
Ha-RasV12-Induced Multilayer Cellular Aggregates Is Mediated by Rac1 Activation Rather Than YAP Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Plasmids, shRNA, siRNA, and Transfection
2.3. RT-PCR
2.4. Western Blot Analyses
2.5. Cell Fractionation
2.6. Pull-Down Assay
2.7. Immunofluorescence Staining and Confocal Microscopy
2.8. Crystal Violet Staining
2.9. Anchorage-Independent Growth Assay
2.10. Statistical Analysis
3. Results
3.1. Ha-RasV12 Facilitates YAP Nuclear Translocation through Downregulated Junctional Myosin IIB
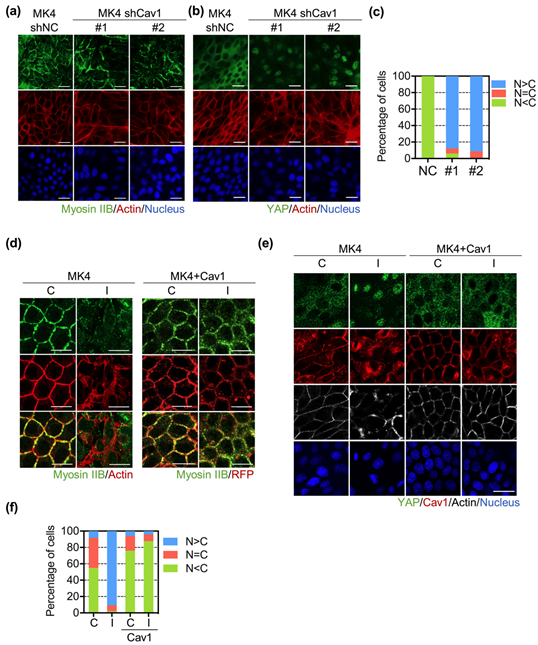
3.2. Cav1 Preserved Junctional Myosin IIB and Restricted Ha-RasV12-Induced YAP Nuclear Translocation
3.3. Ha-RasV12 Downregulated Cav1 Expression and YAP Nuclear Retention through MEK Pathway
3.4. Nuclear YAP Is Not Involved in Ha-RasV12-Induced Cellular Aggregation
3.5. Rac1 Activity Is Required for Ha-RasV12-Induced Cellular Aggregation
3.6. Ha-RasV12-Induced Multilayer Cellular Aggregates through ERK-Rac Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Repasky, G.A.; Chenette, E.J.; Der, C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004, 14, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Tripathi, K.; Garg, M. Mechanistic regulation of epithelial-to-mesenchymal transition through RAS signaling pathway and therapeutic implications in human cancer. J. Cell Commun. Signal 2018, 12, 513–527. [Google Scholar] [CrossRef]
- Chen, C.S.; Tan, J.; Tien, J. Mechanotransduction at cell-matrix and cell-cell contacts. Annu. Rev. Biomed. Eng. 2004, 6, 275–302. [Google Scholar] [CrossRef]
- Betapudi, V. Life without double-headed non-muscle myosin II motor proteins. Front. Chem. 2014, 2, 45. [Google Scholar] [CrossRef]
- Furukawa, K.T.; Yamashita, K.; Sakurai, N.; Ohno, S. The Epithelial Circumferential Actin Belt Regulates YAP/TAZ through Nucleocytoplasmic Shuttling of Merlin. Cell Rep. 2017, 20, 1435–1447. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Katsuno, Y.; Koinuma, D.; Ehata, S.; Morikawa, M. Intracellular and extracellular TGF-beta signaling in cancer: Some recent topics. Front. Med. 2018, 12, 387–411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Gu, Y.; Shea, J.; Slattum, G.; Firpo, M.A.; Alexander, M.; Mulvihill, S.J.; Golubovskaya, V.M.; Rosenblatt, J. Defective apical extrusion signaling contributes to aggressive tumor hallmarks. eLife 2015, 4, e04069. [Google Scholar] [CrossRef]
- Miki, H.; Suetsugu, S.; Takenawa, T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998, 17, 6932–6941. [Google Scholar] [CrossRef]
- Samarin, S.; Nusrat, A. Regulation of epithelial apical junctional complex by Rho family GTPases. Front. Biosci. 2009, 14, 1129–1142. [Google Scholar] [CrossRef]
- Quinlan, M.P. Rac regulates the stability of the adherens junction and its components, thus affecting epithelial cell differentiation and transformation. Oncogene 1999, 18, 6434–6442. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, H.K.; Lin, H.H.; Chiou, Y.W.; Wu, C.L.; Chiu, W.T.; Tang, M.J. Caveolin-1 down-regulation is required for Wnt5a-Frizzled 2 signalling in Ha-Ras(V12) -induced cell transformation. J. Cell Mol. Med. 2018, 22, 2631–2643. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G. The caveolae membrane system. Annu. Rev. Biochem. 1998, 67, 199–225. [Google Scholar] [CrossRef]
- Navarro, A.; Anand-Apte, B.; Parat, M.O. A role for caveolae in cell migration. FASEB J. 2004, 18, 1801–1811. [Google Scholar] [CrossRef]
- Galbiati, F.; Volonte, D.; Brown, A.M.; Weinstein, D.E.; Ben-Ze’ev, A.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 expression inhibits Wnt/beta-catenin/Lef-1 signaling by recruiting beta-catenin to caveolae membrane domains. J. Biol. Chem. 2000, 275, 23368–23377. [Google Scholar] [CrossRef]
- Koleske, A.J.; Baltimore, D.; Lisanti, M.P. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc. Natl. Acad. Sci. USA 1995, 92, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Razandi, M.; Oh, P.; Pedram, A.; Schnitzer, J.; Levin, E.R. ERs associate with and regulate the production of caveolin: Implications for signaling and cellular actions. Mol. Endocrinol. 2002, 16, 100–115. [Google Scholar] [CrossRef]
- Carver, L.A.; Schnitzer, J.E. Caveolae: Mining little caves for new cancer targets. Nat. Rev. Cancer 2003, 3, 571–581. [Google Scholar] [CrossRef]
- Goetz, J.G.; Joshi, B.; Lajoie, P.; Strugnell, S.S.; Scudamore, T.; Kojic, L.D.; Nabi, I.R. Concerted regulation of focal adhesion dynamics by galectin-3 and tyrosine-phosphorylated caveolin-1. J. Cell Biol. 2008, 180, 1261–1275. [Google Scholar] [CrossRef]
- Miao, H.; Strebhardt, K.; Pasquale, E.B.; Shen, T.L.; Guan, J.L.; Wang, B. Inhibition of integrin-mediated cell adhesion but not directional cell migration requires catalytic activity of EphB3 receptor tyrosine kinase. Role of Rho family small GTPases. J. Biol. Chem. 2005, 280, 923–932. [Google Scholar] [CrossRef]
- Chen, W.C.; Lin, H.H.; Tang, M.J. Regulation of proximal tubular cell differentiation and proliferation in primary culture by matrix stiffness and ECM components. Am. J. Physiol. Renal. Physiol. 2014, 307, F695–F707. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Lin, H.K.; Lin, I.H.; Chiou, Y.W.; Chen, H.W.; Liu, C.Y.; Harn, H.I.; Chiu, W.T.; Wang, Y.K.; Shen, M.R.; et al. Mechanical phenotype of cancer cells: Cell softening and loss of stiffness sensing. Oncotarget 2015, 6, 20946–20958. [Google Scholar] [CrossRef] [PubMed]
- Sabra, H.; Brunner, M.; Mandati, V.; Wehrle-Haller, B.; Lallemand, D.; Ribba, A.S.; Chevalier, G.; Guardiola, P.; Block, M.R.; Bouvard, D. beta1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem. 2017, 292, 19179–19197. [Google Scholar] [CrossRef]
- Codelia, V.A.; Sun, G.; Irvine, K.D. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr. Biol. 2014, 24, 2012–2017. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.B.; Morcos, P.A.; Rosenblatt, J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef]
- Tsuji, T.; Ishizaki, T.; Okamoto, M.; Higashida, C.; Kimura, K.; Furuyashiki, T.; Arakawa, Y.; Birge, R.B.; Nakamoto, T.; Hirai, H.; et al. ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J. Cell Biol. 2002, 157, 819–830. [Google Scholar] [CrossRef]
- Ryan, M.B.; Finn, A.J.; Pedone, K.H.; Thomas, N.E.; Der, C.J.; Cox, A.D. ERK/MAPK Signaling Drives Overexpression of the Rac-GEF, PREX1, in BRAF- and NRAS-Mutant Melanoma. Mol. Cancer Res. 2016, 14, 1009–1018. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.S.; Moberg, K.H. Cell-cell junctions: Alpha-catenin and E-cadherin help fence in Yap1. Curr. Biol. 2011, 21, R890–R892. [Google Scholar] [CrossRef] [PubMed]
- Burgermeister, E.; Friedrich, T.; Hitkova, I.; Regel, I.; Einwachter, H.; Zimmermann, W.; Rocken, C.; Perren, A.; Wright, M.B.; Schmid, R.M.; et al. The Ras inhibitors caveolin-1 and docking protein 1 activate peroxisome proliferator-activated receptor gamma through spatial relocalization at helix 7 of its ligand-binding domain. Mol. Cell Biol. 2011, 31, 3497–3510. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, T.; Yap, A.S. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat. Cell Biol. 2015, 17, 533–539. [Google Scholar] [CrossRef]
- Rodgers, L.S.; Fanning, A.S. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton 2011, 68, 653–660. [Google Scholar] [CrossRef]
- Braga, V.M.; Del Maschio, A.; Machesky, L.; Dejana, E. Regulation of cadherin function by Rho and Rac: Modulation by junction maturation and cellular context. Mol. Biol. Cell 1999, 10, 9–22. [Google Scholar] [CrossRef]
- Ebrahim, S.; Fujita, T.; Millis, B.A.; Kozin, E.; Ma, X.; Kawamoto, S.; Baird, M.A.; Davidson, M.; Yonemura, S.; Hisa, Y.; et al. NMII forms a contractile transcellular sarcomeric network to regulate apical cell junctions and tissue geometry. Curr. Biol. 2013, 23, 731–736. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.-Y.; Han, C.-L.; Lin, H.-H.; Tang, M.-J. Ha-RasV12-Induced Multilayer Cellular Aggregates Is Mediated by Rac1 Activation Rather Than YAP Activation. Biomedicines 2022, 10, 977. https://doi.org/10.3390/biomedicines10050977
Wu L-Y, Han C-L, Lin H-H, Tang M-J. Ha-RasV12-Induced Multilayer Cellular Aggregates Is Mediated by Rac1 Activation Rather Than YAP Activation. Biomedicines. 2022; 10(5):977. https://doi.org/10.3390/biomedicines10050977
Chicago/Turabian StyleWu, Li-Ying, Chia-Lin Han, Hsi-Hui Lin, and Ming-Jer Tang. 2022. "Ha-RasV12-Induced Multilayer Cellular Aggregates Is Mediated by Rac1 Activation Rather Than YAP Activation" Biomedicines 10, no. 5: 977. https://doi.org/10.3390/biomedicines10050977
APA StyleWu, L.-Y., Han, C.-L., Lin, H.-H., & Tang, M.-J. (2022). Ha-RasV12-Induced Multilayer Cellular Aggregates Is Mediated by Rac1 Activation Rather Than YAP Activation. Biomedicines, 10(5), 977. https://doi.org/10.3390/biomedicines10050977