
Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
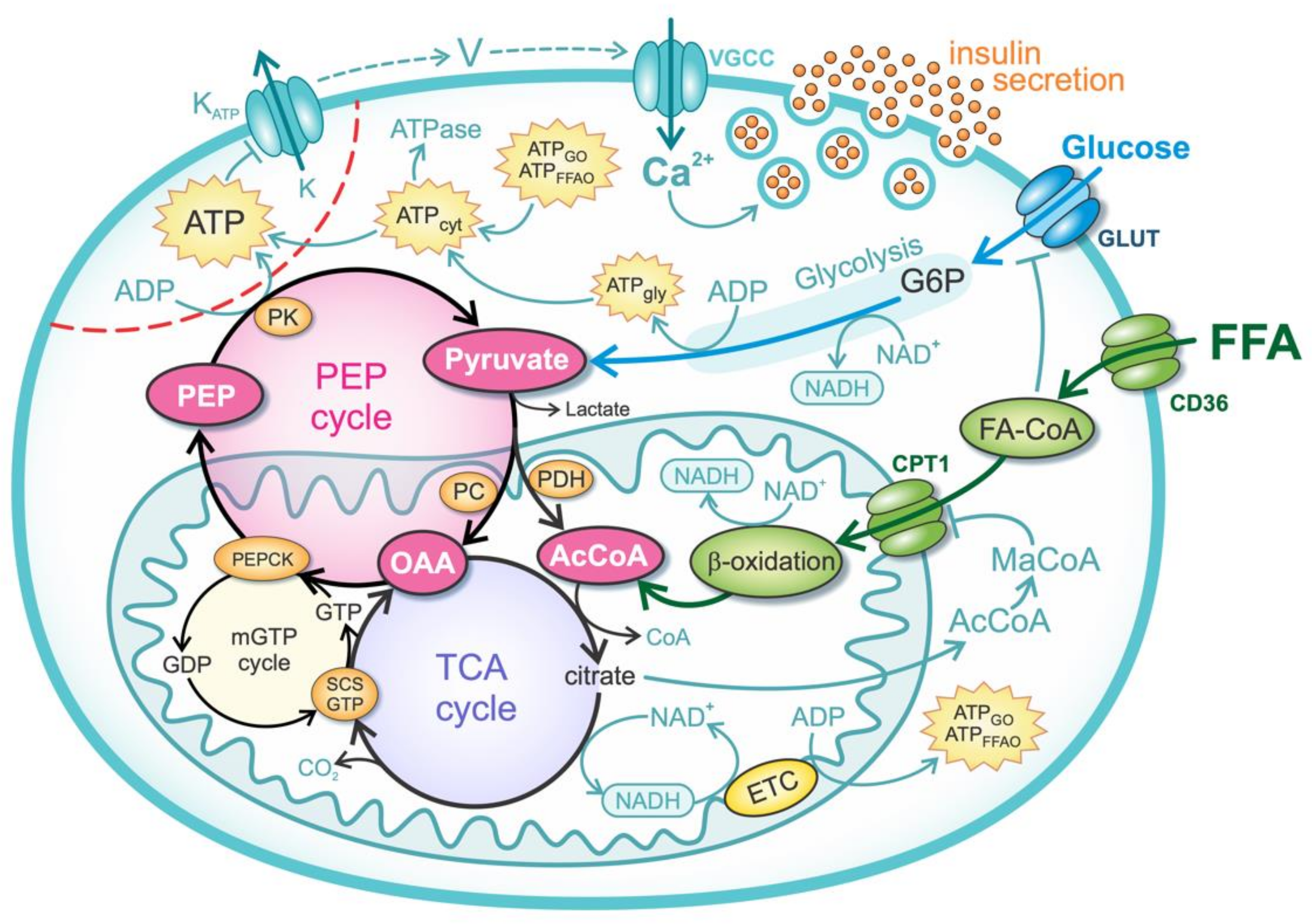
2.1. Glucose Metabolism
2.2. FFA Metabolism
2.3. Anaplerotic Pathway and PEP Cycle
2.4. Mechanisms of Insulin Secretion
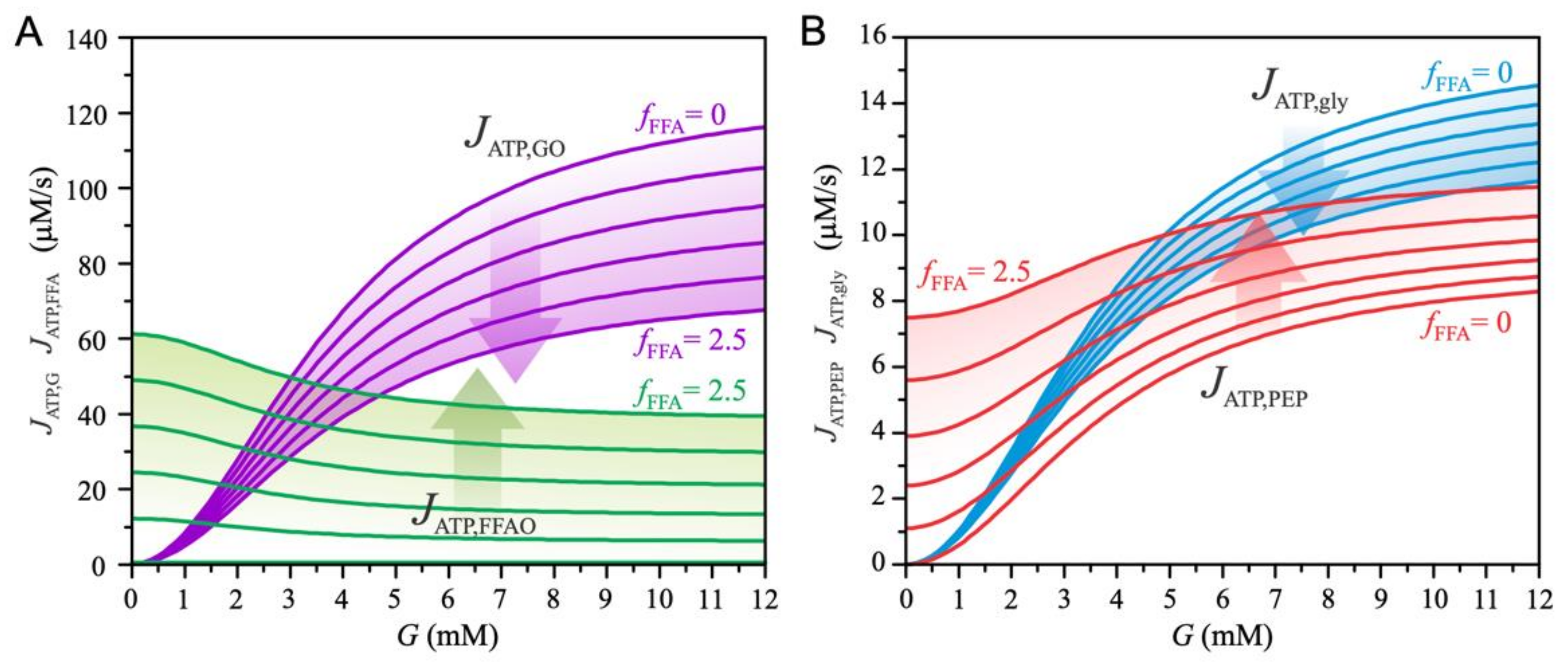
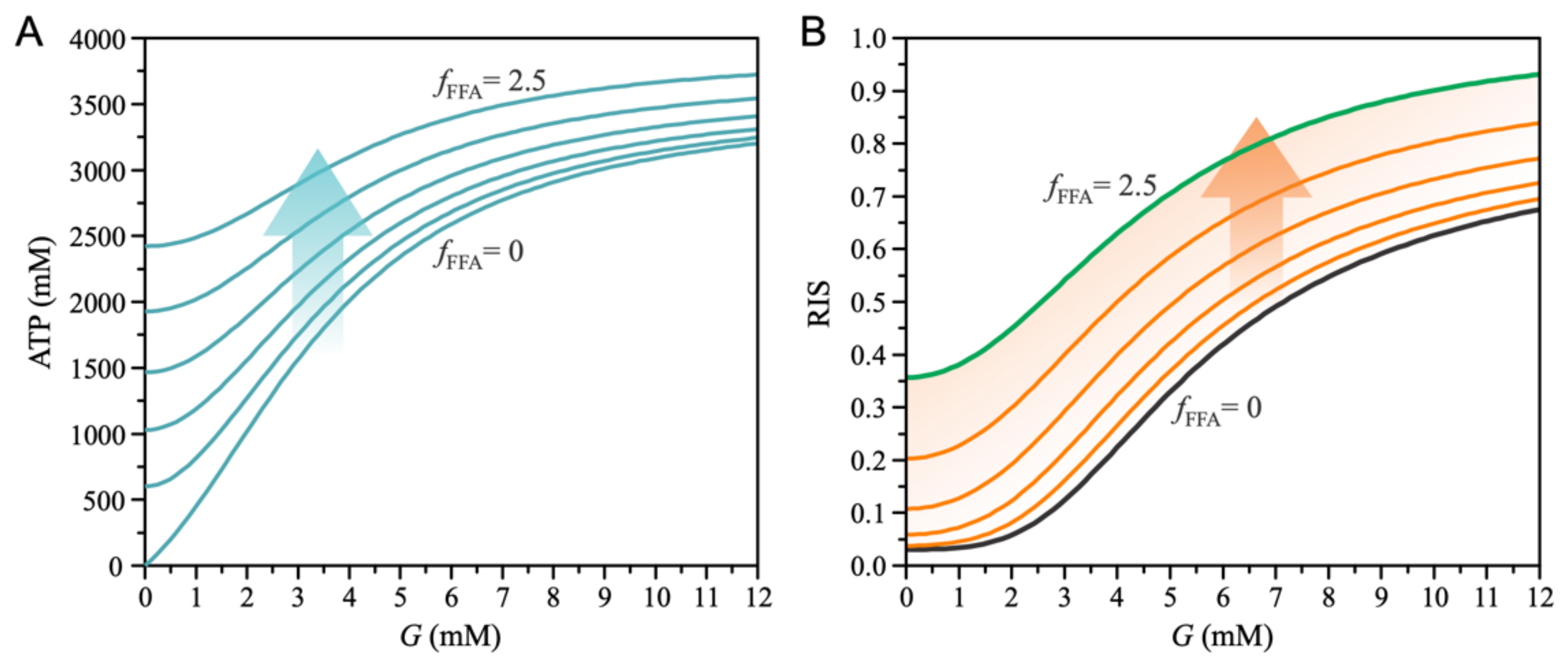
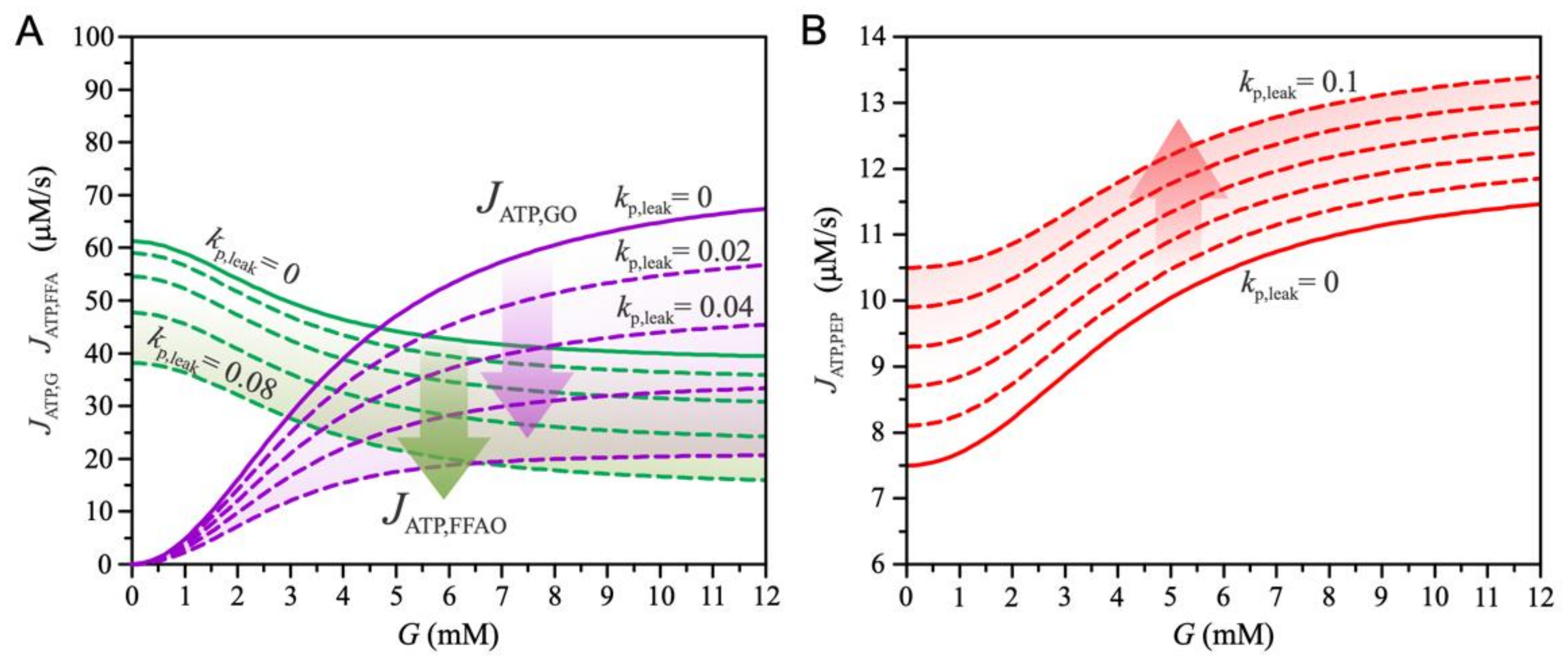
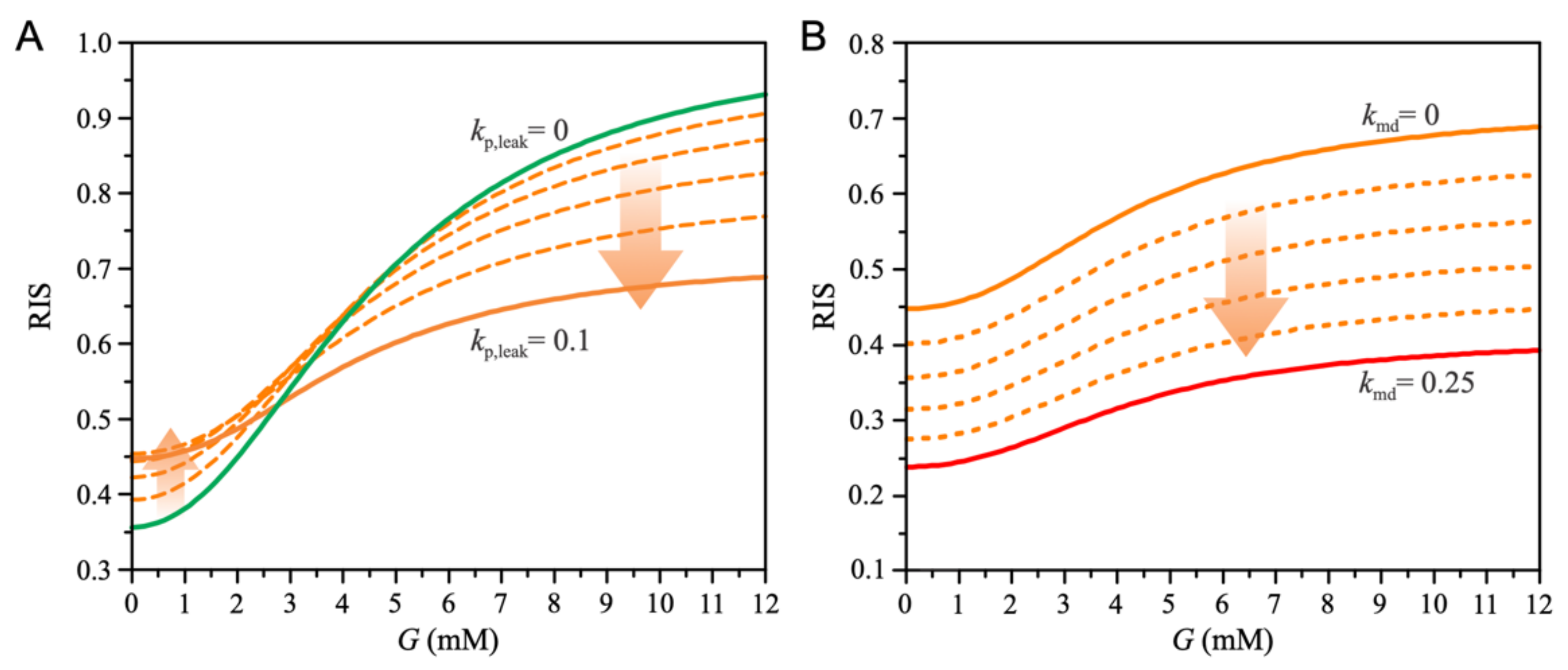
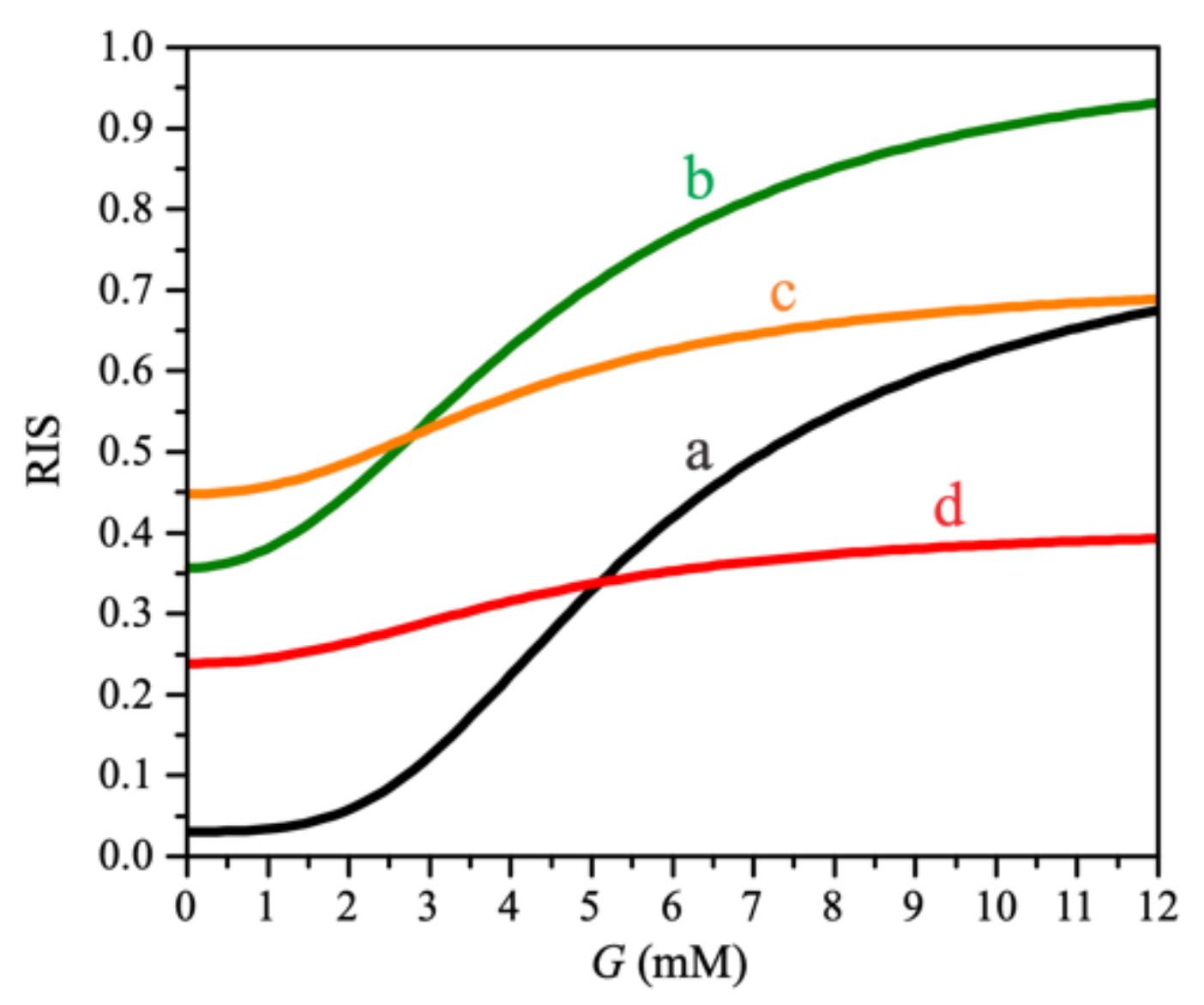
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Kane, J.P.; Pullinger, C.R.; Goldfine, I.D.; Malloy, M.J. Dyslipidemia and diabetes mellitus: Role of lipoprotein species and interrelated pathways of lipid metabolism in diabetes mellitus. Curr. Opin. Pharmacol. 2021, 61, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Diabetic dyslipidaemia. Curr. Opin. Lipidol. 2006, 17, 238–246. [Google Scholar] [CrossRef]
- Markovič, R.; Grubelnik, V.; Vošner, H.B.; Kokol, P.; Završnik, M.; Janša, K.; Zupet, M.; Završnik, J.; Marhl, M. Age-Related Changes in Lipid and Glucose Levels Associated with Drug Use and Mortality: An Observational Study. J. Pers. Med. 2022, 12, 280. [Google Scholar] [CrossRef]
- Taddeo, E.P.; Alsabeeh, N.; Baghdasarian, S.; Wikstrom, J.D.; Ritou, E.; Sereda, S.; Erion, K.; Li, J.; Stiles, L.; Abdulla, M.; et al. Mitochondrial Proton Leak Regulated by Cyclophilin D Elevates Insulin Secretion in Islets at Nonstimulatory Glucose Levels. Diabetes 2019, 69, 131–145. [Google Scholar] [CrossRef]
- Tan, M.; Havel, R.; Gerich, J.; Soeldner, J.; Kane, J. Pancreatic Alpha and Beta Cell Function in Familial Dysbetalipoproteinemia. Horm. Metab. Res. 1980, 12, 421–425. [Google Scholar] [CrossRef]
- Kristinsson, H.; Sargsyan, E.; Manell, H.; Smith, D.M.; Göpel, S.O.; Bergsten, P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 2017, 7, 4657. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Acosta-Montaño, P.; García-González, V. Effects of Dietary Fatty Acids in Pancreatic Beta Cell Metabolism, Implications in Homeostasis. Nutrients 2018, 10, 393. [Google Scholar] [CrossRef]
- Römer, A.; Linn, T.; Petry, S. Lipotoxic Impairment of Mitochondrial Function in β-Cells: A Review. Antioxidants 2021, 10, 293. [Google Scholar] [CrossRef]
- Cnop, M.; Ladrière, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes Obes. Metab. 2010, 12, 76–82. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Lisai, S.; Sirigu, A.; Piras, A.; Collu, M.; Batetta, B.; Gambelli, L.; Banni, S. Dietary Triacylglycerols with Palmitic Acid in the sn-2 Position Modulate Levels of N-Acylethanolamides in Rat Tissues. PLoS ONE 2015, 10, e0120424. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Sargsyan, E.; Artemenko, K.; Manukyan, L.; Bergquist, J.; Bergsten, P. Oleate protects beta-cells from the toxic effect of palmitate by activating pro-survival pathways of the ER stress response. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 1151–1160. [Google Scholar] [CrossRef]
- Kokatnur, M.G.; Oalmann, M.C.; Johnson, W.D.; Malcom, G.T.; Strong, J.P. Fatty acid composition of human adipose tissue from two anatomical sites in a biracial community. Am. J. Clin. Nutr. 1979, 32, 2198–2205. [Google Scholar] [CrossRef]
- Nemecz, M.; Constantin, A.; Dumitrescu, M.; Alexandru, N.; Filippi, A.; Tanko, G.; Georgescu, A. The Distinct Effects of Palmitic and Oleic Acid on Pancreatic Beta Cell Function: The Elucidation of Associated Mechanisms and Effector Molecules. Front. Pharmacol. 2019, 9, 1554. [Google Scholar] [CrossRef]
- Karunakaran, U.; Elumalai, S.; Moon, J.-S.; Won, K.-C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells 2021, 10, 1833. [Google Scholar] [CrossRef]
- Oberhauser, L.; Maechler, P. Lipid-Induced Adaptations of the Pancreatic Beta-Cell to Glucotoxic Conditions Sustain Insulin Secretion. Int. J. Mol. Sci. 2021, 23, 324. [Google Scholar] [CrossRef]
- Reilly, J.M.; Thompson, M.P. Dietary Fatty Acids Up-Regulate the Expression of UCP2 in 3T3-L1 Preadipocytes. Biochem. Biophys. Res. Commun. 2000, 277, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.W.; Koshkin, V.; Saleh, M.C.; Sivitz, W.I.; Zhang, C.-Y.; Lowell, B.B.; Chan, C.B.; Wheeler, M.B. Free Fatty Acid-induced β-Cell Defects Are Dependent on Uncoupling Protein 2 Expression. J. Biol. Chem. 2004, 279, 51049–51056. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Zhao, Y. Uncoupling protein 2 and metabolic diseases. Mitochondrion 2017, 34, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Jaswal, J.; Ussher, J. Myocardial fatty acid utilization as a determinant of cardiac efficiency and function. Clin. Lipidol. 2009, 4, 379–389. [Google Scholar] [CrossRef]
- Sara, M.; Yaser, K.-B.; Maedeh, A.; Mohamadreza, A.; Beitullah, A. The review of the relationship between UCP2 and obesity: Focusing on inflammatory-obesity. New Insights Obes. Genet. Beyond 2021, 5, 001–013. [Google Scholar] [CrossRef]
- Yang, K.; Xu, X.; Nie, L.; Xiao, T.; Guan, X.; He, T.; Yu, Y.; Liu, L.; Huang, Y.; Zhang, J.; et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol. Lett. 2015, 234, 110–119. [Google Scholar] [CrossRef]
- Baffy, G. Mitochondrial uncoupling in cancer cells: Liabilities and opportunities. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 655–664. [Google Scholar] [CrossRef]
- Emre, Y.; Nübel, T. Uncoupling protein UCP2: When mitochondrial activity meets immunity. FEBS Lett. 2010, 584, 1437–1442. [Google Scholar] [CrossRef]
- Bouillaud, F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 377–383. [Google Scholar] [CrossRef]
- Rupprecht, A.; Moldzio, R.; Mödl, B.; Pohl, E.E. Glutamine regulates mitochondrial uncoupling protein 2 to promote glutaminolysis in neuroblastoma cells. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 391–401. [Google Scholar] [CrossRef]
- Manukyan, L.; Ubhayasekera, S.J.K.A.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-Induced Impairments of β-Cell Function Are Linked with Generation of Specific Ceramide Species via Acylation of Sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef]
- Ye, R.; Onodera, T.; Scherer, P.E. Lipotoxicity and β Cell Maintenance in Obesity and Type 2 Diabetes. J. Endocr. Soc. 2019, 3, 617–631. [Google Scholar] [CrossRef]
- Lewandowski, S.L.; Cardone, R.L.; Foster, H.R.; Ho, T.; Potapenko, E.; Poudel, C.; Van Deusen, H.R.; Sdao, S.M.; Alves, T.C.; Zhao, X.; et al. Pyruvate Kinase Controls Signal Strength in the Insulin Secretory Pathway. Cell Metab. 2020, 32, 736–750.e5. [Google Scholar] [CrossRef]
- Ishihara, H. Metabolism-secretion coupling in glucose-stimulated insulin secretion. Diabetol. Int. 2022, 13, 463–470. [Google Scholar] [CrossRef]
- Schuit, F.; De Vos, A.; Farfari, S.; Moens, K.; Pipeleers, D.; Brun, T.; Prentki, M. Metabolic Fate of Glucose in Purified Islet Cells. J. Biol. Chem. 1997, 272, 18572–18579. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.M. Metabolic Signaling in Fuel-Induced Insulin Secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef]
- Sugden, M.C.; Holness, M.J. The pyruvate carboxylase-pyruvate dehydrogenase axis in islet pyruvate metabolism: Going round in circles? Islets 2011, 3, 302–319. [Google Scholar] [CrossRef]
- Corkey, B.E. Targeting Pyruvate Kinase PEPs Up Insulin Secretion and Improves Glucose Homeostasis. Cell Metab. 2020, 32, 693–694. [Google Scholar] [CrossRef]
- Lu, D.; Mulder, H.; Zhao, P.; Burgess, S.C.; Jensen, M.V.; Kamzolova, S.; Newgard, C.B.; Sherry, A.D. 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc. Natl. Acad. Sci. USA 2002, 99, 2708–2713. [Google Scholar] [CrossRef]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Cassell, T.; Smith, P.; Lu, D.; Nam, H.W.; Lane, A.N.; Zhao, Y. UCP2 Overexpression Redirects Glucose into Anabolic Metabolic Pathways. Proteomics 2018, 19, e1800353. [Google Scholar] [CrossRef] [PubMed]
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; et al. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 2019, 28, 759–772.e10. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Kibbey, R.G. The mitochondrial isoform of phosphoenolpyruvate carboxykinase (PEPCK-M) and glucose homeostasis: Has it been overlooked? Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1840, 1313–1330. [Google Scholar] [CrossRef]
- Abulizi, A.; Cardone, R.L.; Stark, R.; Lewandowski, S.L.; Zhao, X.; Hillion, J.; Ma, L.; Sehgal, R.; Alves, T.C.; Thomas, C.; et al. Multi-Tissue Acceleration of the Mitochondrial Phosphoenolpyruvate Cycle Improves Whole-Body Metabolic Health. Cell Metab. 2020, 32, 751–766.e11. [Google Scholar] [CrossRef]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Modelling of energy-driven switch for glucagon and insulin secretion. J. Theor. Biol. 2020, 493, 110213. [Google Scholar] [CrossRef]
- Roden, M. How Free Fatty Acids Inhibit Glucose Utilization in Human Skeletal Muscle. Physiology 2004, 19, 92–96. [Google Scholar] [CrossRef]
- Doliba, N.M.; Fenner, D.; Zelent, B.; Bass, J.; Sarabu, R.; Matschinsky, F.M. Repair of diverse diabetic defects of β-cells in man and mouse by pharmacological glucokinase activation. Diabetes Obes. Metab. 2012, 14, 109–119. [Google Scholar] [CrossRef]
- Liang, Y.; Bai, G.; Doliba, N.; Buettger, C.; Wang, L.; Berner, D.K.; Matschinsky, F.M. Glucose metabolism and insulin release in mouse beta HC9 cells, as model for wild-type pancreatic beta-cells. Am. J. Physiol. Metab. 1996, 270, E846–E857. [Google Scholar] [CrossRef]
- MacDonald, M.J. Feasibility of a Mitochondrial Pyruvate Malate Shuttle in Pancreatic Islets. J. Biol. Chem. 1995, 270, 20051–20058. [Google Scholar] [CrossRef]
- MacDonald, M.J.; Tang, J.; Polonsky, K.S. Low Mitochondrial Glycerol Phosphate Dehydrogenase and Pyruvate Carboxylase in Pancreatic Islets of Zucker Diabetic Fatty Rats. Diabetes 1996, 45, 1626–1630. [Google Scholar] [CrossRef]
- Tan, C.; Tuch, B.E.; Tu, J.; Brown, S.A. Role of NADH Shuttles in Glucose-Induced Insulin Secretion from Fetal β-Cells. Diabetes 2002, 51, 2989–2996. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and Metabolic Evidence for Mitochondrial Defects Associated With β-Cell Dysfunction in a Mouse Model of Type 2 Diabetes. Diabetes 2009, 59, 448–459. [Google Scholar] [CrossRef]
- Heuett, W.J.; Periwal, V. Autoregulation of Free Radicals via Uncoupling Protein Control in Pancreatic β-Cell Mitochondria. Biophys. J. 2010, 98, 207–217. [Google Scholar] [CrossRef][Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef]
- Grubelnik, V.; Markovič, R.; Lipovšek, S.; Leitinger, G.; Gosak, M.; Dolenšek, J.; Valladolid-Acebes, I.; Berggren, P.-O.; Stožer, A.; Perc, M.; et al. Modelling of dysregulated glucagon secretion in type 2 diabetes by considering mitochondrial alterations in pancreatic α-cells. R. Soc. Open Sci. 2020, 7, 191171. [Google Scholar] [CrossRef]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus. Life 2020, 10, 348. [Google Scholar] [CrossRef]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef]
- Nunemaker, C.S.; Zhang, M.; Satin, L.S. Insulin Feedback Alters Mitochondrial Activity Through an ATP-sensitive K+ Channel–Dependent Pathway in Mouse Islets and β-Cells. Diabetes 2004, 53, 1765–1772. [Google Scholar] [CrossRef][Green Version]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.-E.; Corvera, S. The Role of Mitochondria in the Pathogenesis of Type 2 Diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed]
- Wallin, T.; Ma, Z.; Ogata, H.; Jørgensen, I.H.; Iezzi, M.; Wang, H.; Wollheim, C.B.; Björklund, A. Facilitation of fatty acid uptake by CD36 in insulin-producing cells reduces fatty-acid-induced insulin secretion and glucose regulation of fatty acid oxidation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2010, 1801, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Esguerra, J.L.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in β-Cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Moon, J.S.; Karunakaran, U.; Suma, E.; Chung, S.M.; Won, K.C. The Role of CD36 in Type 2 Diabetes Mellitus: β-Cell Dysfunction and Beyond. Diabetes Metab. J. 2020, 44, 222–233. [Google Scholar] [CrossRef]
- McKee, E.E.; Bentley, A.T.; Smith, R.M.; Kraas, J.R.; Ciaccio, C.E. Guanine nucleotide transport by atractyloside-sensitive and -insensitive carriers in isolated heart mitochondria. Am. J. Physiol. Physiol. 2000, 279, C1870–C1879. [Google Scholar] [CrossRef]
- Vozza, A.; Blanco, E.; Palmieri, L.; Palmieri, F. Identification of the Mitochondrial GTP/GDP Transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 20850–20857. [Google Scholar] [CrossRef]
- Nicholls, D.G. The Pancreatic β-Cell: A Bioenergetic Perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef]
- Pedersen, M.G.; Bertram, R.; Sherman, A. Intra- and Inter-Islet Synchronization of Metabolically Driven Insulin Secretion. Biophys. J. 2005, 89, 107–119. [Google Scholar] [CrossRef]
- Magnus, G.; Keizer, J. Model of β-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am. J. Physiol. Physiol. 1998, 274, C1158–C1173. [Google Scholar] [CrossRef]
- Bertram, R.; Sherman, A. A calcium-based phantom bursting model for pancreatic islets. Bull. Math. Biol. 2004, 66, 1313–1344. [Google Scholar] [CrossRef]
- Rorsman, P.; Ramracheya, R.; Rorsman, N.J.G.; Zhang, Q. ATP-regulated potassium channels and voltage-gated calcium channels in pancreatic alpha and beta cells: Similar functions but reciprocal effects on secretion. Diabetologia 2014, 57, 1749–1761. [Google Scholar] [CrossRef]
- Assimacopoulos-Jeannet, F.; Thumelin, S.; Roche, E.; Esser, V.; McGarry, J.D.; Prentki, M. Fatty Acids Rapidly Induce the Carnitine Palmitoyltransferase I Gene in the Pancreatic β-Cell Line INS-1. J. Biol. Chem. 1997, 272, 1659–1664. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Tajiri, Y.; Sako, Y.; Hashimoto, T.; Umeda, F.; Nawata, H. Effects of free fatty acids on β-cell functions: A possible involvement of peroxisome proliferator-activated receptors α or pancreatic/duodenal homeobox. Metabolism 2001, 50, 613–618. [Google Scholar] [CrossRef]
- Lu, B.; Kurmi, K.; Munoz-Gomez, M.; Ambuludi, E.J.J.; Tonne, J.M.; Rakshit, K.; Hitosugi, T.; Kudva, Y.C.; Matveyenko, A.V.; Ikeda, Y. Impaired β-cell glucokinase as an underlying mechanism in diet-induced diabetes. Dis. Models Mech. 2018, 11, dmm033316. [Google Scholar] [CrossRef]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and Molecular Defects of Pancreatic Islets in Human Type 2 Diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef]
- Yaney, G.C.; Corkey, B.E. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 2003, 46, 1297–1312. [Google Scholar] [CrossRef]
- Cen, J.; Sargsyan, E.; Bergsten, P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr. Metab. 2016, 13, 59. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Nolan, C.J.; Madiraju, M.S.; Delghingaro-Augusto, V.; Peyot, M.-L.; Prentki, M. Fatty Acid Signaling in the β-Cell and Insulin Secretion. Diabetes 2006, 55, S16–S23. [Google Scholar] [CrossRef]
- Kristinsson, H.; Smith, D.M.; Bergsten, P.; Sargsyan, E. FFAR1 Is Involved in Both the Acute and Chronic Effects of Palmitate on Insulin Secretion. Endocrinology 2013, 154, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Corkey, B.E.; Madiraju, S.R.M. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 2019, 63, 10–20. [Google Scholar] [CrossRef]
- Busch, A.K.; Cordery, D.; Denyer, G.S.; Biden, T.J. Expression Profiling of Palmitate- and Oleate-Regulated Genes Provides Novel Insights into the Effects of Chronic Lipid Exposure on Pancreatic β-Cell Function. Diabetes 2002, 51, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, V.; Wang, X.; Scherer, P.E.; Chan, C.; Wheeler, M.B. Mitochondrial Functional State in Clonal Pancreatic β-Cells Exposed to Free Fatty Acids. J. Biol. Chem. 2003, 278, 19709–19715. [Google Scholar] [CrossRef] [PubMed]
- Erion, K.; Corkey, B.E. β-Cell Failure or β-Cell Abuse? Front. Endocrinol. 2018, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Kim-Muller, J.Y.; Kim, Y.; Fan, J.; Zhao, S.; Banks, A.; Prentki, M.; Accili, D. FoxO1 Deacetylation Decreases Fatty Acid Oxidation in β-Cells and Sustains Insulin Secretion in Diabetes. J. Biol. Chem. 2016, 291, 10162–10172. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Talchai, S.C.; Kim-Muller, J.Y.; Cinti, F.; Ishida, E.; Ordelheide, A.M.; Kuo, T.; Fan, J.; Son, J. When β-cells fail: Lessons from dedifferentiation. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 117–122. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grubelnik, V.; Zmazek, J.; Završnik, M.; Marhl, M. Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion. Biomedicines 2022, 10, 1627. https://doi.org/10.3390/biomedicines10071627
Grubelnik V, Zmazek J, Završnik M, Marhl M. Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion. Biomedicines. 2022; 10(7):1627. https://doi.org/10.3390/biomedicines10071627
Chicago/Turabian StyleGrubelnik, Vladimir, Jan Zmazek, Matej Završnik, and Marko Marhl. 2022. "Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion" Biomedicines 10, no. 7: 1627. https://doi.org/10.3390/biomedicines10071627
APA StyleGrubelnik, V., Zmazek, J., Završnik, M., & Marhl, M. (2022). Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion. Biomedicines, 10(7), 1627. https://doi.org/10.3390/biomedicines10071627