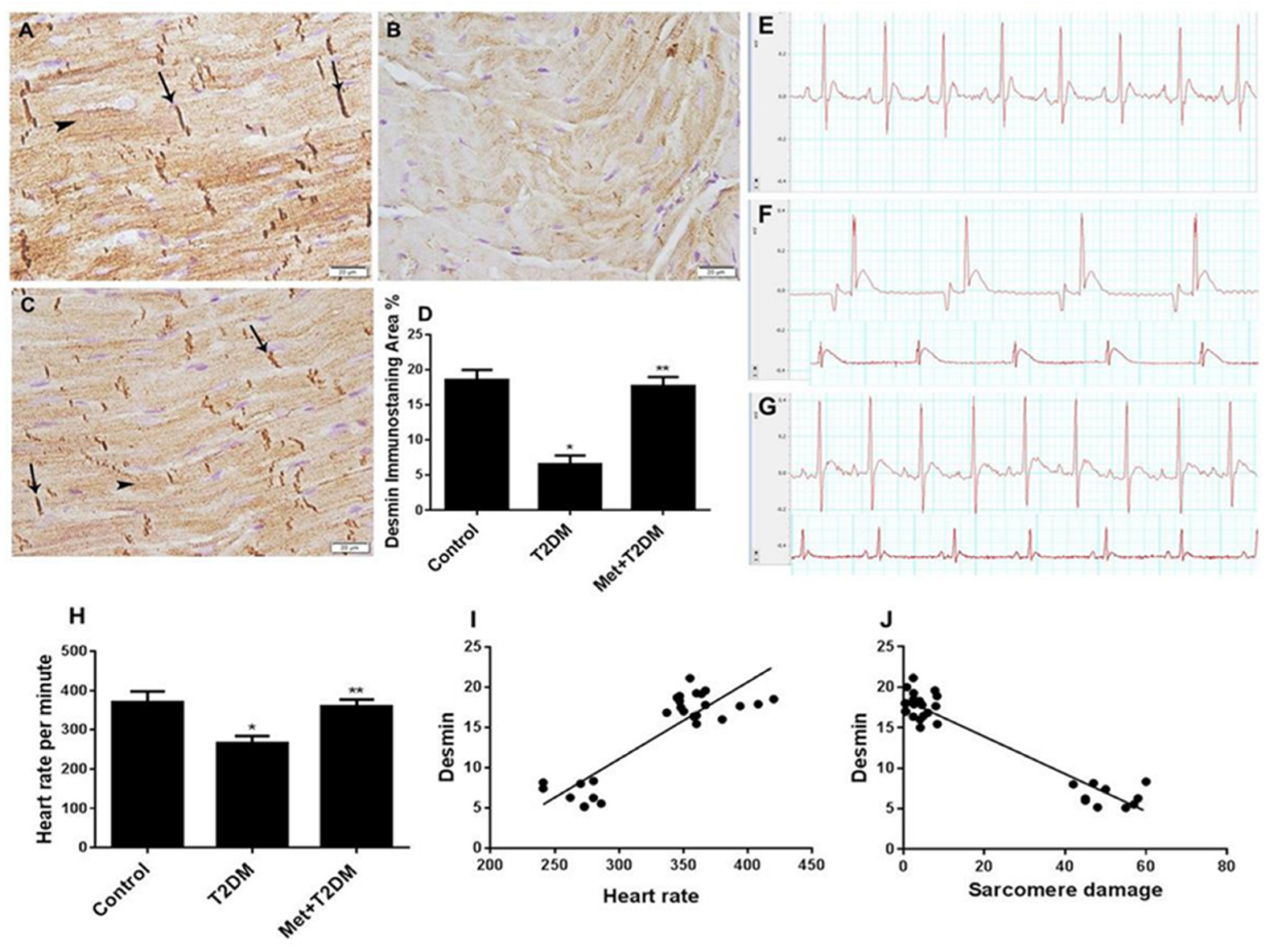
3.2. Downregulation of Cardiac Desmin Expression in Diabetic Rats Is Inhibited by Metformin
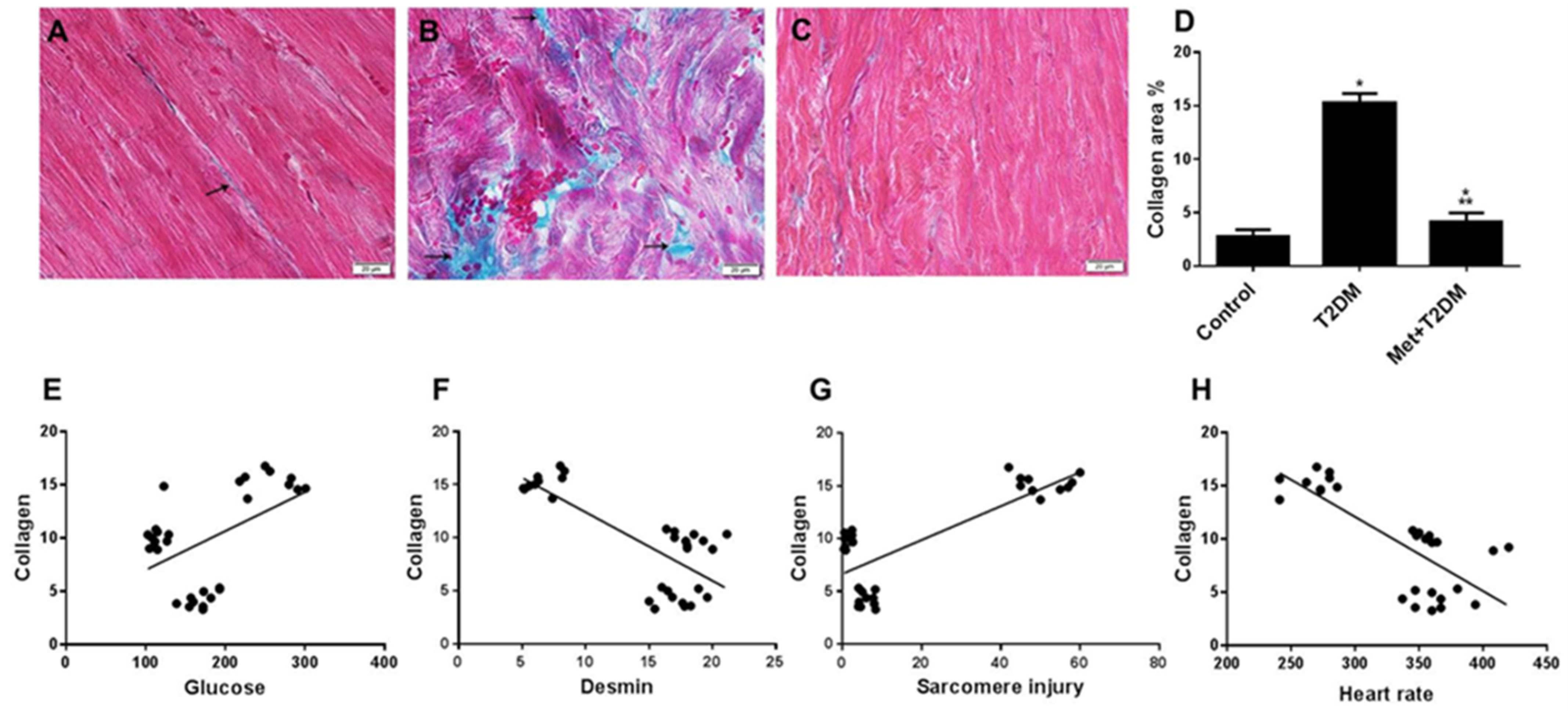
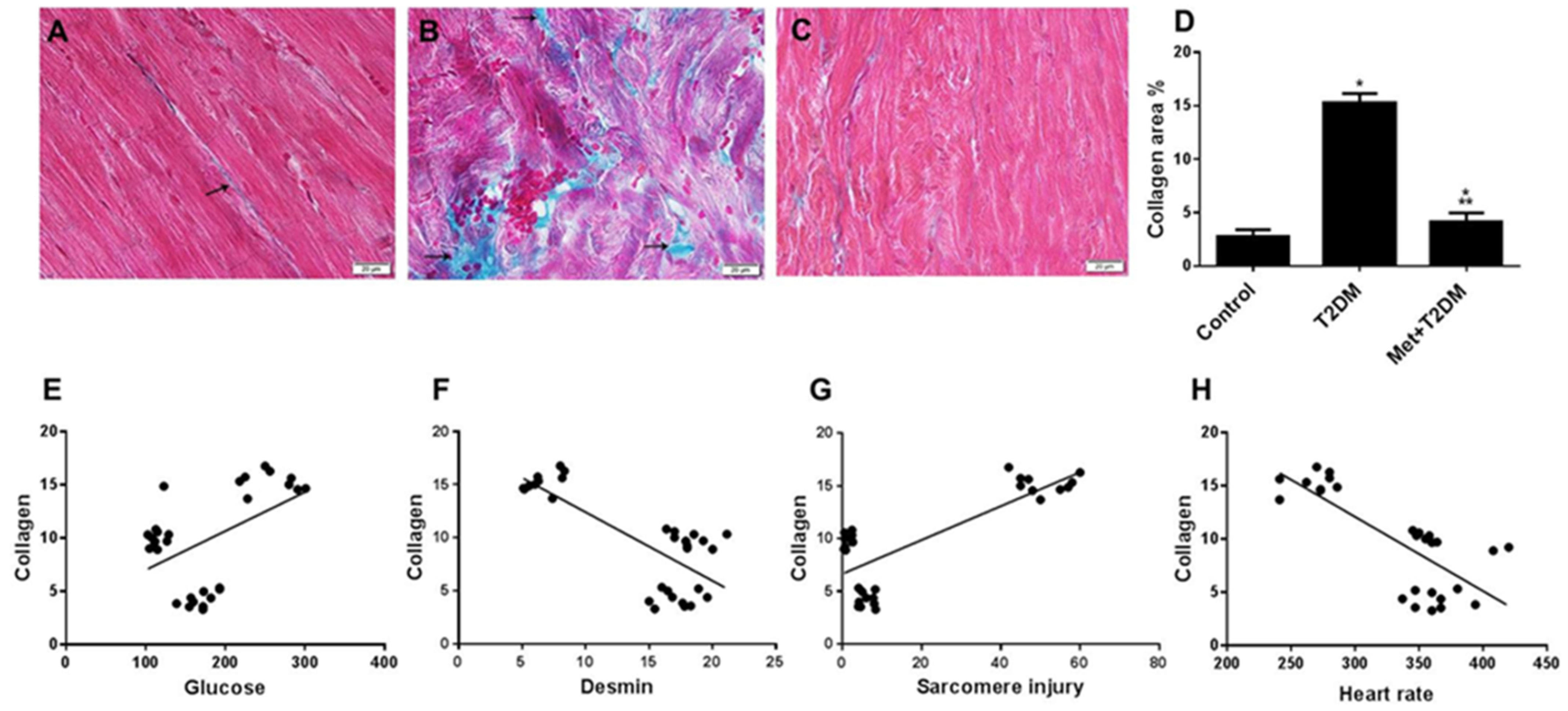
3.3. Induction of Cardiac Fibrosis by Diabetes Is Inhibited by Metformin
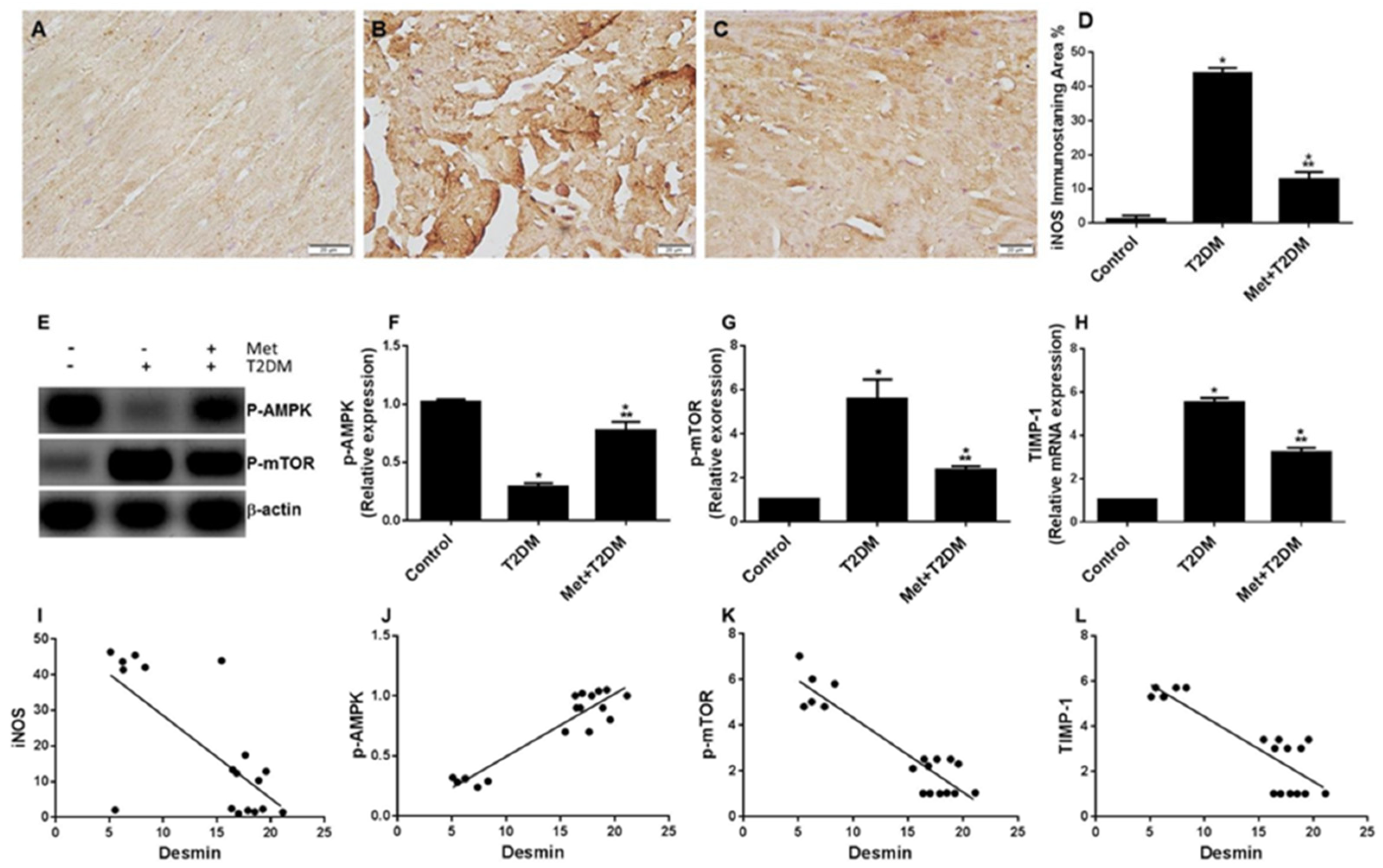
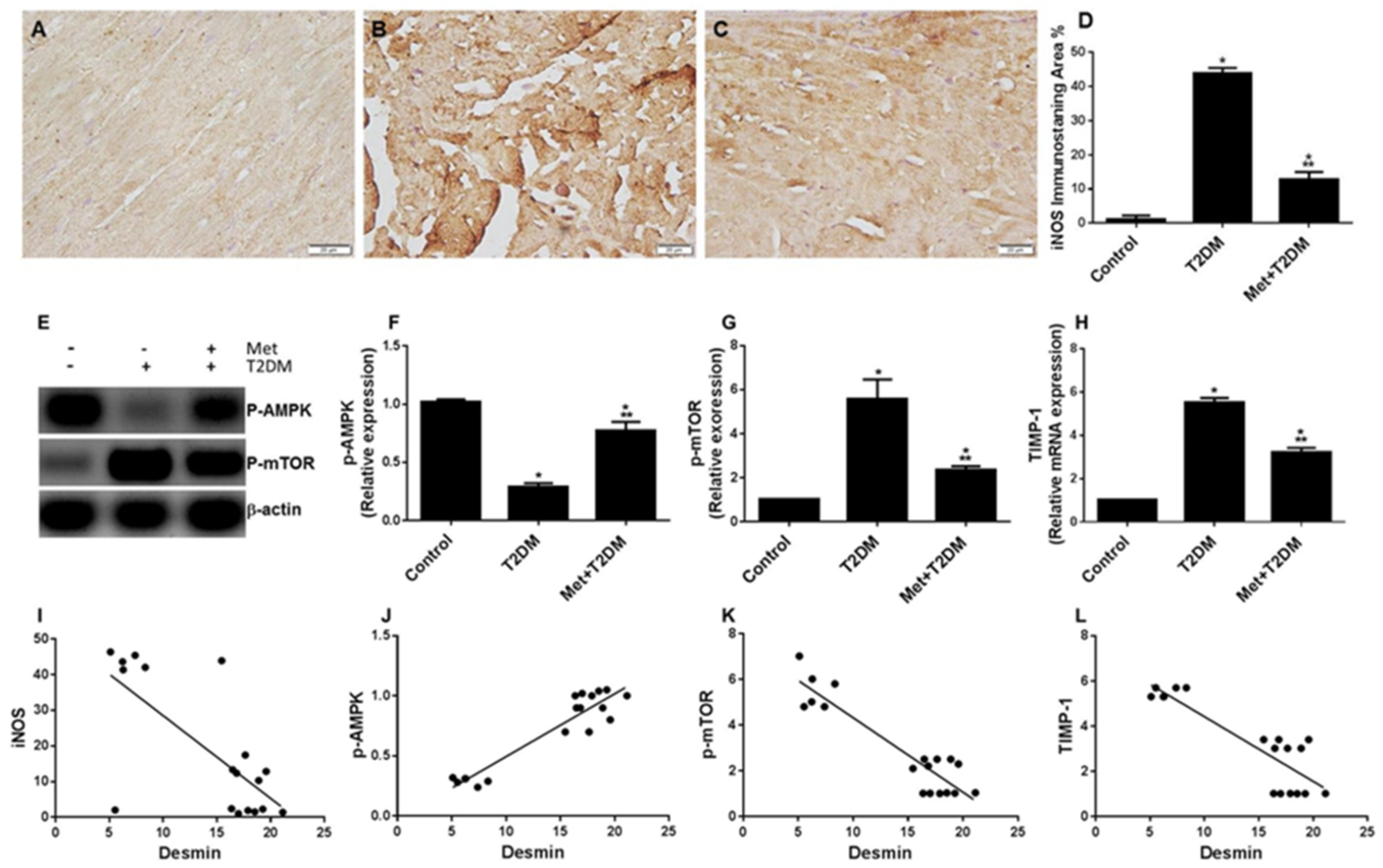
3.4. Diabetes Induces the iNOS–mTOR–TIMP-1 Axis of Fibrosis Is Protected by Metformin
4. Discussion
Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeFronzo, R.A. Pharmacologic Therapy for Type 2 Diabetes Mellitus. Ann. Intern. Med. 1999, 131, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.C.K. The diabesity epidemic in the light of evolution: Insights from the capacity–load model. Diabetologia 2019, 62, 1740–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laakso, M. Cardiovascular disease in type 2 diabetes from population to man to mechanisms: The Kelly West Award Lecture 2008. Diabetes Care 2010, 33, 442–449. [Google Scholar] [PubMed] [Green Version]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- Petrie, J.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2017, 34, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [PubMed]
- Ronti, T.; Lupattelli, G.; Mannarino, E. The endocrine function of adipose tissue: An update. Clin. Endocrinol. 2006, 64, 355–365. [Google Scholar] [CrossRef]
- Barazzoni, R.; Aleksova, A.; Armellini, I.; Cattin, M.R.; Zanetti, M.; Carriere, C. Adipokines, Ghrelin and Obesity-Associated Insulin Resistance in Nondiabetic Patients with Acute Coronary Syndrome. Obesity 2012, 20, 2348–2353. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Chuang, L.-M. The role of oxidative stress in the pathogenesis of type 2 diabetes: From molecular mechanism to clinical implication. Am. J. Transl. Res. 2010, 2, 316–331. [Google Scholar]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Vargas-Robles, H.; Rios, A.; Arellano-Mendoza, M.; Escalante, B.A.; Schnoor, M.M. Antioxidative Diet Supplementation Reverses High-Fat Diet-Induced Increases of Cardiovascular Risk Factors in Mice. Oxidative Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nafisa, A.; Gray, S.G.; Cao, Y.; Wang, T.; Xu, S.; Wattoo, F.H.; Barras, M.; Cohen, N.; Kamato, D.; Little, P.J. Endothelial function and dysfunction: Impact of metformin. Pharmacol. Ther. 2018, 192, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2015, 90, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Aragno, M.M.; Mastrocola, R.; Alloatti, G.; Vercellinatto, I.; Bardini, P.; Geuna, S. Oxidative Stress Triggers Cardiac Fibrosis in the Heart of Diabetic Rats. Endocrinology 2008, 149, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Gogiraju, R.; Xu, X.; Bochenek, M.L.; Steinbrecher, J.H.; Lehnart, S.E.; Wenzel, P. Endothelial p53 Deletion Improves Angiogenesis and Prevents Cardiac Fibrosis and Heart Failure Induced by Pressure Overload in Mice. J. Am. Heart Assoc. 2015, 4, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, L.G.; Dalakas, M.C. Tragedy in a heartbeat: Malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Investig. 2009, 119, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.H.; de Walle, H.E.K.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2010, 80, 354–366. [Google Scholar] [CrossRef]
- Singh, S.R.; Robbins, J. Desmin and Cardiac Disease: An Unfolding Story. Circ. Res. 2018, 122, 1324–1326. [Google Scholar] [CrossRef] [Green Version]
- Al-Ani, B.; Alzamil, N.M.; Hewett, P.W.; Al-Hashem, F.; Bin-Jaliah, I.; Shatoor, A.S. Metformin ameliorates ROS-p53-collagen axis of fibrosis and dyslipidemia in type 2 diabetes mellitus-induced left ventricular injury. Arch. Physiol. Biochem. 2021, 1–7. [Google Scholar] [CrossRef]
- Reed, M.J.; Meszaros, K.; Entes, L.J.; Claypool, M.D.; Pinkett, J.G.; Gadbois, T.M. A new rat model of type 2 diabetes: The fat-fed, streptozotocin-treated rat. Metabolism 2000, 49, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Dallak, M.; Dawood, A.F.; Haidara, M.A.; Abdel Kader, D.H.; Eid, R.A.; Kamar, S.S. Suppression of glomerular damage and apoptosis and biomarkers of acute kidney injury induced by acetaminophen toxicity using a combination of resveratrol and quercetin. Drug Chem. Toxicol. 2020, 45, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Al-Hashem, F.; Al-Humayed, S.; Amin, S.N.; Kamar, S.S.; Mansy, S.S.; Hassan, S. Metformin inhibits mTOR-HIF-1α axis and profibrogenic and inflammatory biomarkers in thioacetamide-induced hepatic tissue alterations. J. Cell. Physiol. 2019, 234, 9328–9337. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord 2010, 110, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Gaertner, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Diermeier, S.; Iberl, J.; Vetter, K.; Haug, M.; Pollmann, C.; Reischl, B.; Buttgereit, A.; Schurmann, S.; Sporrer, M.; Goldmann, W.H.; et al. Early signs of architectural and biomechanical failure in isolated myofibers and immortalized myoblasts from desmin-mutant knock-in mice. Sci. Rep. 2017, 7, 1391. [Google Scholar] [CrossRef] [Green Version]
- Meyer, G.A.; Lieber, R.L. Skeletal muscle fibrosis develops in response to desmin deletion. Am. J. Physiol. Physiol. 2012, 302, C1609–C1620. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Kim, Y.; Park, C.H.; Kim, J.Y.; Min, P.K.; Yoon, Y.W. Effect of sarcomere and mitochondria-related mutations on myocardial fibrosis in patients with hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2021, 23, 1–11. [Google Scholar] [CrossRef]
- Timm, K.N.; Tyler, D. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Mericskay, M.; Agbulut, O.; Butler-Browne, G.; Carlsson, L.; Thornell, L.E. Desmin Is Essential for the Tensile Strength and Integrity of Myofibrils but Not for Myogenic Commitment, Differentiation, and Fusion of Skeletal Muscle. J. Cell Biol. 1997, 139, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Osinska, H.; DornII, G.W.; Nieman, M.; Lorenz, J.N.; Gerdes, A.M. Mouse Model of Desmin-Related Cardiomyopathy. Circulation 2001, 103, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Uchida, S.; Masuo, O.; Cejna, M.; Park, J.S.; Gwon, H.C. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: Restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 2005, 111, 2073–2085. [Google Scholar] [PubMed] [Green Version]
- Marchini, G.S.; Cestari, I.N.; Salemi, V.M.C.; Irigoyen, M.C.; Arnold, A.; Kakoi, A. Early changes in myocyte contractility and cardiac function in streptozotocin-induced type 1 diabetes in rats. PLoS ONE 2020, 15, e0237305. [Google Scholar] [CrossRef] [PubMed]
- Vesentini, G.; Marini, G.; Piculo, F.; Damasceno, D.C.; Matheus, S.M.M.; Felisbino, S.L. Morphological changes in rat rectus abdominis muscle induced by diabetes and pregnancy. Braz. J. Med Biol. Res. 2018, 51, e7035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flagg, T.P.; Cazorla, O.; Remedi, M.S.; Haim, T.E.; Tones, M.A.; Bahinski, A. Ca2+-Independent Alterations in Diastolic Sarcomere Length and Relaxation Kinetics in a Mouse Model of Lipotoxic Diabetic Cardiomyopathy. Circ. Res. 2009, 104, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Haywood, G.A.; Tsao, P.S.; von der Leyen, H.E.; Mann, M.J.; Keeling, P.J.; Trindade, P.T. Expression of Inducible Nitric Oxide Synthase in Human Heart Failure. Circulation 1996, 93, 1087–1094. [Google Scholar] [CrossRef]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Ma, X.; Feng, W.; Fu, Y.; Lu, Z.; Xu, M.; Shen, Q.; Zhu, Y.; Zhang, Y. Metformin attenuates cardiac fibrosis by inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc. Res. 2010, 87, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Hattori, K.; Hayashi, T. Pleiotropic Benefits of Metformin: Macrophage Targeting Its Anti-inflammatory Mechanisms. Diabetes 2015, 64, 1907–1909. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kwak, H.J.; Cha, J.Y.; Jeong, Y.S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin Suppresses Lipopolysaccharide (LPS)-induced Inflammatory Response in Murine Macrophages via Activating Transcription Factor-3 (ATF-3) Induction. J. Biol. Chem. 2014, 289, 23246–23255. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.; Gilbert, H.; Kirstine, D.; Frandsen, B.; Kristensen, P.; Johannes, F.E.; Nauck, M.; Nissen, S.; Buse, J. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthew, D. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Colhoun, H.; Dagenais, G.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeye, J.S.; Riddle, M.C.; Ryden, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.; Buttler, G.; Filippatos, G.; Pocock, S.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawood, A.F.; Alzamil, N.M.; Hewett, P.W.; Momenah, M.A.; Dallak, M.; Kamar, S.S.; Abdel Kader, D.H.; Yassin, H.; Haidara, M.A.; Maarouf, A.; et al. Metformin Protects against Diabetic Cardiomyopathy: An Association between Desmin–Sarcomere Injury and the iNOS/mTOR/TIMP-1 Fibrosis Axis. Biomedicines 2022, 10, 984. https://doi.org/10.3390/biomedicines10050984
Dawood AF, Alzamil NM, Hewett PW, Momenah MA, Dallak M, Kamar SS, Abdel Kader DH, Yassin H, Haidara MA, Maarouf A, et al. Metformin Protects against Diabetic Cardiomyopathy: An Association between Desmin–Sarcomere Injury and the iNOS/mTOR/TIMP-1 Fibrosis Axis. Biomedicines. 2022; 10(5):984. https://doi.org/10.3390/biomedicines10050984
Chicago/Turabian StyleDawood, Amal F., Norah M. Alzamil, Peter W. Hewett, Maha A. Momenah, Mohammad Dallak, Samaa S. Kamar, Dina H. Abdel Kader, Hanaa Yassin, Mohamed A. Haidara, Amro Maarouf, and et al. 2022. "Metformin Protects against Diabetic Cardiomyopathy: An Association between Desmin–Sarcomere Injury and the iNOS/mTOR/TIMP-1 Fibrosis Axis" Biomedicines 10, no. 5: 984. https://doi.org/10.3390/biomedicines10050984
APA StyleDawood, A. F., Alzamil, N. M., Hewett, P. W., Momenah, M. A., Dallak, M., Kamar, S. S., Abdel Kader, D. H., Yassin, H., Haidara, M. A., Maarouf, A., & Al-Ani, B. (2022). Metformin Protects against Diabetic Cardiomyopathy: An Association between Desmin–Sarcomere Injury and the iNOS/mTOR/TIMP-1 Fibrosis Axis. Biomedicines, 10(5), 984. https://doi.org/10.3390/biomedicines10050984