
Ziprasidone Induces Rabbit Atrium Arrhythmogenesis via Modification of Oxidative Stress and Sodium/Calcium Homeostasis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Preparation of Atrial Tissues for Electromechanical and Pharmacological Tests
2.2. Cardiomyocyte Isolation
2.3. Electrophysiological Measurement
ICa,L, and INa,L
2.4. Intracellular Ca2+ Monitoring
2.5. Measurement of ROS Production and Cytosolic Na+ Levels
2.6. Western Blot Analysis
2.7. Data Analysis
3. Results
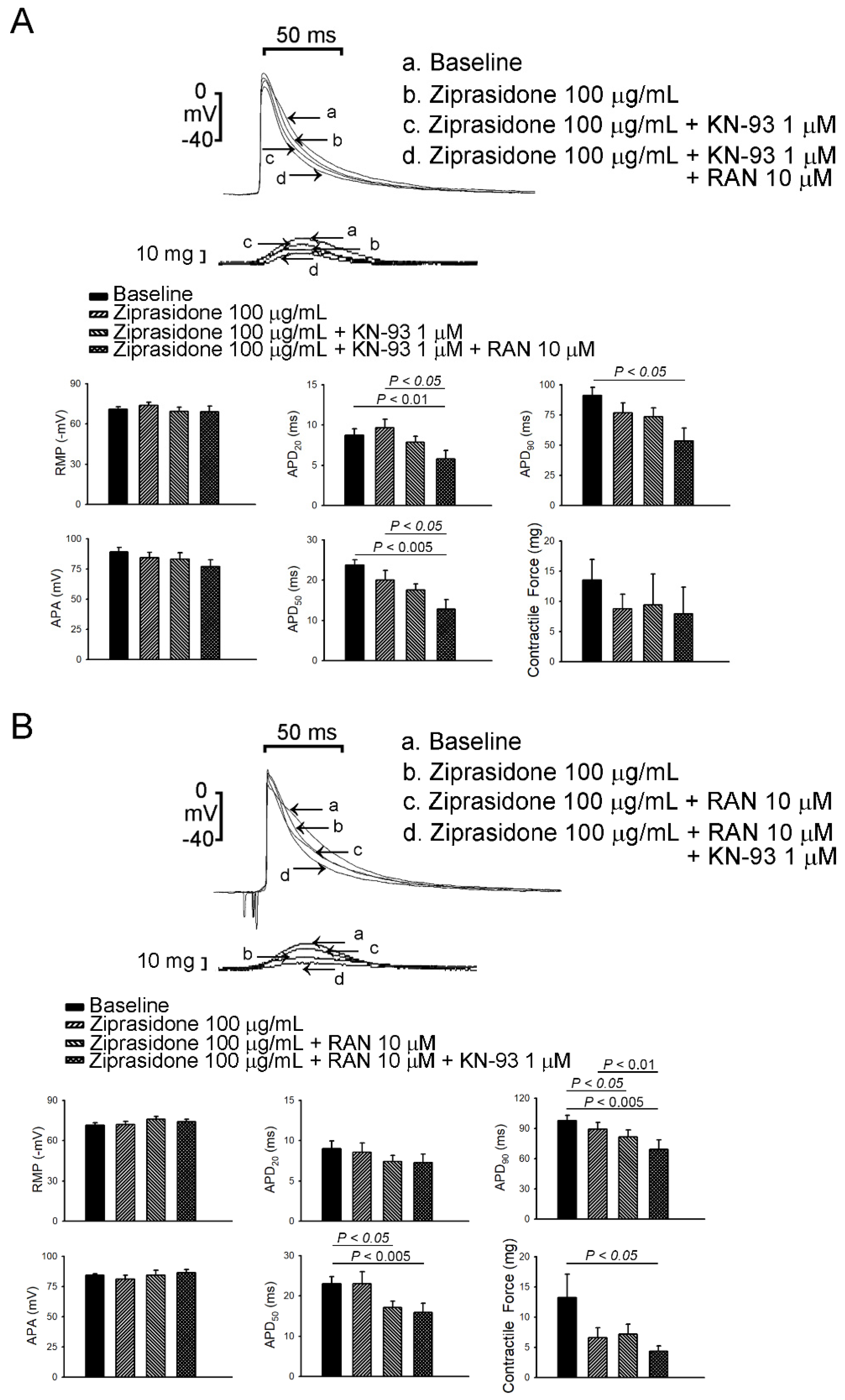
3.1. Atrial-Tissue Electrical Activity
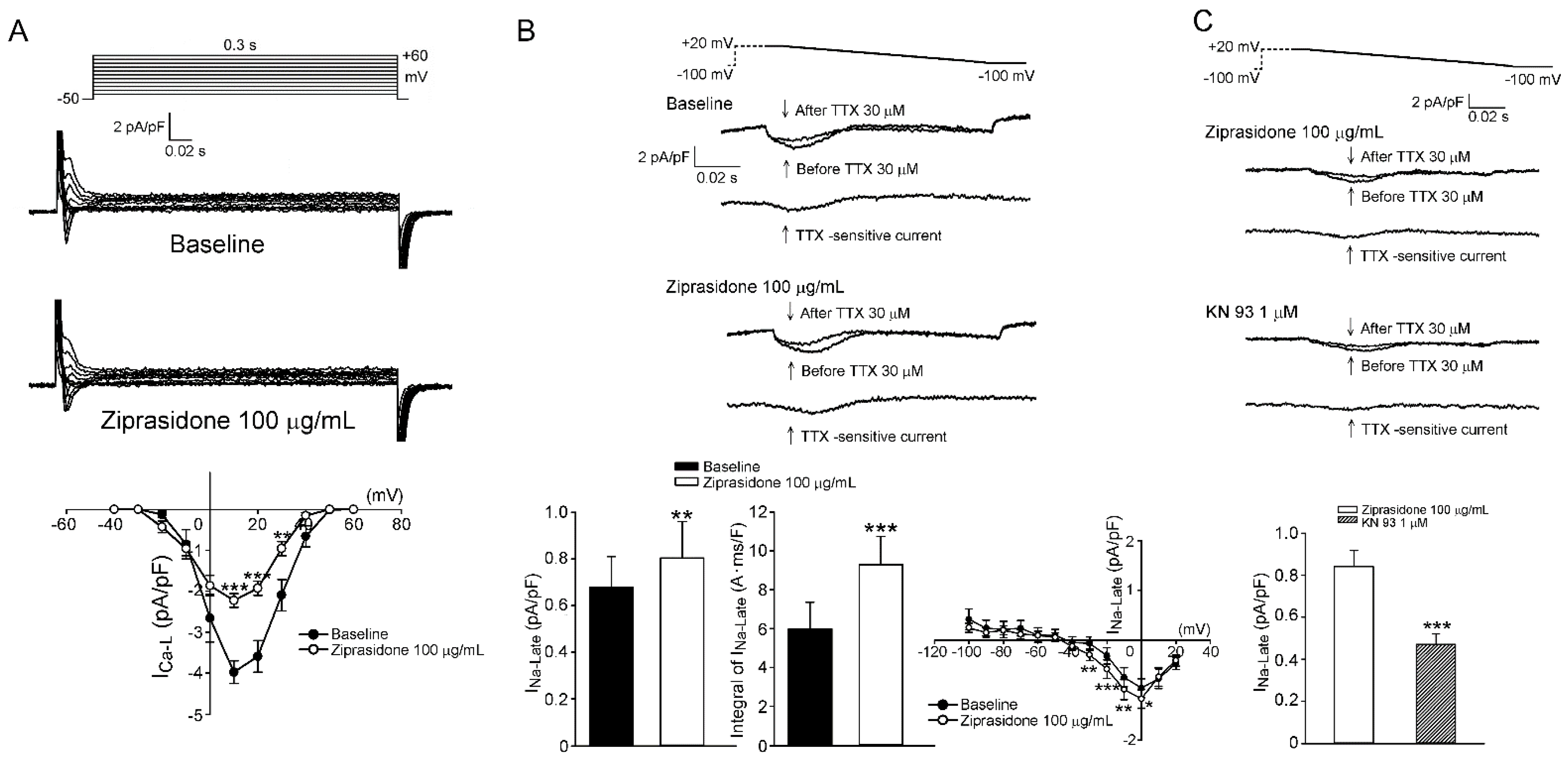
3.2. ICa,L, INa,L, and Cytosolic Na+ Levels
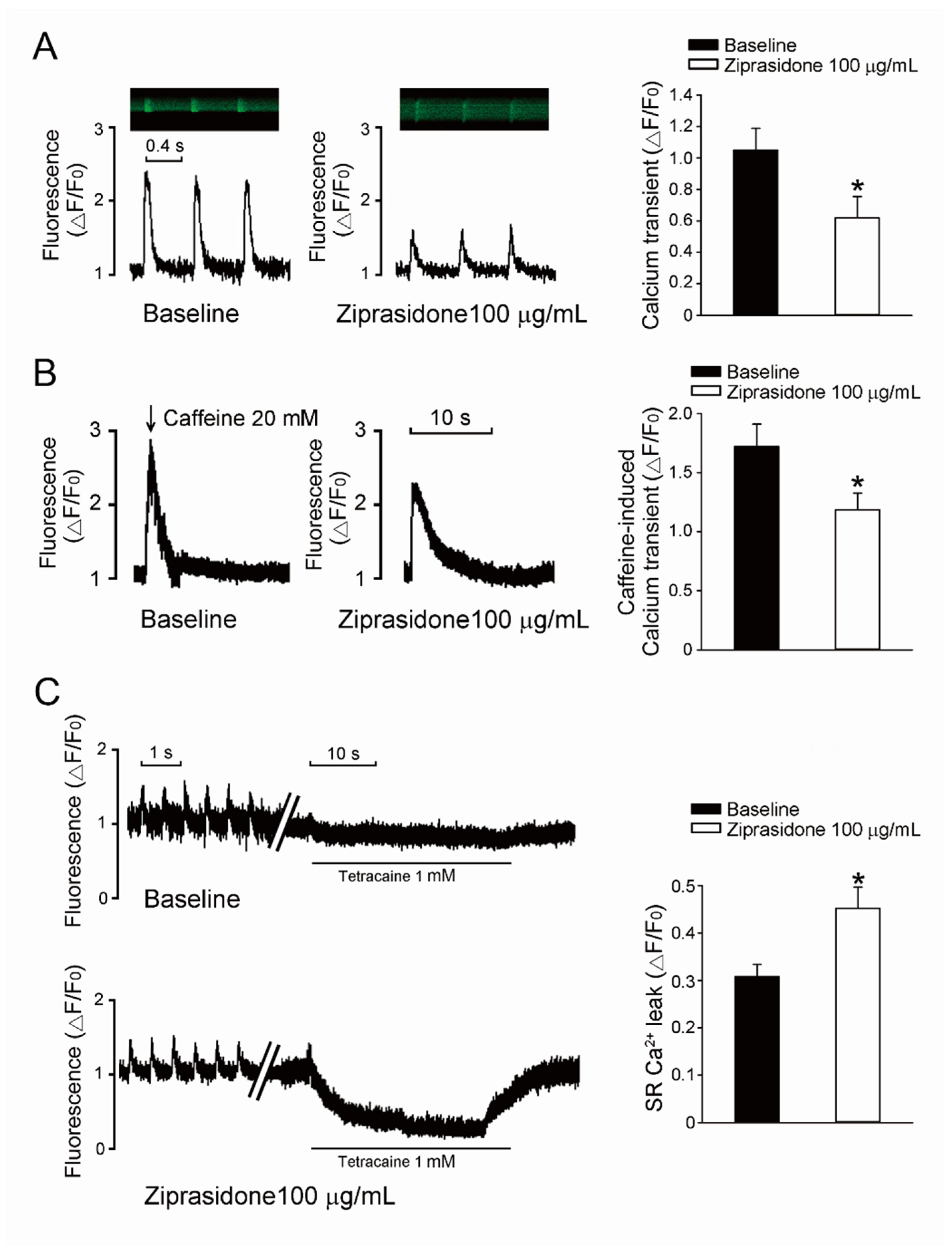
3.3. Ca2+ Transient Amplitudes, SR Ca2+ Stores, and SR Ca2+ Leak
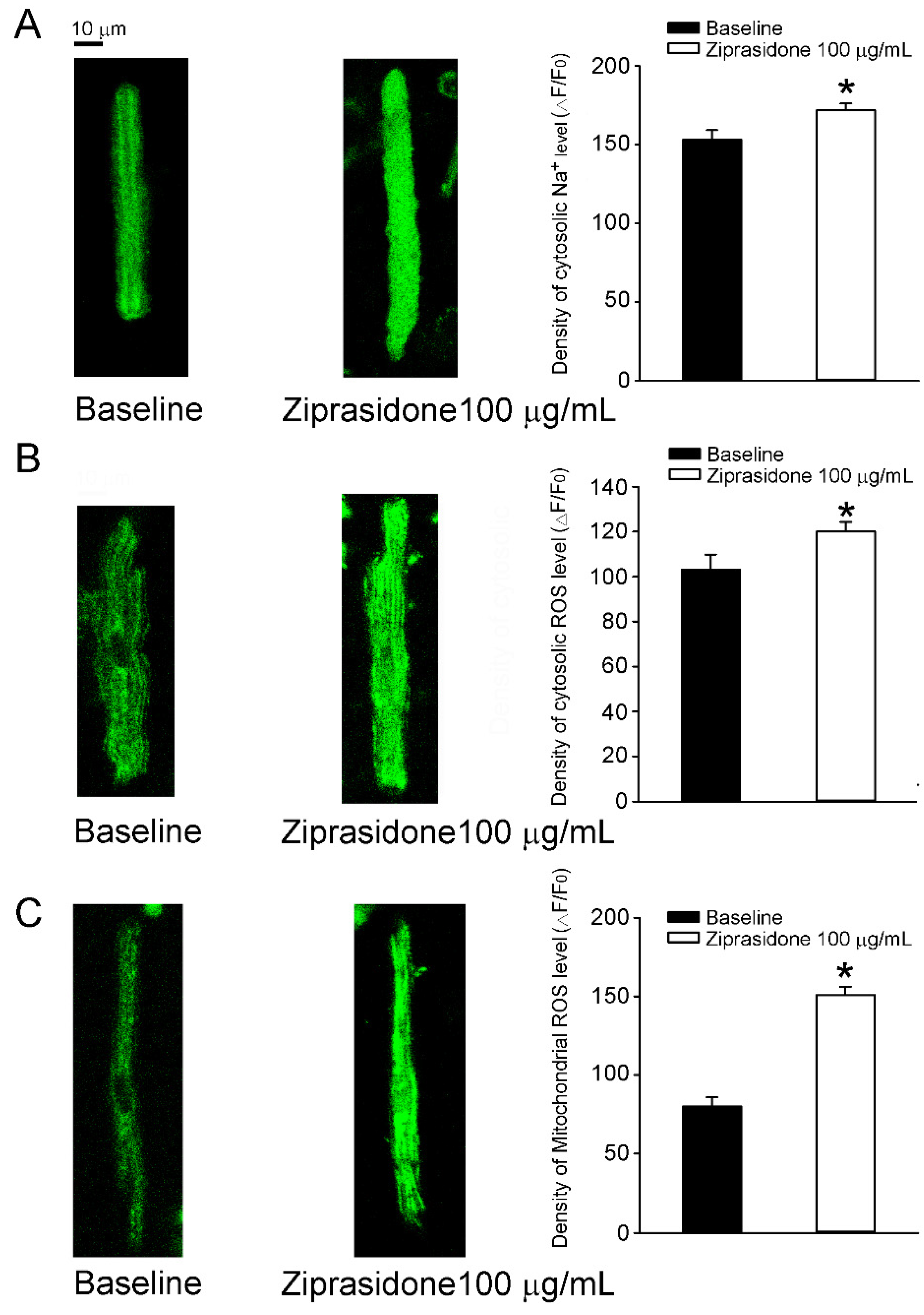
3.4. Oxidative Stress
3.5. Levels of Proinflammatory Cytokines, Inflammasome Markers, and Ca2+ Regulatory Proteins
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daniel, D.G.; Zimbroff, D.L.; Potkin, S.G.; Reeves, K.R.; Harrigan, E.P.; Lakshminarayanan, M. Ziprasidone 80 mg/day and 160 mg/day in the acute exacerbation of schizophrenia and schizoaffective disorder: A 6-week placebo-controlled trial. Ziprasidone Study Group. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 20, 491–505. [Google Scholar] [CrossRef]
- Ray, W.A.; Meredith, S.; Thapa, P.B.; Meador, K.G.; Hall, K.; Murray, K.T. Antipsychotics and the risk of sudden cardiac death. Arch. Gen. Psychiatry 2001, 58, 1161–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, W.A.; Chung, C.P.; Murray, K.T.; Hall, K.; Stein, C.M. Atypical antipsychotic drugs and the risk of sudden cardiac death. N. Engl. J. Med. 2009, 360, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.S.; Tsai, Y.T.; Tsai, H.J. Antipsychotic drugs and the risk of ventricular arrhythmia and/or sudden cardiac death: A nation-wide case-crossover study. J. Am. Heart Assoc. 2015, 4, e001568. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.E.; Winkler, D.; Konstantinidis, A.; Huf, W.; Engel, R.; Toto, S.; Grohmann, R.; Kasper, S. Cardiovascular Adverse Reactions During Antipsychotic Treatment: Results of AMSP, A Drug Surveillance Program between 1993 and 2013. Int. J. Neuropsychopharmacol. 2020, 23, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Beach, S.R.; Celano, C.M.; Sugrue, A.M.; Adams, C.; Ackerman, M.J.; Noseworthy, P.A.; Huffman, J.C. QT Prolongation, Torsades de Pointes, and Psychotropic Medications: A 5-Year Update. Psychosomatics 2018, 59, 105–122. [Google Scholar] [CrossRef]
- Loebel, A.; Miceli, J.; Chappell, P.; Siu, C. Electrocardiographic changes with ziprasidone. J. Am. Acad. Child Adolesc. Psychiatry 2006, 45, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.-H.; Lo, L.-W.; Liou, Y.-J.; Shu, J.-H.; Hsu, H.-C.; Liang, Y.; Huang, C.-C.; Huang, P.-H.; Lin, S.-J.; Chen, J.-W.; et al. Antipsychotic treatment is associated with risk of atrial fibrillation: A nationwide nested case-control study. Int. J. Cardiol. 2017, 227, 134–140. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Wolf, P.A.; D’Agostino, R.B.; Silbershatz, H.; Kannel, W.B.; Levy, D. Impact of atrial fibrillation on the risk of death: The Framingham Heart Study. Circulation 1998, 98, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, O.D.; Abildstrøm, S.Z.; Ottesen, M.M.; Rask-Madsen, C.; Bagger, H.; Køber, L. Increased risk of sudden and non-sudden cardiovascular death in patients with atrial fibrillation/flutter following acute myocardial infarction. Eur. Heart J. 2005, 27, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-S.; Lin, Y.-S.; Lin, Y.-K.; Chen, Y.-C.; Kao, Y.-H.; Hsu, C.-C.; Chen, S.-A.; Chen, Y.-J. Atrial arrhythmogenesis in a rabbit model of chronic obstructive pulmonary disease. Transl. Res. J. Lab. Clin. Med. 2020, 223, 25–39. [Google Scholar] [CrossRef]
- Lee, T.-I.; Chen, Y.-C.; Lin, Y.-K.; Chung, C.-C.; Lu, Y.-Y.; Kao, Y.-H.; Chen, Y.-J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.K.; Chen, Y.C.; Huang, J.H.; Lin, Y.-J.; Huang, S.-S.; Chen, S.-A.; Chen, Y.-J. Leptin modulates electrophysiological characteristics and isoproterenol-induced arrhythmogenesis in atrial myocytes. J. Biomed. Sci. 2013, 20, 94. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Chen, S.A.; Chen, Y.C.; Yeh, H.I.; Chan, P.; Chang, M.S.; Lin, C.I. Effects of rapid atrial pacing on the arrhythmogenic activity of single cardiomyocytes from pulmonary veins: Implication in initiation of atrial fibrillation. Circulation 2001, 104, 2849–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-Y.; Chen, Y.-C.; Kao, Y.-H.; Hsieh, M.-H.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. Redox and Activation of Protein Kinase a Dysregulates Calcium Homeostasis in Pulmonary Vein Cardiomyocytes of Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005701. [Google Scholar] [CrossRef]
- Linley, J.E. Perforated whole-cell patch-clamp recording. Methods Mol. Biol. 2013, 998, 149–157. [Google Scholar] [PubMed]
- Chen, Y.-C.; Kao, Y.-H.; Huang, C.-F.; Cheng, C.-C.; Chen, Y.-J.; Chen, S.-A. Heat stress responses modulate calcium regulations and electrophysiological characteristics in atrial myocytes. J. Mol. Cell. Cardiol. 2010, 48, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Wongcharoen, W.; Chen, Y.-C.; Chen, Y.-J.; Chang, C.-M.; Yeh, H.-I.; Lin, C.-I.; Chen, S.-A. Effects of a Na+/Ca2+ exchanger inhibitor on pulmonary vein electrical activity and ouabain-induced arrhythmogenicity. Cardiovasc. Res. 2006, 70, 497–508. [Google Scholar] [CrossRef]
- Shannon, T.R.; Ginsburg, K.S.; Bers, D.M. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 2002, 91, 594–600. [Google Scholar] [CrossRef] [Green Version]
- Viatchenko-Karpinski, S.; Kornyeyev, D.; El-Bizri, N.; Budas, G.; Fan, P.; Jiang, Z.; Yang, J.; Anderson, M.E.; Shryock, J.C.; Chang, C.-P. Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 2014, 76, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-Y.; Chen, Y.-C.; Kao, Y.-H.; Hsieh, M.-H.; Lin, Y.-K.; Chung, C.-C.; Lee, T.-I.; Tsai, W.-C.; Chen, S.-A.; Chen, Y.-J. Fibroblast growth factor 23 dysregulates late sodium current and calcium homeostasis with enhanced arrhythmogenesis in pulmonary vein cardiomyocytes. Oncotarget 2016, 7, 69231–69242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Herting, J.; Tirilomis, T.; Renner, A.; Neef, S.; Toischer, K.; Ellenberger, D.; Förster, A.; Schmitto, J.D.; Gummer, J.; et al. Ca/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 2013, 128, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Chiang, D.Y.; Wang, S.; Wang, Q.; Sun, L.; Voigt, N.; Respress, J.L.; Ather, S.; Skapura, D.G.; Jordan, V.K.; et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 2014, 129, 1276–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003, 92, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelu, M.G.; Sarma, S.; Sood, S.; Wang, S.; van Oort, R.J.; Skapura, D.G.; Li, N.; Santonastasi, M.; Müller, F.U.; Schmitz, W.; et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Investig. 2009, 119, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Shannon, T.R.; Pogwizd, S.M.; Bers, D.M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 2003, 93, 592–594. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M.; Eisner, D.A.; Valdivia, H.H. Sarcoplasmic reticulum Ca2+ and heart failure: Roles of diastolic leak and Ca2+ transport. Circ. Res. 2003, 93, 487–490. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.S.; Bers, D.M. Role of Ca/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sag, C.M.; Wadsack, D.P.; Khabbazzadeh, S.; Abesser, M.; Grefe, C.; Neumann, K.; Opiela, M.-K.; Backs, J.; Olson, E.N.; Brown, J.H. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ. Heart Fail. 2009, 2, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrens, X.H.; Lehnart, S.E.; Reiken, S.R.; Marks, A.R. Ca/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004, 94, e61–e70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer-Short, A.; Musa, H.; Alsina, K.M.; Ni, L.; Word, T.A.; Reynolds, J.O.; Gratz, D.; Lane, C.; El-Refaey, M.; Unudurthi, S. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2020, 17, 503–511. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 1998, 98, 2545–2552. [Google Scholar] [CrossRef]
- Armoundas, A.A.; Hobai, I.A.; Tomaselli, G.F.; Winslow, R.L.; O’Rourke, B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 2003, 93, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Shryock, J.C.; Belardinelli, L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2031–H2039. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.A.; Reznikov, V.; Undrovinas, N.A.; Sabbah, H.N.; Undrovinas, A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: Similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1597–H1608. [Google Scholar] [CrossRef] [Green Version]
- Sapia, L.; Palomeque, J.; Mattiazzi, A.; Petroff, M.V. Na/K ATPase inhibition by ouabain induces CaMKII-dependent apoptosis in adult rat cardiac myocytes. J. Mol. Cell. Cardiol. 2010, 49, 459–468. [Google Scholar] [CrossRef]
- Koval, O.M.; Snyder, J.S.; Wolf, R.M.; Pavlovicz, R.E.; Glynn, P.; Curran, J.; Leymaster, N.D.; Dun, W.; Wright, P.J.; Cardona, N.; et al. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 2012, 126, 2084–2094. [Google Scholar] [CrossRef] [Green Version]
- Belardinelli, L.; Shryock, J.C.; Fraser, H. Inhibition of the late sodium current as a potential cardioprotective principle: Effects of the late sodium current inhibitor ranolazine. Heart 2006, 92 (Suppl. S4), iv6–iv14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasenfuss, G.; Maier, L.S. Mechanism of action of the new anti-ischemia drug ranolazine. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2008, 97, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K.; Davies, S.S.; Nakajima, T.; Ong, B.-H.; Kupershmidt, S.; Fessel, J.; Amarnath, V.; Anderson, M.E.; Boyden, P.A.; Viswanathan, P.C.; et al. Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ. Res. 2005, 97, 1262–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Kolettis, T.M.; Galaris, D.; Goudevenos, J.A. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int. J. Cardiol. 2007, 115, 135–143. [Google Scholar] [CrossRef]
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855. [Google Scholar] [CrossRef]
- Nishio, S.; Teshima, Y.; Takahashi, N.; Thuc, L.C.; Saito, S.; Fukui, A.; Kume, O.; Fukunaga, N.; Hara, M.; Nakagawa, M.; et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J. Mol. Cell. Cardiol. 2012, 52, 1103–1111. [Google Scholar] [CrossRef]
- Wagner, S.; Ruff, H.M.; Weber, S.L.; Bellmann, S.; Sowa, T.; Schulte, T.; Anderson, M.E.; Grandi, E.; Bers, D.M.; Backs, J.; et al. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 2011, 108, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Lee, C.H.; Lee, J.G.; Kim, L.W.; Shin, B.S.; Lee, B.J.; Kim, Y.H. Protective effects of atypical antipsychotic drugs against MPP(+)-induced oxidative stress in PC12 cells. Neurosci. Res. 2011, 69, 283–290. [Google Scholar] [CrossRef]
- Terada, K.; Murata, A.; Toki, E.; Goto, S.; Yamakawa, H.; Setoguchi, S.; Watase, D.; Koga, M.; Takata, J.; Matsunaga, K.; et al. Atypical Antipsychotic Drug Ziprasidone Protects against Rotenone-Induced Neurotoxicity: An In Vitro Study. Molecules 2020, 25, 4206. [Google Scholar] [CrossRef]
- Nikolić-Kokić, A.; Tatalović, N.; Nestorov, J.; Mijović, M.; Mijusković, A.; Miler, M.; Oreščanin-Dušić, Z.; Nikolić, M.; Milošević, V.; Blagojević, D.; et al. Clozapine, ziprasidone, and sertindole-induced morphological changes in the rat heart and their relationship to antioxidant enzymes function. J. Toxicol. Environ. Health Part A 2018, 81, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Brundel, B. Inflammasomes and Proteostasis Novel Molecular Mechanisms Associated with Atrial Fibrillation. Circ. Res. 2020, 127, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.; Barbisan, F.; do Prado-Lima, P.A.S.; Azzolin, V.F.; Jung, I.E.d.C.; Duarte, M.M.M.F.; Teixeira, C.F.; Mastella, M.H.; da Cruz, I.B.M. Ziprasidone, a second-generation antipsychotic drug, triggers a macrophage inflammatory response in vitro. Cytokine 2018, 106, 101–107. [Google Scholar] [CrossRef]
- Rayner-Hartley, E.; Sedlak, T. Ranolazine: A Contemporary Review. J. Am. Heart Assoc. 2016, 5, e003196. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C.; Belardinelli, L.; Zygmunt, A.C.; Burashnikov, A.; di Diego, J.M.; Fish, J.M.; Cordeiro, J.M.; Thomas, G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 2004, 110, 904–910. [Google Scholar] [CrossRef]
- Gao, L.; Blair, L.A.; Marshall, J. CaMKII-independent effects of KN93 and its inactive analog KN92: Reversible inhibition of L-type calcium channels. Biochem. Biophys. Res. Commun. 2006, 345, 1606–1610. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tai, B.-Y.; Lu, M.-K.; Yang, H.-Y.; Tsai, C.-S.; Lin, C.-Y. Ziprasidone Induces Rabbit Atrium Arrhythmogenesis via Modification of Oxidative Stress and Sodium/Calcium Homeostasis. Biomedicines 2022, 10, 976. https://doi.org/10.3390/biomedicines10050976
Tai B-Y, Lu M-K, Yang H-Y, Tsai C-S, Lin C-Y. Ziprasidone Induces Rabbit Atrium Arrhythmogenesis via Modification of Oxidative Stress and Sodium/Calcium Homeostasis. Biomedicines. 2022; 10(5):976. https://doi.org/10.3390/biomedicines10050976
Chicago/Turabian StyleTai, Buh-Yuan, Ming-Kun Lu, Hsiang-Yu Yang, Chien-Sung Tsai, and Chih-Yuan Lin. 2022. "Ziprasidone Induces Rabbit Atrium Arrhythmogenesis via Modification of Oxidative Stress and Sodium/Calcium Homeostasis" Biomedicines 10, no. 5: 976. https://doi.org/10.3390/biomedicines10050976
APA StyleTai, B.-Y., Lu, M.-K., Yang, H.-Y., Tsai, C.-S., & Lin, C.-Y. (2022). Ziprasidone Induces Rabbit Atrium Arrhythmogenesis via Modification of Oxidative Stress and Sodium/Calcium Homeostasis. Biomedicines, 10(5), 976. https://doi.org/10.3390/biomedicines10050976