
Incorporation of Oxidized Phenylalanine Derivatives into Insulin Signaling Relevant Proteins May Link Oxidative Stress to Signaling Conditions Underlying Chronic Insulin Resistance

 , ,
, ,  , and
, and
Abstract
1. Introduction
1.1. Pathogenesis of Insulin Resistance
1.2. Acute Insulin Resistance
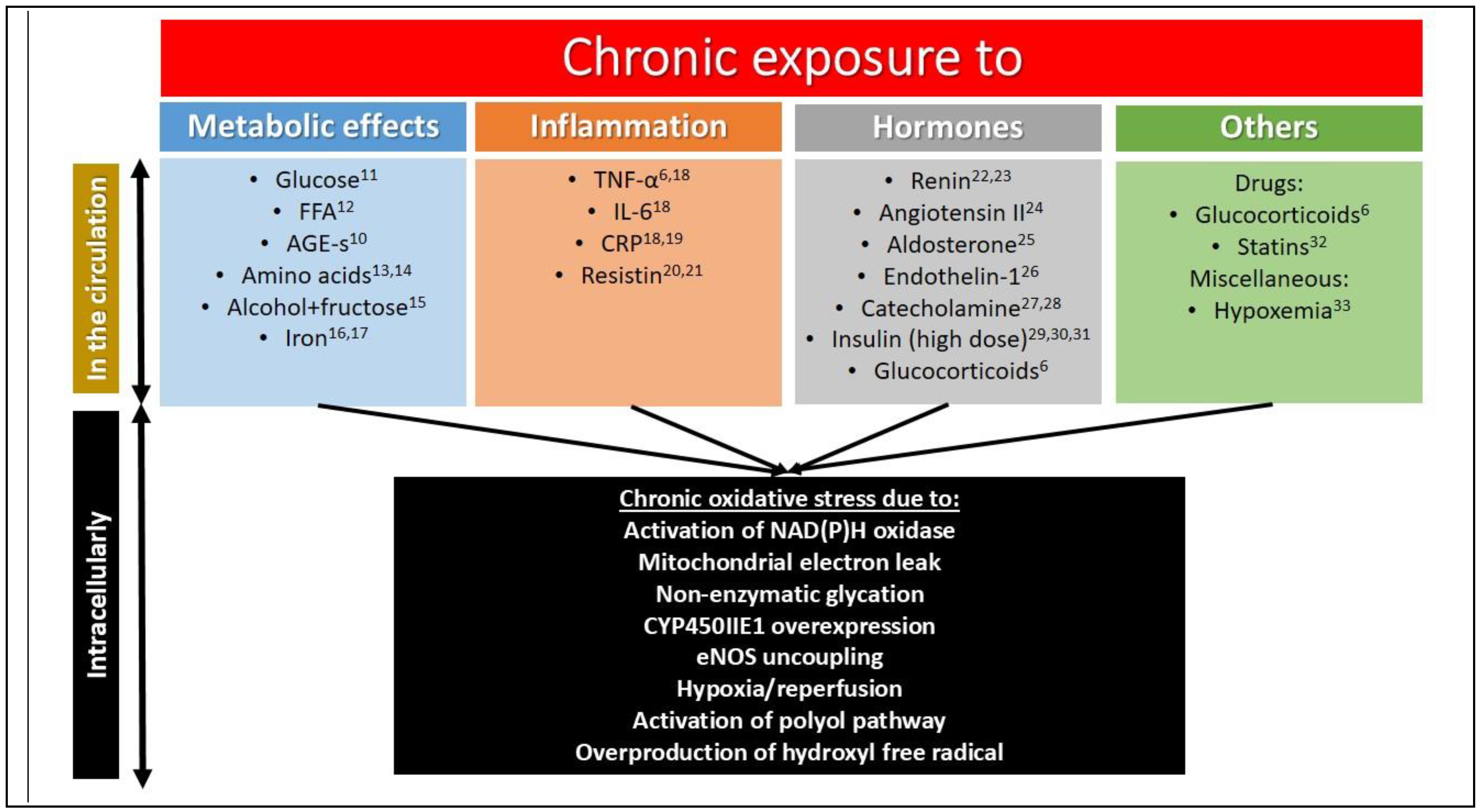
1.3. Chronic Insulin Resistance
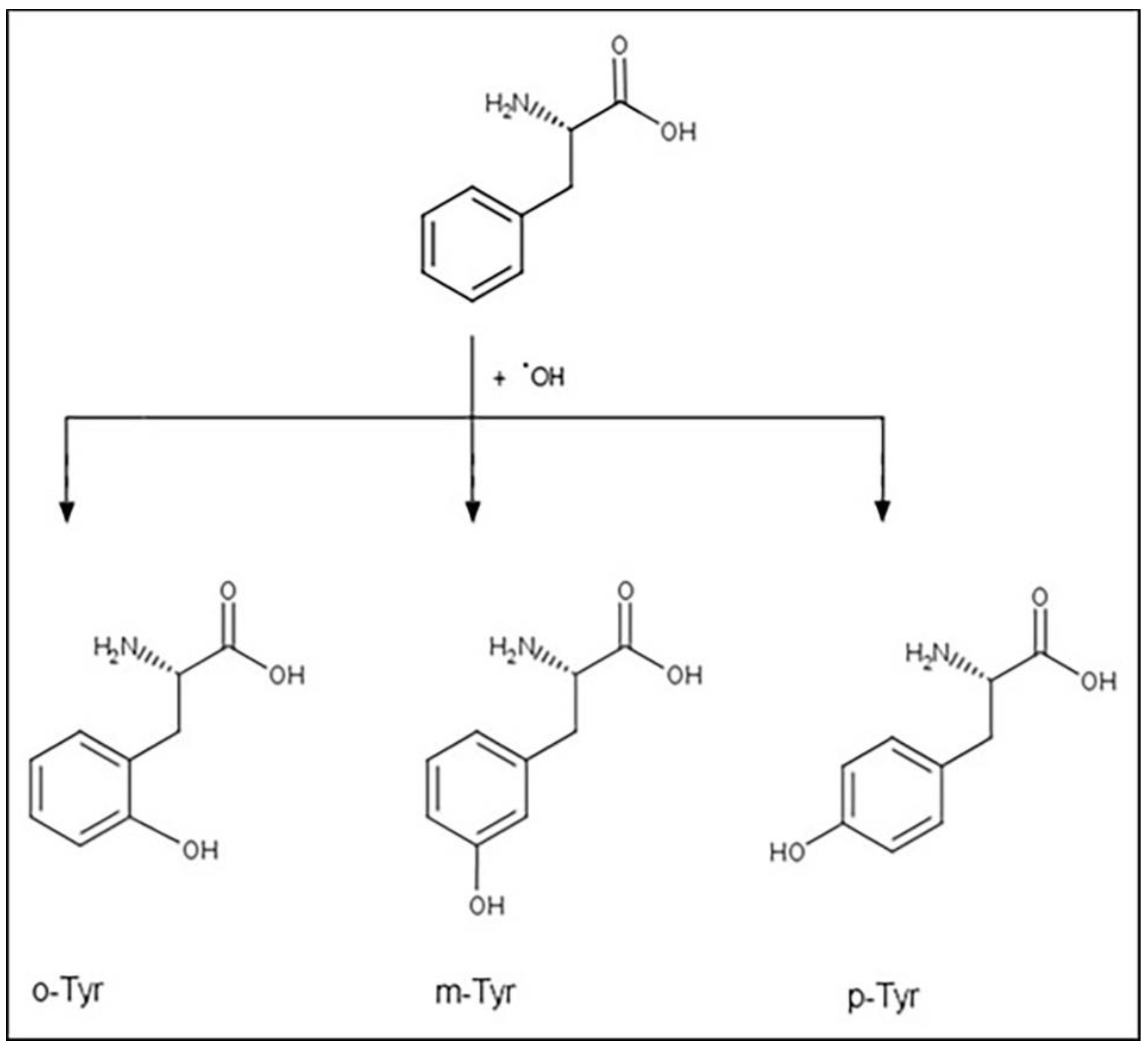
1.4. Tyrosine Isomers and Hydroxyl Free Radical
1.5. Non-Physiological Tyrosine Isomers as Markers
1.6. Non-Physiological Tyrosine Isomers as “Makers”
2. Materials and Methods
2.1. Cell Culture
2.2. Isotope Glucose Uptake
2.3. HPLC Analysis
2.4. Western Blot Analysis
2.5. Protein Expression
2.6. Peptide Synthesis
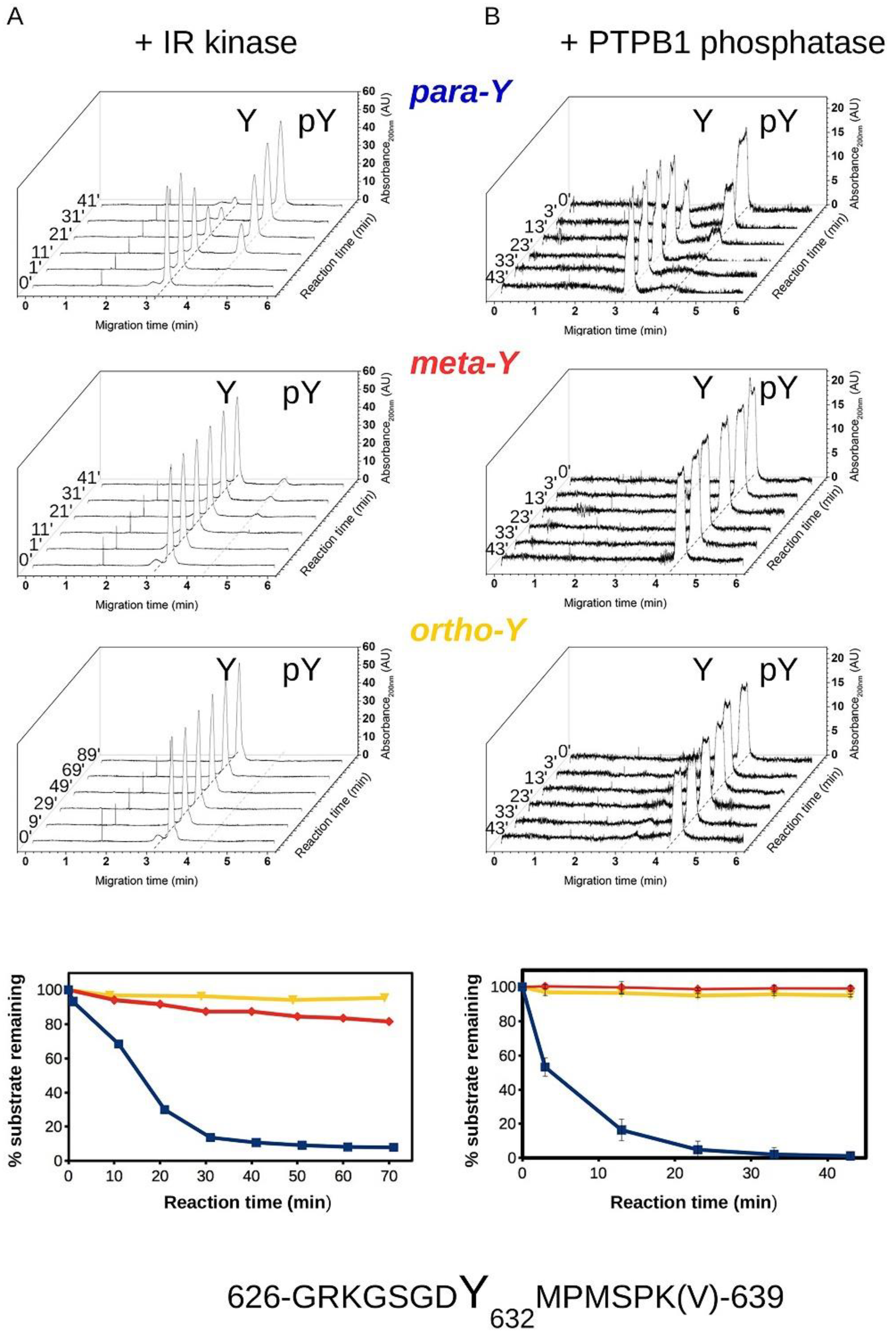
2.7. Phosphorylation and Dephosphorylation Assays—Capillary Electrophoresis
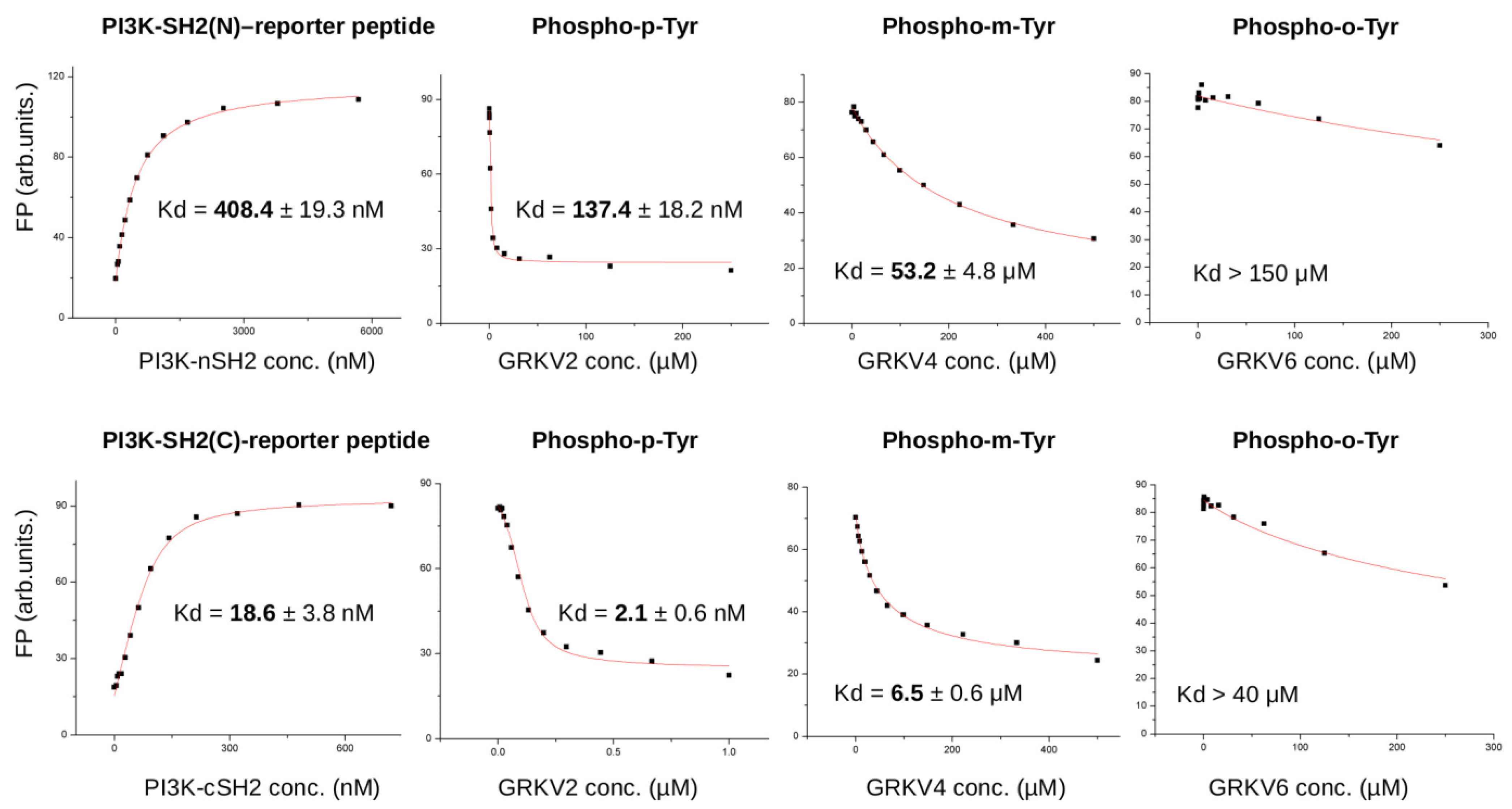
2.8. Protein-Peptide Binding Assays
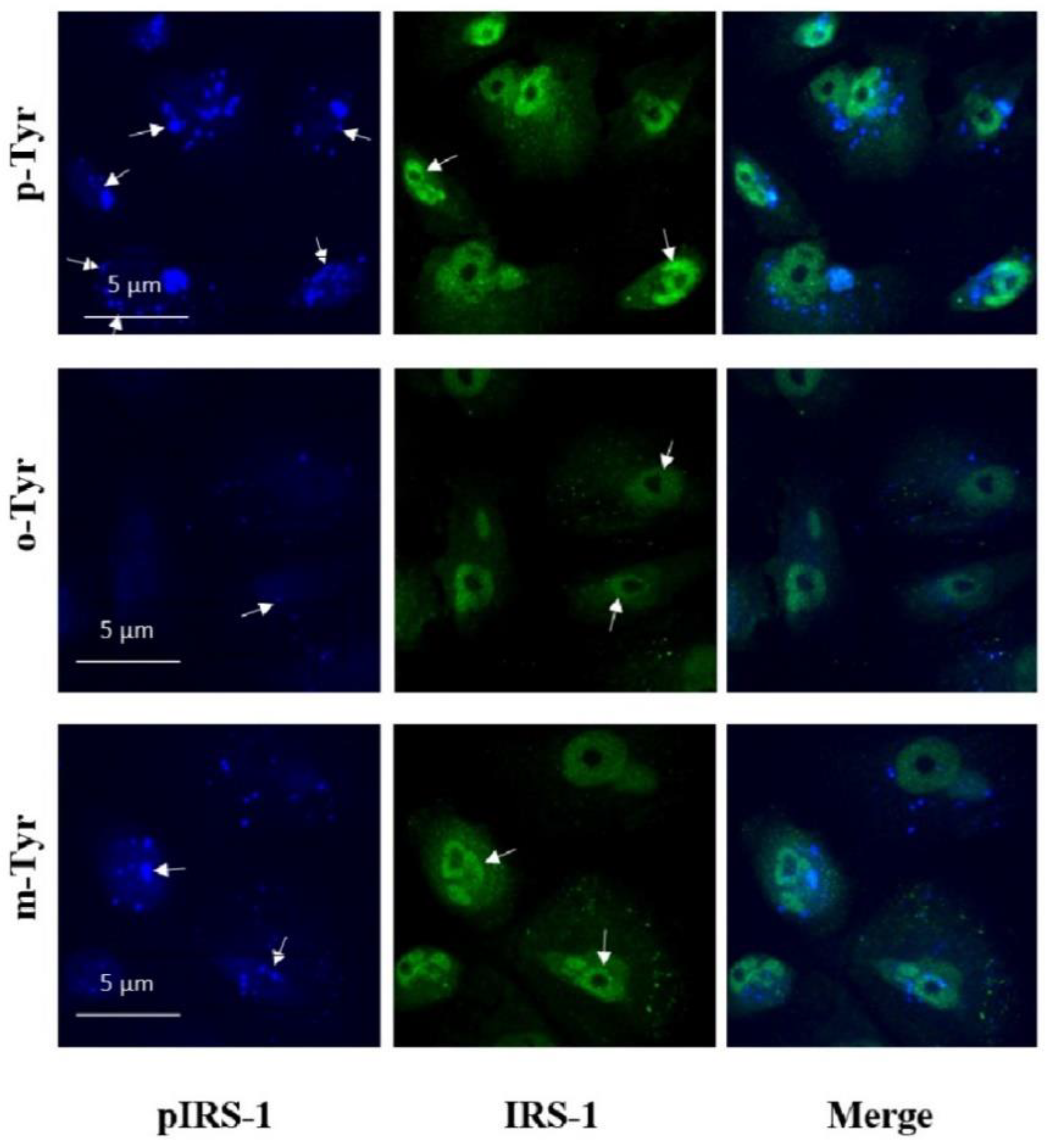
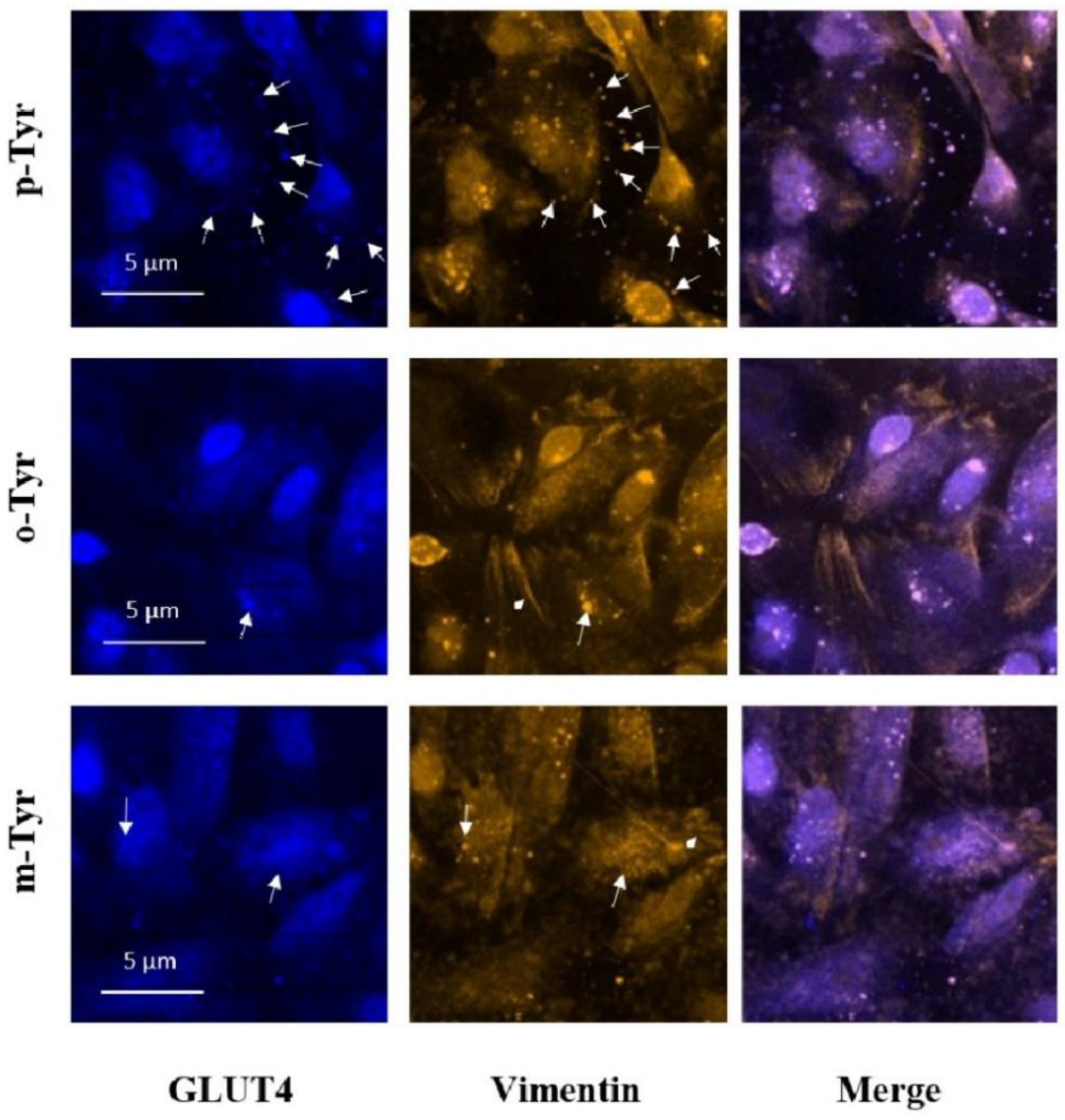
2.9. Immunofluorescence
2.10. Statistical Analysis
3. Results
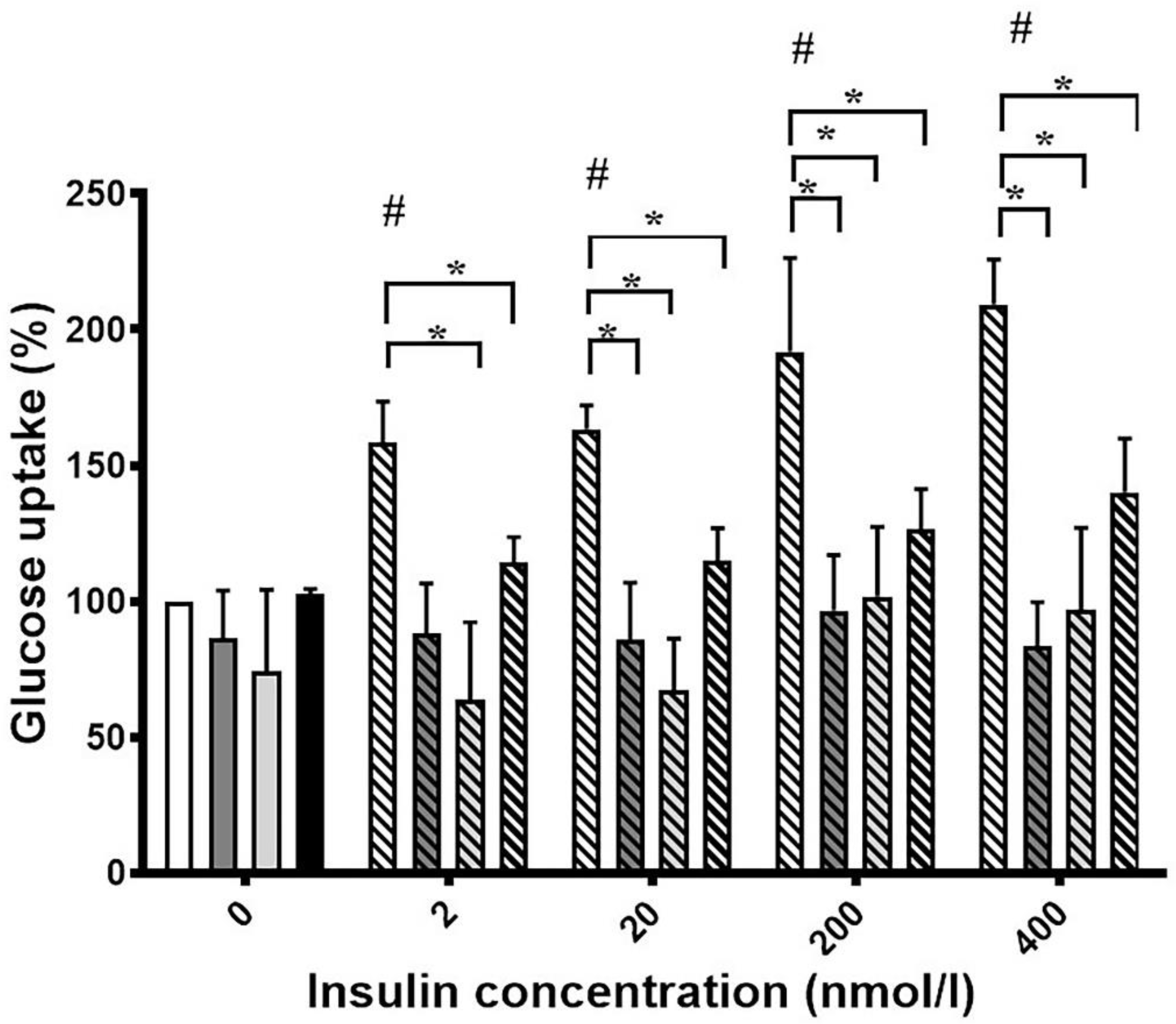
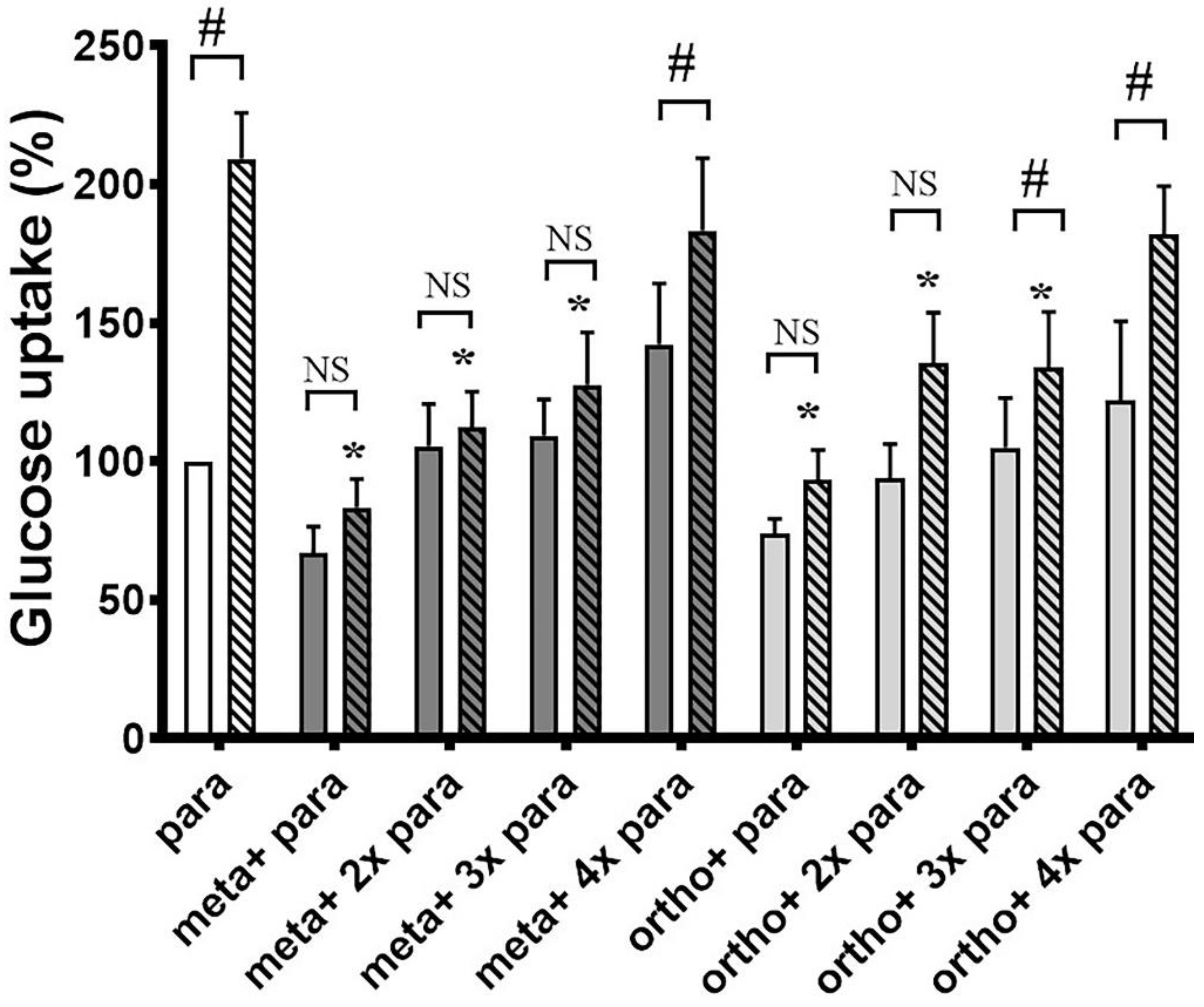
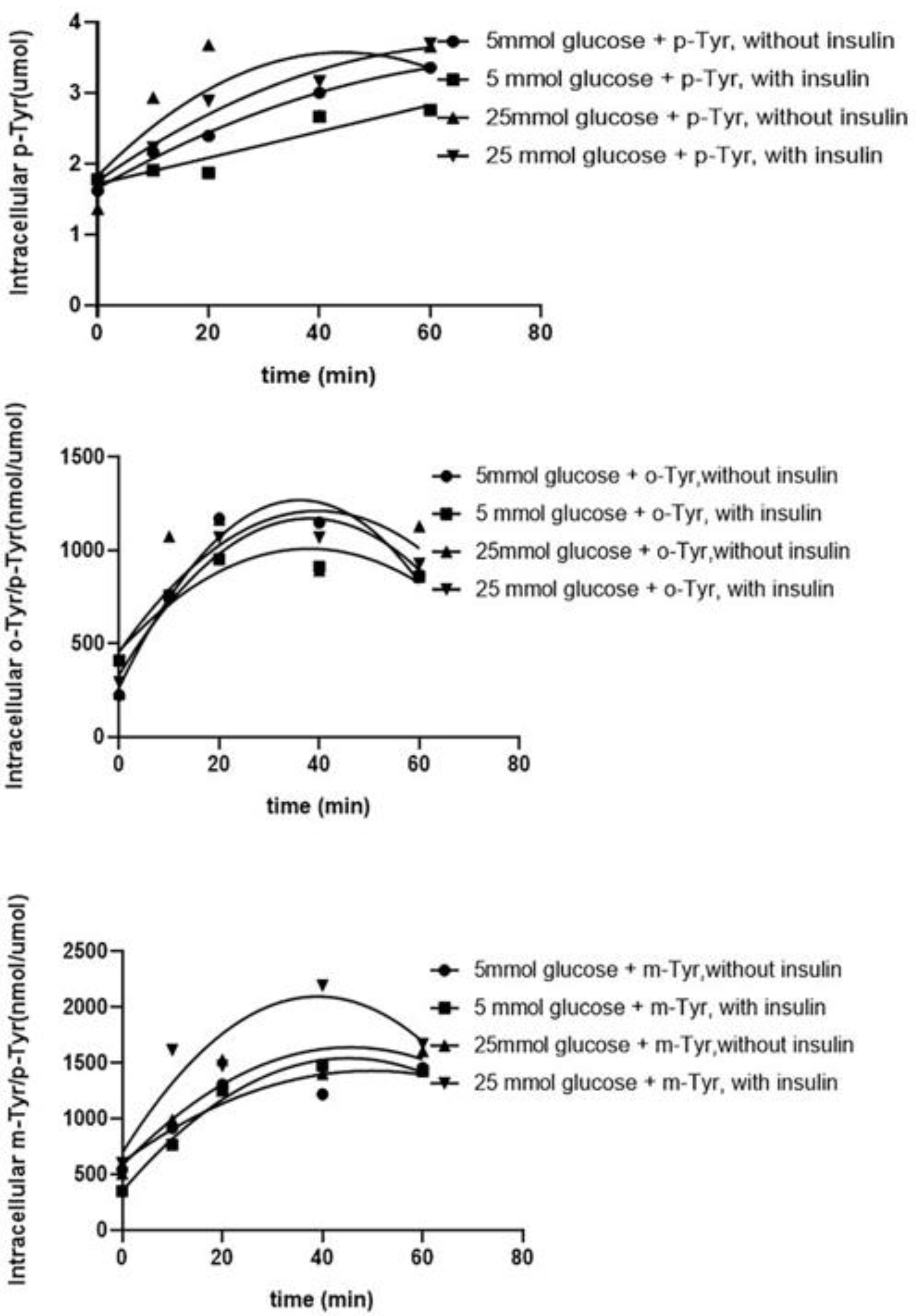
3.1. Ortho- and Meta-Tyrosine Inhibit Insulin-Induced Glucose Uptake
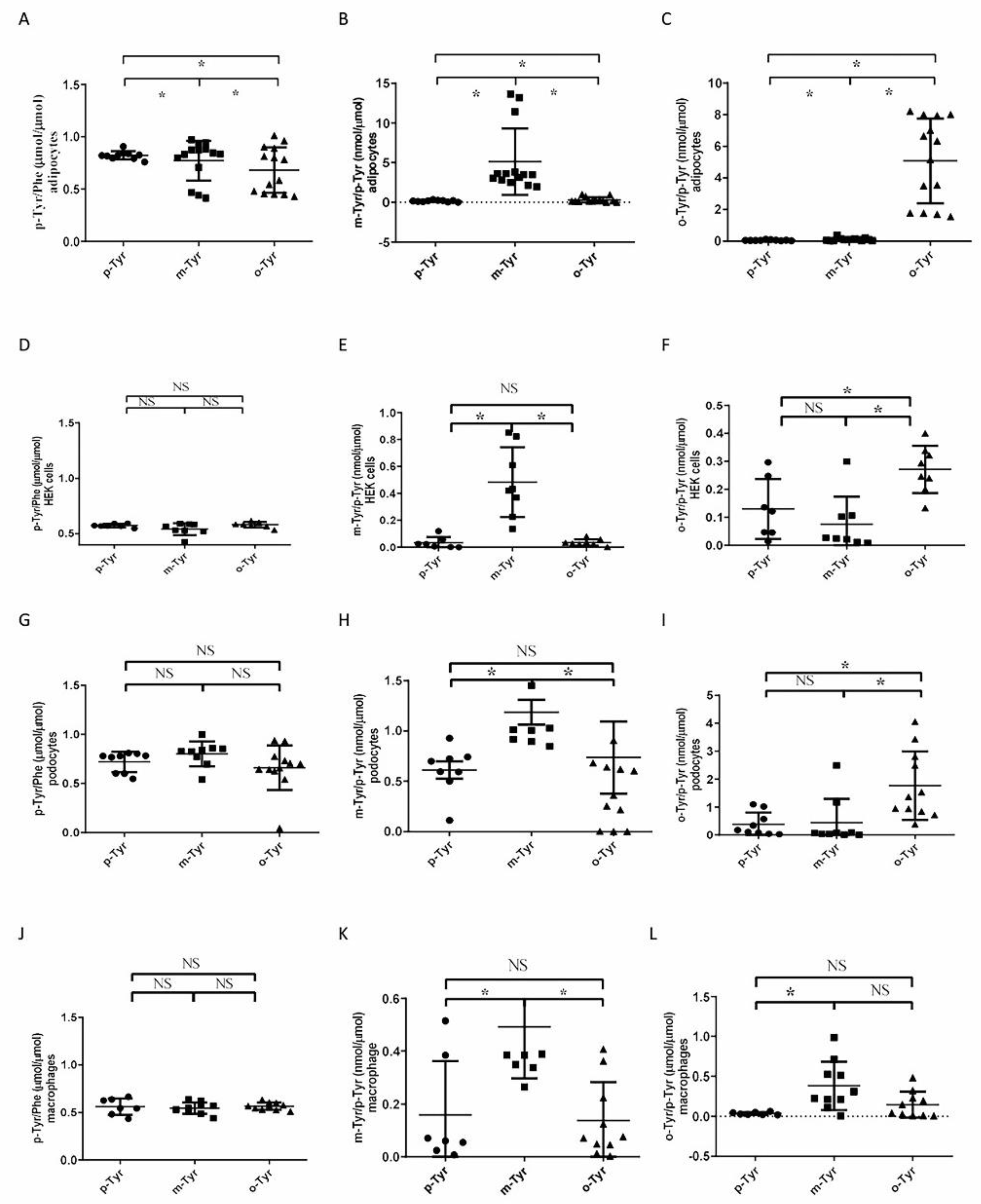
3.1.1. Both o- and m-Tyr Can Be Taken Up by Fat Cells within Several Minutes and Are Incorporated into Cellular Proteins
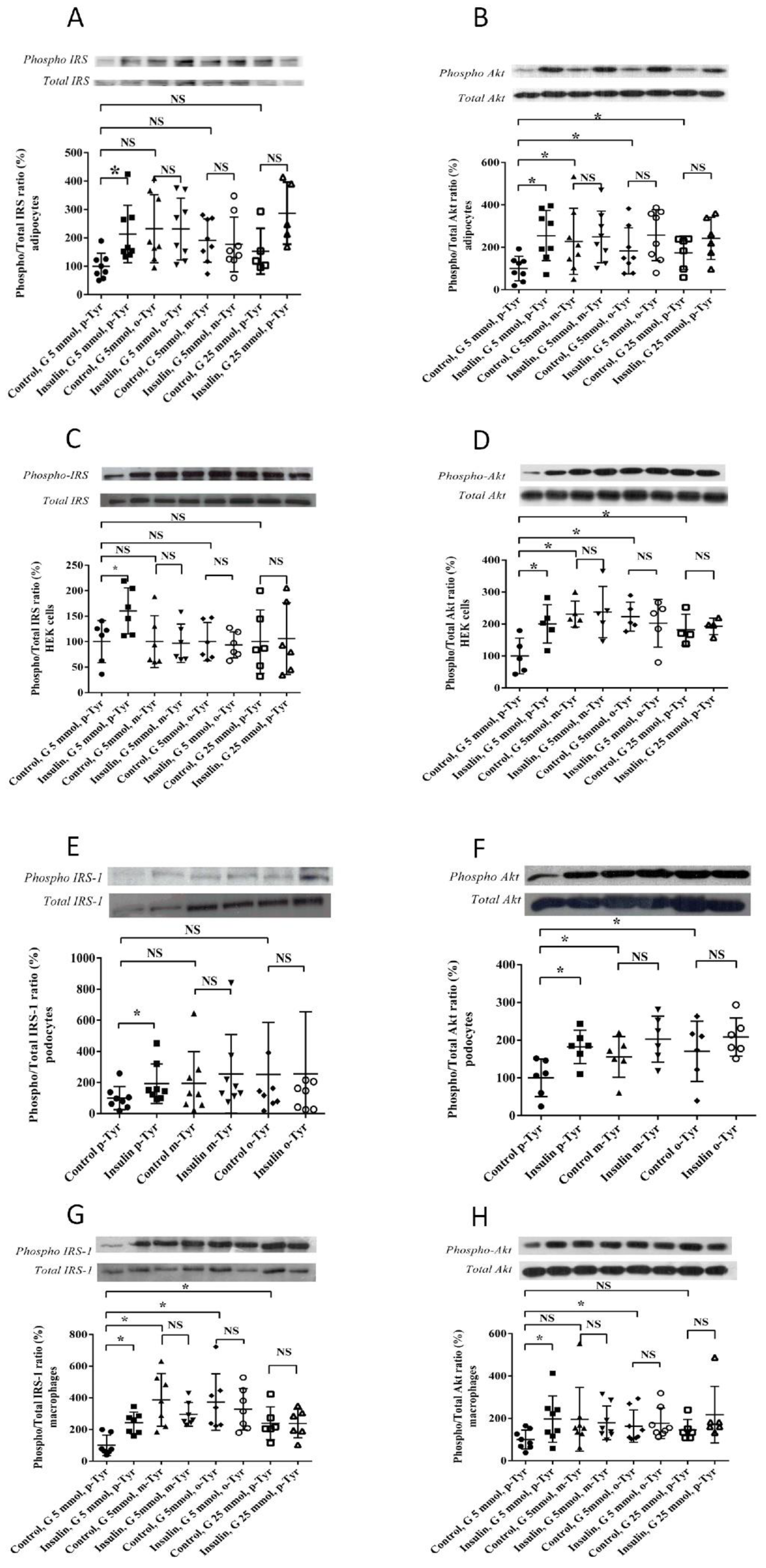
3.1.2. Phosphorylation of IRS-1 in Cells Grown on o- or m-Tyr
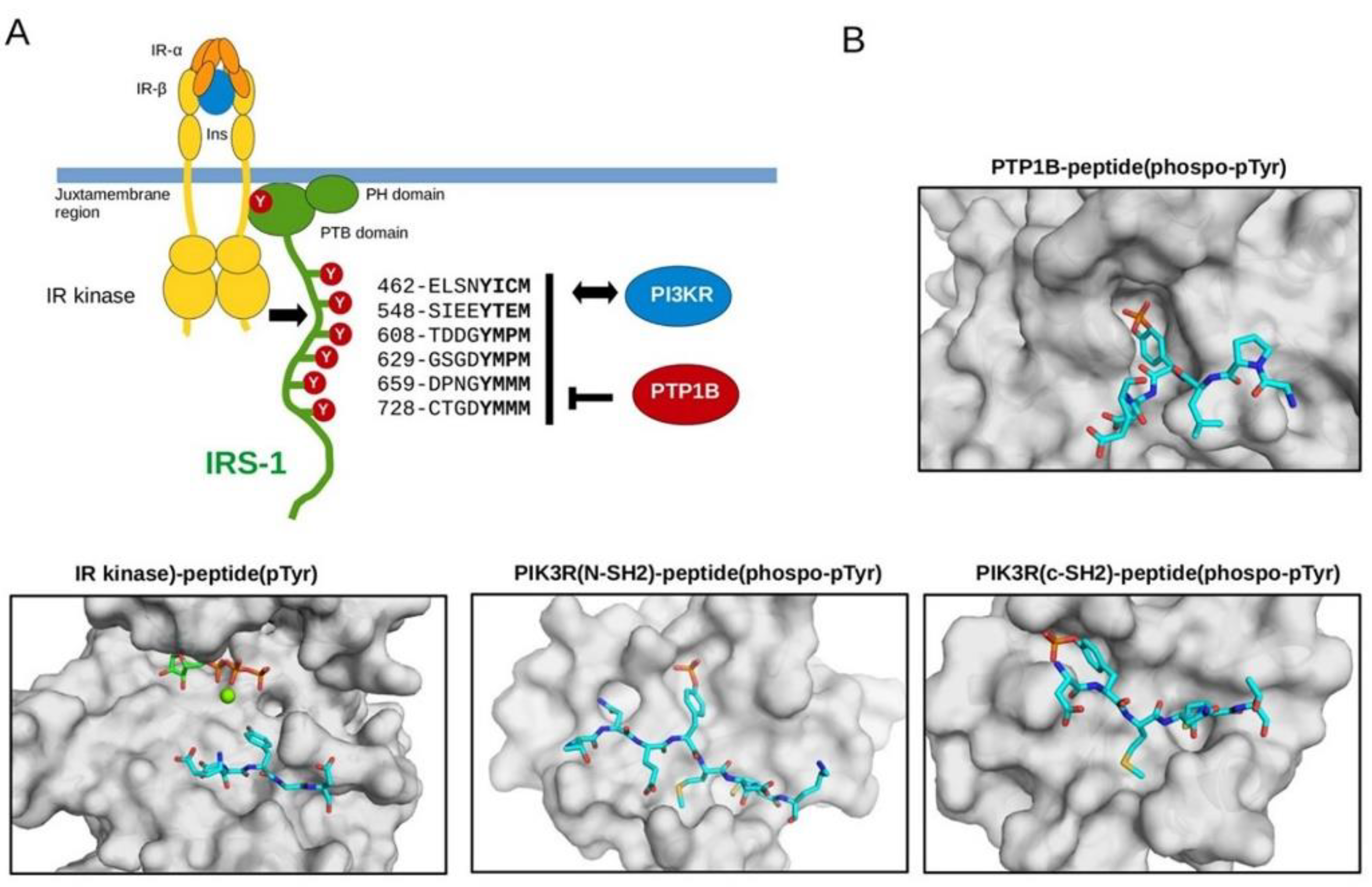
3.2. Biochemical Characterization of IRS Peptides Containing Different Forms of Tyrosine
3.3. Microscopical Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Fronzo, R.A. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.A.; Gavin, J.R.; Aguilar, R.B. The time is right for a new classification system for diabetes: Rationale and implications of the beta-cell-centric classification schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Pessin, J.E. Insulin regulation of glucose uptake: A complex interplay of intracellular signalling pathways. Diabetologia 2002, 45, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Galiero, R.; Caturano, A.; Vetrano, E.; Cesaro, A.; Rinaldi, L.; Salvatore, T.; Marfella, R.; Sardu, C.; Moscarella, E.; Gragnano, F.; et al. Pathophysiological mechanisms and clinical evidence of relationship between Nonalcoholic fatty liver disease (NAFLD) and cardiovascular disease. Rev. Cardiovasc. Med. 2021, 22, 755–768. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Akbari, M.; Ostadmohammadi, V.; Lankarani, K.B.; Tabrizi, R.; Kolahdooz, F.; Khatibi, S.R.; Asemi, Z. The effects of alpha-lipoic acid supplementation on glucose control and lipid profiles among patients with metabolic diseases: A systematic review and meta-analysis of randomized controlled trials. Metabolism 2018, 87, 56–69. [Google Scholar] [CrossRef]
- Garvey, W.T.; Olefsky, J.M.; Griffin, J.; Hamman, R.F.; Kolterman, O.G. The effect of insulin treatment on insulin secretion and insulin action in type, I.I.; diabetes mellitus. Diabetes 1985, 34, 222–234. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Xu, W.; Shi, L.; Zhang, Q.; Zhu, D.; Hu, Y.; Zhou, Z.; Yan, X.; Tian, H.; et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel-group trial. Lancet 2008, 371, 1753–1760. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A.; et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: A randomised controlled trial. Diabetologia 2016, 59, 2181–2192. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. FFA-induced hepatic insulin resistance in vivo is mediated by, P.K.;Cδ, NADPH oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E34–E46. [Google Scholar] [CrossRef]
- Li, H.; Lee, J.; He, C.; Zou, M.-H.; Xie, Z. Suppression of the mTORC1/STAT3/Notch1 pathway by activated, A.M.;PK prevents hepatic insulin resistance induced by excess amino acids. Am. J. Physiol. Metab. 2014, 306, E197–E209. [Google Scholar] [CrossRef]
- Gu, C.; Shi, Y.; Le, G. Effect of dietary protein level and origin on the redox status in the digestive tract of mice. Int. J. Mol. Sci. 2008, 9, 464–475. [Google Scholar] [CrossRef]
- Alwahsh, S.M.; Xu, M.; Schultze, F.C.; Wilting, J.; Mihm, S.; Raddatz, D.; Ramadori, G. Combination of alcohol and fructose exacerbates metabolic imbalance in terms of hepatic damage, dyslipidemia, and insulin resistance in rats. PLoS ONE 2014, 9, e104220. [Google Scholar] [CrossRef]
- Wlazlo, N.; van Greevenbroek, M.M.; Ferreira, I.; Jansen, E.H.; Feskens, E.J.; van der Kallen, C.J.; Schalkwijk, C.G.; Bravenboer, B.; Stehouwer, C.D. Iron metabolism is associated with adipocyte insulin resistance and plasma adiponectin: The Cohort on Diabetes and Atherosclerosis Maastricht (CODAM) study. Diabetes Care 2013, 36, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Messner, D.J.; Rhieu, B.H.; Kowdley, K.V. Iron Overload Causes Oxidative Stress and Impaired Insulin Signaling in AML-12 Hepatocytes. Am. J. Dig. Dis. 2013, 58, 1899–1908. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Qamirani, E.; Ren, Y.; Kuo, L.; Hein, T.W. C-reactive protein inhibits endothelium-dependent, N.O.;-mediated dilation in coronary arterioles by activating p38 kinase and, N.A.;D(P)H oxidase. Arter. Thromb. Vasc. Biol. 2005, 25, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Piya, M.K.; McTernan, P.G.; Kumar, S. Adipokine inflammation and insulin resistance: The role of glucose, lipids and endotoxin. J. Endocrinol. 2013, 216, T1–T15. [Google Scholar] [CrossRef]
- Raghuraman, G.; Zuniga, M.C.; Yuan, H.; Zhou, W. PKCε mediates resistin-induced, N.A.;DPH oxidase activation and inflammation leading to smooth muscle cell dysfunction and intimal hyperplasia. Atherosclerosis 2016, 253, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Li, W.; Seth, D.M.; Nair, A.R.; Francis, J.; Feng, Y. (Pro)renin receptor mediates both angiotensin, I.I.;-dependent and -independent oxidative stress in neuronal cells. PLoS ONE 2013, 8, e58339. [Google Scholar] [CrossRef]
- Habibi, J.; Whaley-Connell, A.; Hayden, M.R.; DeMarco, V.; Schneider, R.; Sowers, S.D.; Karuparthi, P.; Ferrario, C.M.; Sowers, J.R. Renin inhibition attenuates insulin resistance, oxidative stress, and pancreatic remodeling in the transgenic Ren2 rat. Endocrinology 2008, 149, 5643–5653. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Whaley-Connell, A.; Sowers, J.R. Oxidative stress in the cardiorenal metabolic syndrome. Curr. Hypertens. Rep. 2012, 14, 360–365. [Google Scholar] [CrossRef]
- Ishibashi, K.I.; Imamura, T.; Sharma, P.M.; Huang, J.; Ugi, S.; Olefsky, J.M. Chronic endothelin-1 treatment leads to heterologous desensitization of insulin signaling in 3T3-L1 adipocytes. J. Clin. Investig. 2001, 107, 1193–1202. [Google Scholar] [CrossRef][Green Version]
- Häring, H.; Kirsch, D.; Obermaier, B.; Ermel, B.; Machicao, F. Decreased tyrosine kinase activity of insulin receptor isolated from rat adipocytes rendered insulin-resistant by catecholamine treatment in vitro. Biochem. J. 1986, 234, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Bleeke, T.; Zhang, H.; Madamanchi, N.; Patterson, C.; Faber, J.E. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ. Res. 2004, 94, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Raman, P.; Bhonagiri, P.; Strawbridge, A.B.; Pattar, G.R.; Elmendorf, J.S. Protective Effect of Phosphatidylinositol 4,5-Bisphosphate against Cortical Filamentous Actin Loss and Insulin Resistance Induced by Sustained Exposure of 3T3-L1 Adipocytes to Insulin. J. Biol. Chem. 2004, 279, 39705–39709. [Google Scholar] [CrossRef] [PubMed]
- Catalano, K.J.; Maddux, B.A.; Szary, J.; Youngren, J.F.; Goldfine, I.D.; Schaufele, F. Insulin resistance induced by hyperinsulinemia coincides with a persistent alteration at the insulin receptor tyrosine kinase domain. PLoS ONE 2014, 9, e108693. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Mahadev, K.; Wu, X.; Zhu, L.; Motoshima, H. Role of Insulin-Induced Reactive Oxygen Species in the Insulin Signaling Pathway. Antioxidants Redox Signal. 2005, 7, 1021–1031. [Google Scholar] [CrossRef]
- Kain, V.; Kapadia, B.; Misra, P.; Saxena, U. Simvastatin may induce insulin resistance through a novel fatty acid mediated cholesterol independent mechanism. Sci. Rep. 2015, 5, srep13823. [Google Scholar] [CrossRef] [PubMed]
- Olea, E.; Agapito, M.T.; Gallego-Martin, T.; Rocher, A.; Gomez-Niño, A.; Obeso, A.; Gonzalez, C.; Yubero, S. Intermittent hypoxia and diet-induced obesity: Effects on oxidative status, sympathetic tone, plasma glucose and insulin levels, and arterial pressure. J. Appl. Physiol. 2014, 117, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.B. On the metabolism of phenylalanine and tyrosine. J. Biol. Chem. 1949, 181, 281–294. [Google Scholar] [CrossRef]
- Molnár, G.A.; Kun, S.; Sélley, E.; Kertész, M.; Szélig, L.; Csontos, C.; Böddi, K.; Bogár, L.; Miseta, A. Role of tyrosine isomers in acute and chronic diseases leading to oxidative stress-A Review. Curr. Med. Chem. 2016, 23, 667–685. [Google Scholar] [CrossRef]
- Pennathur, S.; Wagner, J.D.; Leeuwenburgh, C.; Litwak, K.N.; Heinecke, J.W. A hydroxyl radical-like species oxidizes cynomolgus monkey artery wall proteins in early diabetic vascular disease. J. Clin. Investig. 2001, 107, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Dean, R.T.; Southan, M.; Truscott, R. The hydroxyl radical in lens nuclear cataractogenesis. J. Biol. Chem. 1998, 273, 28603–28609. [Google Scholar] [CrossRef] [PubMed]
- Molnár, G.A.; Wagner, Z.; Markó, L.; Kőszegi, T.; Mohás, M.; Kocsis, B.; Matus, Z.; Wagner, L.; Tamaskó, M.; Mazák, I.; et al. Urinary ortho-tyrosine excretion in diabetes mellitus and renal failure: Evidence for hydroxyl radical production. Kidney Int. 2005, 68, 2281–2287. [Google Scholar] [CrossRef]
- Brasnyó, P.; Molnar, G.A.; Mohás, M.; Markó, L.; Laczy, B.; Cseh, J.; Mikolás, E.; Szijártó, I.A.; Mérei, A.; Halmai, R.; et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br. J. Nutr. 2011, 106, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Duke, S.O. The emergence of grass root chemical ecology. Proc. Natl. Acad. Sci. USA 2007, 104, 16729–16730. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, R.A.; Bruzzo, J.; Chiarella, P.; Di Gianni, P.; Isturiz, M.A.; Linskens, S.; Speziale, N.; Meiss, R.P.; Bustuoabad, O.D.; Pasqualini, C.D. Tyrosine isomers mediate the classical phenomenon of concomitant tumor resistance. Cancer Res. 2011, 71, 7113–7124. [Google Scholar] [CrossRef] [PubMed]
- Mikolás, E.; Kun, S.; Laczy, B.; Molnár, G.A.; Sélley, E.; Wittmann, I.; Koszegi, T. Incorporation of ortho- and meta-tyrosine into cellular proteins leads to erythropoietin-resistance in an erythroid cell line. Kidney Blood Press. Res. 2013, 38, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Kaddai, V.; Gonzalez, T.; Keslair, F.; Grémeaux, T.; Bonnafous, S.; Gugenheim, J.; Tran, A.; Gual, P.; Le Marchand-Brustel, Y.; Cormont, M. Rab4b is a small, G.T.;Pase involved in the control of the glucose transporter, G.L.;UT4 localization in adipocyte. PLoS ONE 2009, 4, e5257. [Google Scholar] [CrossRef] [PubMed]
- Schiwek, D.; Endlich, N.; Holzman, L.; Holthöfer, H.; Kriz, W.; Endlich, K. Stable expression of nephrin and localization to cell-cell contacts in novel murine podocyte cell lines. Kidney Int. 2004, 66, 91–101. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Chatterjee, S.; Chaudhuri, M.; White, M.F. YMXM motifs of, I.R.;S-1 define substrate specificity of the insulin receptor kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 2027–2031. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Bittner-Kowalczyk, A.; White, M.F.; Harbeck, M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J. Biol. Chem. 2000, 275, 4283–4289. [Google Scholar] [CrossRef]
- Sánchez-Margalet, V.; Goldfine, I.D.; Truitt, K.; Imboden, J.; Sung, C.K. Role of p85 subunit of phosphatidylinositol-3-kinase as an adaptor molecule linking the insulin receptor to insulin receptor substrate 1. Mol. Endocrinol. 1995, 9, 435–442. [Google Scholar] [CrossRef]
- Selner, N.G.; Luechapanichkul, R.; Chen, X.; Neel, B.G.; Zhang, Z.Y.; Knapp, S.; Bell, C.E.; Pei, D. Diverse levels of sequence selectivity and catalytic efficiency of protein-tyrosine phosphatases. Biochemistry 2014, 53, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tseng, Y.D.; Xu, C.F.; Neubert, T.A.; White, M.F.; Hubbard, S.R. Structural and biochemical characterization of the, K.R.;LB region in insulin receptor substrate-2. Nat. Struct. Mol. Biol. 2008, 15, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Eck, M.J.; Schlessinger, J.; Shoelson, S.E.; Harrison, S.C. Crystal structure of the, P.I.; 3-kinase p85 amino-terminal, S.H.;2 domain and its phosphopeptide complexes. Nat. Struct. Mol. Biol. 1996, 3, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Inaba, S.; Numoto, N.; Ogawa, S.; Morii, H.; Ikura, T.; Abe, R.; Ito, N.; Oda, M. Crystal Structures and Thermodynamic Analysis Reveal Distinct Mechanisms of CD28 Phosphopeptide Binding to the Src Homology 2 (SH2) Domains of Three Adaptor Proteins. J. Biol. Chem. 2017, 292, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Gross, M.; Lee, D.H.; Holvoet, P.; Himes, J.H.; Shikany, J.M.; Jacobs, D.R. Oxidative stress and insulin resistance: The coronary artery risk development in young adults study. Diabetes Care 2009, 32, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Karin, M. JNK1 and IKKβ: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611. [Google Scholar] [CrossRef] [PubMed]
- Sitte, N. Oxidative damage to proteins. In Ageing at the Molecular Level; von Thomas, Z., Ed.; Springer: Dordrecht, The Netherlands, 2003; pp. 27–45. [Google Scholar]
- Torres-Cuevas, I.; Kuligowski, J.; Escobar, J.; Vento, M. Determination of biomarkers of protein oxidation in tissue and plasma. Free Radic. Biol. Med. 2014, 75 (Suppl. S1), S51. [Google Scholar] [CrossRef]
- Ledo, A.; Arduini, A.; A Asensi, M.; Sastre, J.; Escrig, R.; Brugada, M.; Aguar, M.; Saenz, P.; Vento, M. Human milk enhances antioxidant defenses against hydroxyl radical aggression in preterm infants. Am. J. Clin. Nutr. 2008, 89, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Park, J.L.; Byun, J.; Pennathur, S.; Kollmeyer, J.; Shayman, J.A. Decreased Nitric Oxide Bioavailability in a Mouse Model of Fabry Disease. J. Am. Soc. Nephrol. 2009, 20, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.; Creed, J.; Schears, G.; Zaitseva, T.; Greeley, W.; Wilson, D.F.; Pastuszko, A. Comparison of low-flow cardiopulmonary bypass and circulatory arrest on brain oxygen and metabolism. Ann. Thorac. Surg. 2004, 77, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- Ishimitsu, S.; Fujimoto, S.; Ohara, A. High-performance liquid chromatographic determination of m-tyrosine and o-tyrosine in rat urine. J. Chromatogr. B Biomed. Sci. Appl. 1989, 489, 377–383. [Google Scholar] [CrossRef]
- Huang, C.; Wu, M.; Du, J.; Liu, D.; Chan, C. Systematic modeling for the insulin signaling network mediated by IRS1 and IRS2. J. Theor. Biol. 2014, 355, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yang, X.; Chan, C. A Dynamic Analysis of IRS-PKR Signaling in Liver Cells: A Discrete Modeling Approach. PLoS ONE 2009, 4, e8040. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Liuwantara, D.; Gurlo, T.; Kirakossian, D.; Man, C.D.; Cobelli, C.; White, M.F.; Copps, K.D.; Volpi, E.; Fujita, S.; et al. Pulsatile Portal Vein Insulin Delivery Enhances Hepatic Insulin Action and Signaling. Diabetes 2012, 61, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Szijártó, I.A.; Molnár, G.A.; Mikolás, E.; Fisi, V.; Cseh, J.; Laczy, B.; Kovács, T.; Böddi, K.; Takátsy, A.; Gollasch, M.; et al. Elevated Vascular Level of ortho-Tyrosine Contributes to the Impairment of Insulin-Induced Arterial Relaxation. Horm. Metab. Res. 2014, 46, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Selley, E.; Kun, S.; Kürthy, M.; Kovacs, T.; Wittmann, I.; Molnar, G. Para-Tyrosine Supplementation Improves Insulin- and Liraglutide- Induced Vasorelaxation in Cholesterol-Fed Rats. Protein Pept. Lett. 2015, 22, 736–742. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | tR (min) |
|---|---|
| GRKGSGDF(4-hydroxy)MPMSPKV | tR = 9.96 |
| GRKGSGDF(3-hydroxy)MPMSPKV | tR = 9.19 |
| GRKGSGDF(2-hydroxy)MPMSPKV | tR = 12.08 |
| GRKGSGDF(4-PO4H2)MPMSPKV | tR = 9.06 |
| GRKGSGDF(3-PO4H2)MPMSPKV | tR = 8.24 |
| GRKGSGDF(2-PO4H2)MPMSPKV | tR = 8.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohás-Cseh, J.; Molnár, G.A.; Pap, M.; Laczy, B.; Vas, T.; Kertész, M.; Németh, K.; Hetényi, C.; Csikós, O.; Tóth, G.K.; et al. Incorporation of Oxidized Phenylalanine Derivatives into Insulin Signaling Relevant Proteins May Link Oxidative Stress to Signaling Conditions Underlying Chronic Insulin Resistance. Biomedicines 2022, 10, 975. https://doi.org/10.3390/biomedicines10050975
Mohás-Cseh J, Molnár GA, Pap M, Laczy B, Vas T, Kertész M, Németh K, Hetényi C, Csikós O, Tóth GK, et al. Incorporation of Oxidized Phenylalanine Derivatives into Insulin Signaling Relevant Proteins May Link Oxidative Stress to Signaling Conditions Underlying Chronic Insulin Resistance. Biomedicines. 2022; 10(5):975. https://doi.org/10.3390/biomedicines10050975
Chicago/Turabian StyleMohás-Cseh, Judit, Gergő Attila Molnár, Marianna Pap, Boglárka Laczy, Tibor Vas, Melinda Kertész, Krisztina Németh, Csaba Hetényi, Orsolya Csikós, Gábor K. Tóth, and et al. 2022. "Incorporation of Oxidized Phenylalanine Derivatives into Insulin Signaling Relevant Proteins May Link Oxidative Stress to Signaling Conditions Underlying Chronic Insulin Resistance" Biomedicines 10, no. 5: 975. https://doi.org/10.3390/biomedicines10050975
APA StyleMohás-Cseh, J., Molnár, G. A., Pap, M., Laczy, B., Vas, T., Kertész, M., Németh, K., Hetényi, C., Csikós, O., Tóth, G. K., Reményi, A., & Wittmann, I. (2022). Incorporation of Oxidized Phenylalanine Derivatives into Insulin Signaling Relevant Proteins May Link Oxidative Stress to Signaling Conditions Underlying Chronic Insulin Resistance. Biomedicines, 10(5), 975. https://doi.org/10.3390/biomedicines10050975