
Genetic Alterations in Benign Adrenal Tumors


Abstract
1. Introduction
2. Benign Adrenocortical Tumors Producing Cortisol
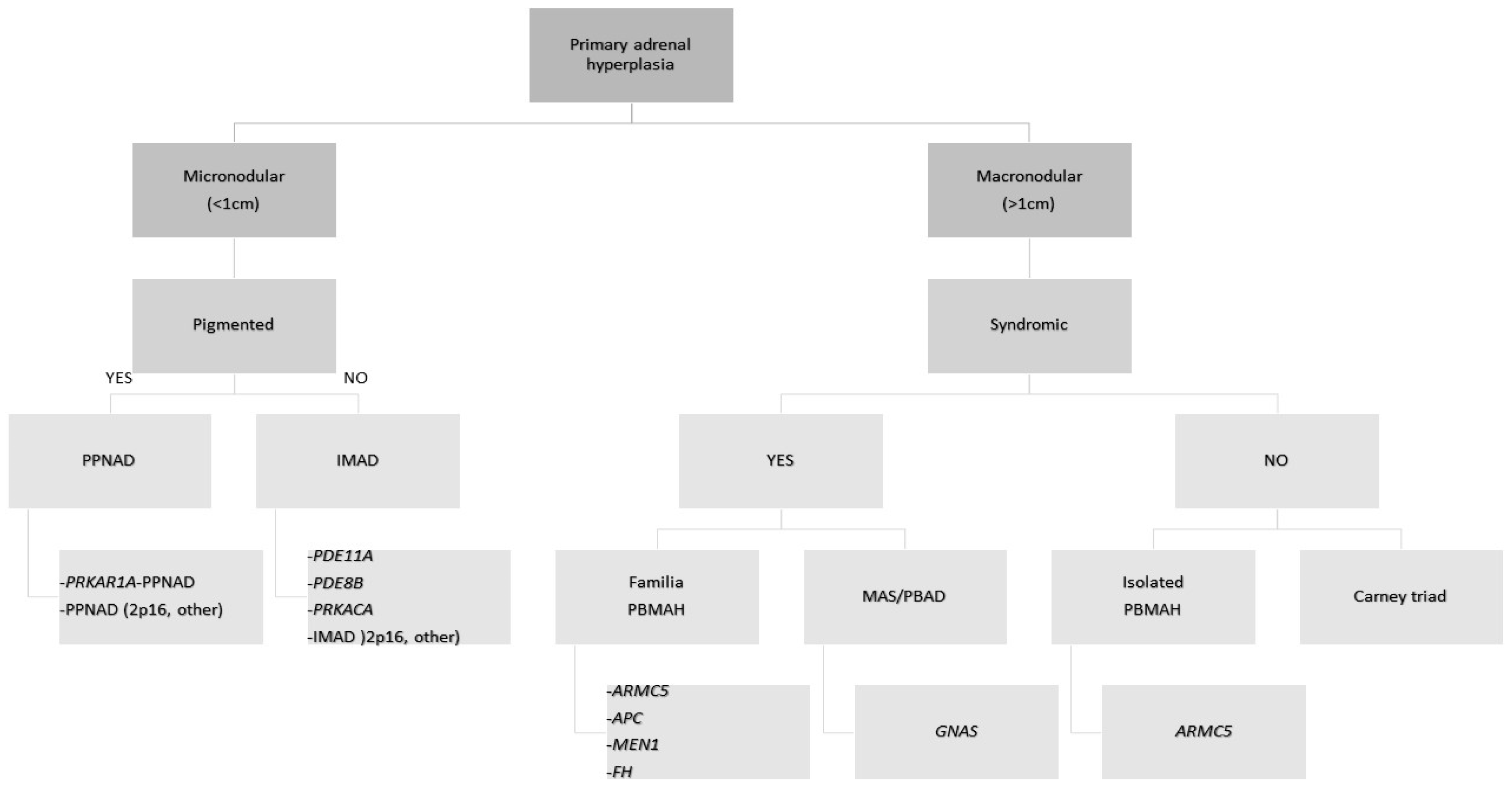
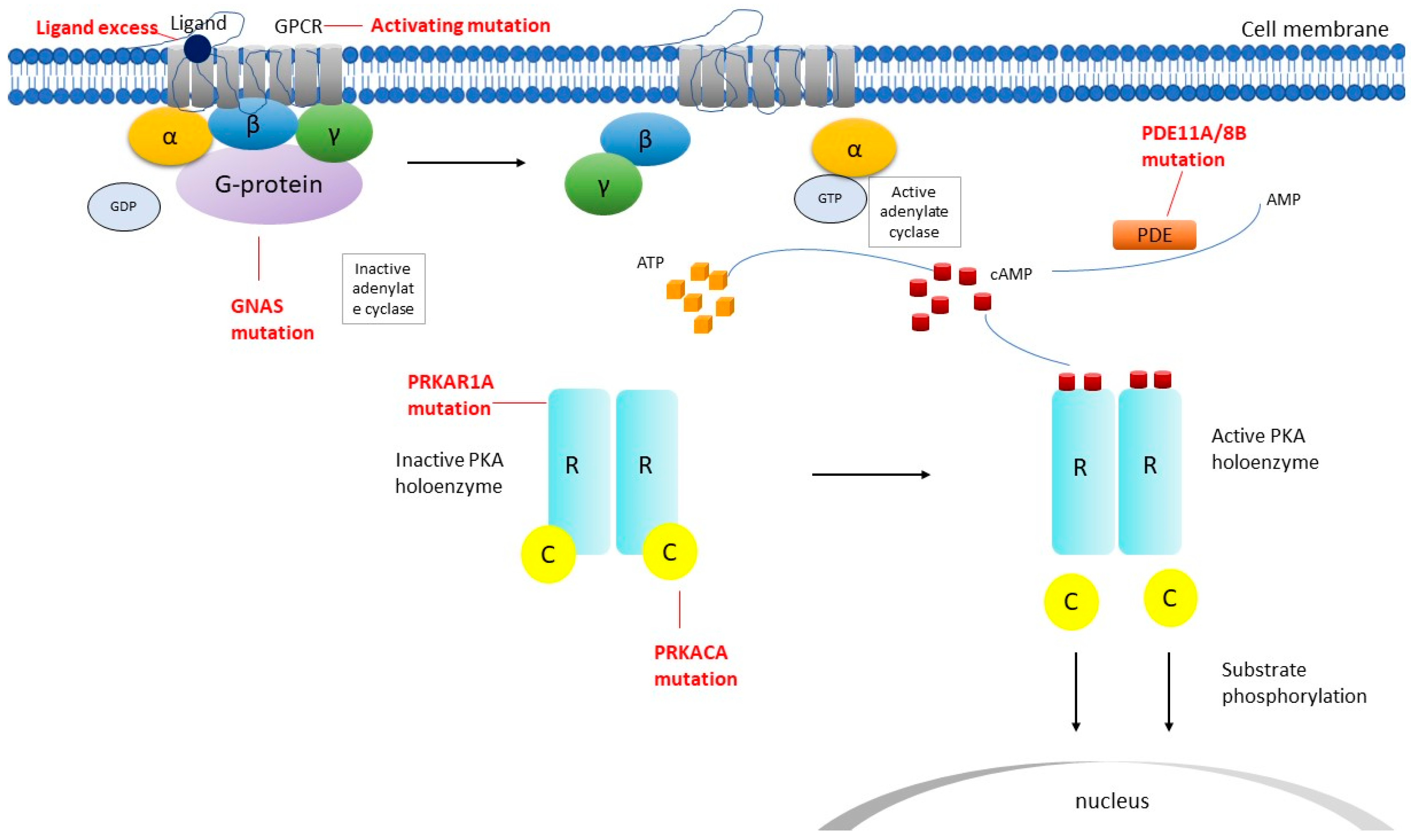
2.1. Micronodular Adrenocortical Hyperplasia
2.2. Macronodular Adrenal Hyperplasia (Pbmah)
2.3. Adrenocortical Adenomas Producing Cortisol
3. Benign Adrenocortical Tumors Producing Aldosterone (Adenomas and Hyperplasias)
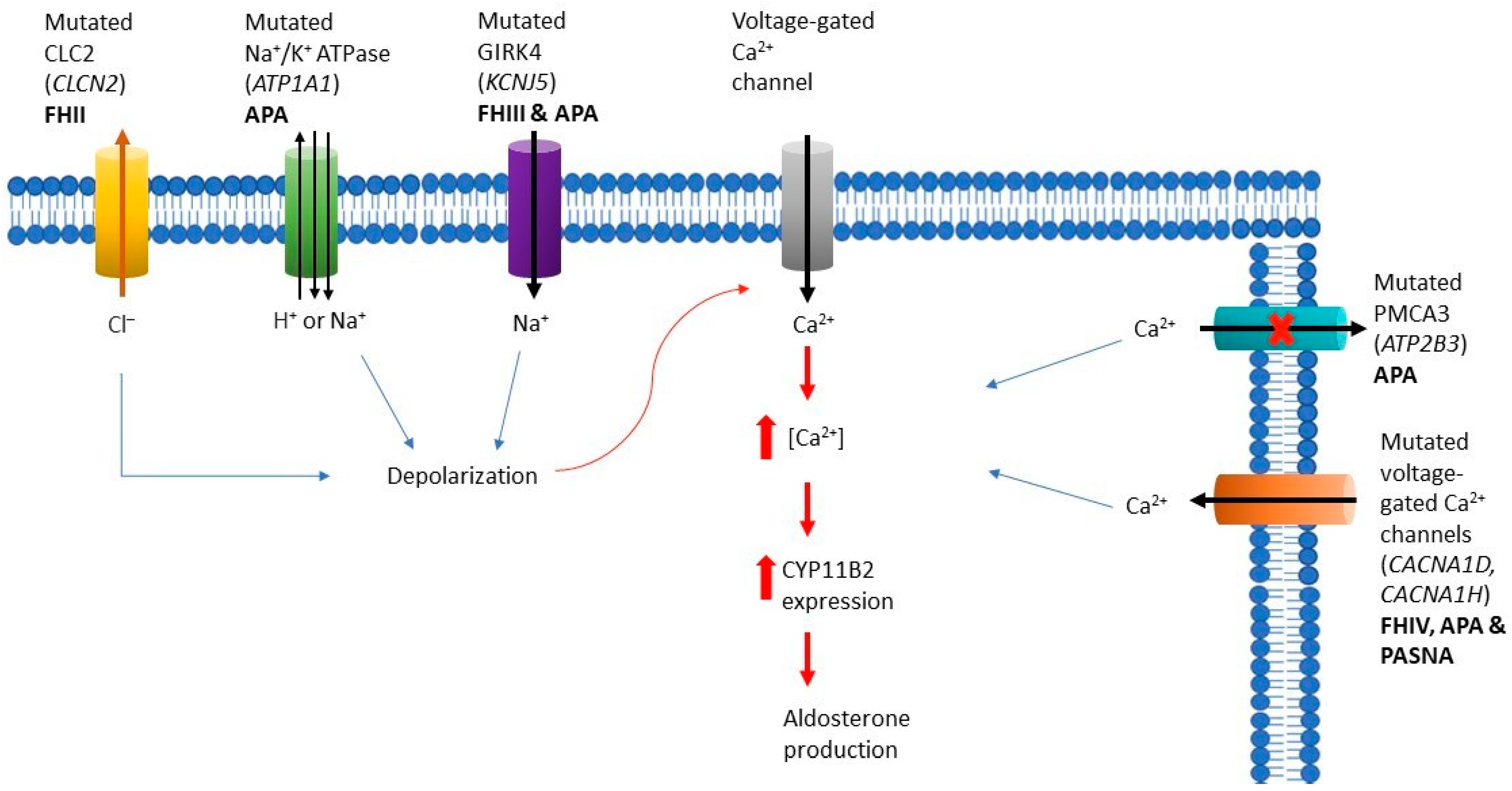
3.1. Familial Hyperaldosteronism
3.1.1. Familial Hyperaldosteronism Type I
3.1.2. Familial Hyperaldosteronism Type II
3.1.3. Familial Hyperaldosteronism Type III
3.1.4. Familial Hyperaldosteronism Type IV
3.2. Aldosterone-Producing Adrenocortical Adenomas
4. Conclusions
Funding
Conflicts of Interest
References
- Val, P.; Martinez, A. Editorial: Adrenal Cortex: From Physiology to Disease. Front. Endocrinol. 2016, 7, 51. [Google Scholar] [CrossRef][Green Version]
- Bertherat, J.; Mosnier-Pudar, H.; Bertagna, X. Adrenal incidentalomas. Curr. Opin. Oncol. 2002, 14, 58–63. [Google Scholar] [CrossRef]
- Grumbach, M.M.; Biller, B.M.; Braunstein, G.D.; Campbell, K.K.; Carney, J.A.; Godley, P.A.; Harris, E.L.; Lee, J.K.; Oertel, Y.C.; Posner, M.C.; et al. Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann. Intern. Med. 2003, 138, 424–429. [Google Scholar] [CrossRef]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef]
- Zeiger, M.A.; Thompson, G.B.; Duh, Q.Y.; Hamrahian, A.H.; Angelos, P.; Elaraj, D.; Fishman, E.; Kharlip, J.; Garber, J.R.; Mechanick, J.I.; et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: Executive summary of recommendations. Endocr. Pract. 2009, 15, 450–453. [Google Scholar] [CrossRef]
- Mansmann, G.; Lau, J.; Balk, E.; Rothberg, M.; Miyachi, Y.; Bornstein, S.R. The clinically inapparent adrenal mass: Update in diagnosis and management. Endocr. Rev. 2004, 25, 309–340. [Google Scholar] [CrossRef]
- Bonnet-Serrano, F.; Bertherat, J. Genetics of tumors of the adrenal cortex. Endocr. Relat. Cancer 2018, 25, R131–R152. [Google Scholar] [CrossRef]
- Kebebew, E.; Reiff, E.; Duh, Q.Y.; Clark, O.H.; McMillan, A. Extent of disease at presentation and outcome for adrenocortical carcinoma: Have we made progress? World J. Surg. 2006, 30, 872–878. [Google Scholar] [CrossRef]
- Kerkhofs, T.M.; Verhoeven, R.H.; Van der Zwan, J.M.; Dieleman, J.; Kerstens, M.N.; Links, T.P.; Van de Poll-Franse, L.V.; Haak, H.R. Adrenocortical carcinoma: A population-based study on incidence and survival in the Netherlands since 1993. Eur. J. Cancer 2013, 49, 2579–2586. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assie, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef]
- Carney, J.A.; Young, W.F.; Stratakis, C.A. Primary bimorphic adrenocortical disease: Cause of hypercortisolism in McCune-Albright syndrome. Am. J. Surg. Pathol. 2011, 35, 1311–1326. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Assie, G.; Libe, R.; Espiard, S.; Rizk-Rabin, M.; Guimier, A.; Luscap, W.; Barreau, O.; Lefevre, L.; Sibony, M.; Guignat, L.; et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N. Engl. J. Med. 2013, 369, 2105–2114. [Google Scholar] [CrossRef]
- Gicquel, C.; Raffin-Sanson, M.L.; Gaston, V.; Bertagna, X.; Plouin, P.F.; Schlumberger, M.; Louvel, A.; Luton, J.P.; Le Bouc, Y. Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: Study on a series of 82 tumors. J. Clin. Endocrinol. Metab. 1997, 82, 2559–2565. [Google Scholar] [CrossRef]
- Reincke, M.; Karl, M.; Travis, W.H.; Mastorakos, G.; Allolio, B.; Linehan, H.M.; Chrousos, G.P. p53 mutations in human adrenocortical neoplasms: Immunohistochemical and molecular studies. J. Clin. Endocrinol. Metab. 1994, 78, 790–794. [Google Scholar] [CrossRef][Green Version]
- Lindholm, J.; Juul, S.; Jorgensen, J.O.; Astrup, J.; Bjerre, P.; Feldt-Rasmussen, U.; Hagen, C.; Jorgensen, J.; Kosteljanetz, M.; Kristensen, L.; et al. Incidence and late prognosis of cushing’s syndrome: A population-based study. J. Clin. Endocrinol. Metab. 2001, 86, 117–123. [Google Scholar] [CrossRef]
- Steffensen, C.; Bak, A.M.; Rubeck, K.Z.; Jorgensen, J.O. Epidemiology of Cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 1–5. [Google Scholar] [CrossRef]
- Bolland, M.J.; Holdaway, I.M.; Berkeley, J.E.; Lim, S.; Dransfield, W.J.; Conaglen, J.V.; Croxson, M.S.; Gamble, G.D.; Hunt, P.J.; Toomath, R.J. Mortality and morbidity in Cushing’s syndrome in New Zealand. Clin. Endocrinol. 2011, 75, 436–442. [Google Scholar] [CrossRef]
- Valassi, E.; Santos, A.; Yaneva, M.; Toth, M.; Strasburger, C.J.; Chanson, P.; Wass, J.A.; Chabre, O.; Pfeifer, M.; Feelders, R.A.; et al. The European Registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur. J. Endocrinol. 2011, 165, 383–392. [Google Scholar] [CrossRef]
- Lacroix, A.; Feelders, R.A.; Stratakis, C.A.; Nieman, L.K. Cushing’s syndrome. Lancet 2015, 386, 913–927. [Google Scholar] [CrossRef]
- Hannah-Shmouni, F.; Stratakis, C.A. A Gene-Based Classification of Primary Adrenocortical Hyperplasias. Horm. Metab. Res. 2020, 52, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Boikos, S.A. Genetics of adrenal tumors associated with Cushing’s syndrome: A new classification for bilateral adrenocortical hyperplasias. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 748–757. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Stratakis, C.A.; Hannah-Shmouni, F. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101428. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Ilouz, R.; Zhang, P.; Kornev, A.P. Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012, 13, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.; Salpea, P.; Stratakis, C.A. Carney complex: An update. Eur. J. Endocrinol. 2015, 173, M85–M97. [Google Scholar] [CrossRef]
- Pitsava, G.; Zhu, C.; Sundaram, R.; Mills, J.L.; Stratakis, C.A. Predicting the risk of cardiac myxoma in Carney complex. Genet. Med. 2021, 23, 80–85. [Google Scholar] [CrossRef]
- Bertherat, J.; Horvath, A.; Groussin, L.; Grabar, S.; Boikos, S.; Cazabat, L.; Libe, R.; Rene-Corail, F.; Stergiopoulos, S.; Bourdeau, I.; et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J. Clin. Endocrinol. Metab. 2009, 94, 2085–2091. [Google Scholar] [CrossRef]
- Stratakis, C.A. Genetics of Carney complex and related familial lentiginoses, and other multiple tumor syndromes. Front. Biosci. 2000, 5, D353–D366. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Cazabat, L.; Ragazzon, B.; Groussin, L.; Bertherat, J. PRKAR1A mutations in primary pigmented nodular adrenocortical disease. Pituitary 2006, 9, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Carney, J.A.; Lin, J.P.; Papanicolaou, D.A.; Karl, M.; Kastner, D.L.; Pras, E.; Chrousos, G.P. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J. Clin. Investig. 1996, 97, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Matyakhina, L.; Pack, S.; Kirschner, L.S.; Pak, E.; Mannan, P.; Jaikumar, J.; Taymans, S.E.; Sandrini, F.; Carney, J.A.; Stratakis, C.A. Chromosome 2 (2p16) abnormalities in Carney complex tumours. J. Med. Genet. 2003, 40, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Bertherat, J.; Groussin, L.; Sandrini, F.; Matyakhina, L.; Bei, T.; Stergiopoulos, S.; Papageorgiou, T.; Bourdeau, I.; Kirschner, L.S.; Vincent-Dejean, C.; et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res. 2003, 63, 5308–5319. [Google Scholar] [PubMed]
- Sahut-Barnola, I.; de Joussineau, C.; Val, P.; Lambert-Langlais, S.; Damon, C.; Lefrancois-Martinez, A.M.; Pointud, J.C.; Marceau, G.; Sapin, V.; Tissier, F.; et al. Cushing’s syndrome and fetal features resurgence in adrenal cortex-specific Prkar1a knockout mice. PLoS Genet. 2010, 6, e1000980. [Google Scholar] [CrossRef]
- Horvath, A.; Boikos, S.; Giatzakis, C.; Robinson-White, A.; Groussin, L.; Griffin, K.J.; Stein, E.; Levine, E.; Delimpasi, G.; Hsiao, H.P.; et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat. Genet. 2006, 38, 794–800. [Google Scholar] [CrossRef]
- Libe, R.; Horvath, A.; Vezzosi, D.; Fratticci, A.; Coste, J.; Perlemoine, K.; Ragazzon, B.; Guillaud-Bataille, M.; Groussin, L.; Clauser, E.; et al. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with Carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J. Clin. Endocrinol. Metab. 2011, 96, E208–E214. [Google Scholar] [CrossRef]
- Horvath, A.; Mericq, V.; Stratakis, C.A. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N. Engl. J. Med. 2008, 358, 750–752. [Google Scholar] [CrossRef]
- Libe, R.; Fratticci, A.; Coste, J.; Tissier, F.; Horvath, A.; Ragazzon, B.; Rene-Corail, F.; Groussin, L.; Bertagna, X.; Raffin-Sanson, M.L.; et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin. Cancer Res. 2008, 14, 4016–4024. [Google Scholar] [CrossRef]
- Vezzosi, D.; Libe, R.; Baudry, C.; Rizk-Rabin, M.; Horvath, A.; Levy, I.; Rene-Corail, F.; Ragazzon, B.; Stratakis, C.A.; Vandecasteele, G.; et al. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): Functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J. Clin. Endocrinol. Metab. 2012, 97, E2063–E2069. [Google Scholar] [CrossRef]
- Rothenbuhler, A.; Horvath, A.; Libe, R.; Faucz, F.R.; Fratticci, A.; Raffin Sanson, M.L.; Vezzosi, D.; Azevedo, M.; Levy, I.; Almeida, M.Q.; et al. Identification of novel genetic variants in phosphodiesterase 8B (PDE8B), a cAMP-specific phosphodiesterase highly expressed in the adrenal cortex, in a cohort of patients with adrenal tumours. Clin. Endocrinol. 2012, 77, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Forlino, A.; Vetro, A.; Garavelli, L.; Ciccone, R.; London, E.; Stratakis, C.A.; Zuffardi, O. PRKACB and Carney complex. N. Engl. J. Med. 2014, 370, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Lodish, M.B.; Yuan, B.; Levy, I.; Braunstein, G.D.; Lyssikatos, C.; Salpea, P.; Szarek, E.; Karageorgiadis, A.S.; Belyavskaya, E.; Raygada, M.; et al. Germline PRKACA amplification causes variable phenotypes that may depend on the extent of the genomic defect: Molecular mechanisms and clinical presentations. Eur. J. Endocrinol. 2015, 172, 803–811. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Tadjine, M.; Lampron, A.; Ouadi, L.; Horvath, A.; Stratakis, C.A.; Bourdeau, I. Detection of somatic beta-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin. Endocrinol. 2008, 69, 367–373. [Google Scholar] [CrossRef]
- Gaujoux, S.; Tissier, F.; Groussin, L.; Libe, R.; Ragazzon, B.; Launay, P.; Audebourg, A.; Dousset, B.; Bertagna, X.; Bertherat, J. Wnt/beta-catenin and 3′,5′-cyclic adenosine 5′-monophosphate/protein kinase A signaling pathways alterations and somatic beta-catenin gene mutations in the progression of adrenocortical tumors. J. Clin. Endocrinol. Metab. 2008, 93, 4135–4140. [Google Scholar] [CrossRef]
- Louiset, E.; Duparc, C.; Young, J.; Renouf, S.; Tetsi Nomigni, M.; Boutelet, I.; Libe, R.; Bram, Z.; Groussin, L.; Caron, P.; et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N. Engl. J. Med. 2013, 369, 2115–2125. [Google Scholar] [CrossRef]
- Hsiao, H.P.; Kirschner, L.S.; Bourdeau, I.; Keil, M.F.; Boikos, S.A.; Verma, S.; Robinson-White, A.J.; Nesterova, M.; Lacroix, A.; Stratakis, C.A. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J. Clin. Endocrinol. Metab. 2009, 94, 2930–2937. [Google Scholar] [CrossRef]
- Libe, R.; Coste, J.; Guignat, L.; Tissier, F.; Lefebvre, H.; Barrande, G.; Ajzenberg, C.; Tauveron, I.; Clauser, E.; Dousset, B.; et al. Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: A frequent finding in a prospective study of 32 patients with overt or subclinical Cushing’s syndrome. Eur. J. Endocrinol. 2010, 163, 129–138. [Google Scholar] [CrossRef]
- Hofland, J.; Hofland, L.J.; van Koetsveld, P.M.; Steenbergen, J.; de Herder, W.W.; van Eijck, C.H.; de Krijger, R.R.; van Nederveen, F.H.; van Aken, M.O.; de Groot, J.W.; et al. ACTH-independent macronodular adrenocortical hyperplasia reveals prevalent aberrant in vivo and in vitro responses to hormonal stimuli and coupling of arginine-vasopressin type 1a receptor to 11beta-hydroxylase. Orphanet J. Rare Dis. 2013, 8, 142. [Google Scholar] [CrossRef]
- Lacroix, A.; Bolte, E.; Tremblay, J.; Dupre, J.; Poitras, P.; Fournier, H.; Garon, J.; Garrel, D.; Bayard, F.; Taillefer, R.; et al. Gastric inhibitory polypeptide-dependent cortisol hypersecretion—A new cause of Cushing’s syndrome. N. Engl. J. Med. 1992, 327, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Hamet, P.; Boutin, J.M. Leuprolide acetate therapy in luteinizing hormone—Dependent Cushing’s syndrome. N. Engl. J. Med. 1999, 341, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hwang, R.; Lee, J.; Rhee, Y.; Kim, D.J.; Chung, U.I.; Lim, S.K. Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of familial ACTH-independent macronodular adrenal hyperplasia. Clin. Endocrinol. 2005, 63, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Mircescu, H.; Jilwan, J.; N’Diaye, N.; Bourdeau, I.; Tremblay, J.; Hamet, P.; Lacroix, A. Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing’s syndrome? J. Clin. Endocrinol. Metab. 2000, 85, 3531–3536. [Google Scholar] [CrossRef]
- Miyamura, N.; Taguchi, T.; Murata, Y.; Taketa, K.; Iwashita, S.; Matsumoto, K.; Nishikawa, T.; Toyonaga, T.; Sakakida, M.; Araki, E. Inherited adrenocorticotropin-independent macronodular adrenal hyperplasia with abnormal cortisol secretion by vasopressin and catecholamines: Detection of the aberrant hormone receptors on adrenal gland. Endocrine 2002, 19, 319–326. [Google Scholar] [CrossRef]
- Reznik, Y.; Allali-Zerah, V.; Chayvialle, J.A.; Leroyer, R.; Leymarie, P.; Travert, G.; Lebrethon, M.C.; Budi, I.; Balliere, A.M.; Mahoudeau, J. Food-dependent Cushing’s syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N. Engl. J. Med. 1992, 327, 981–986. [Google Scholar] [CrossRef]
- Vezzosi, D.; Cartier, D.; Regnier, C.; Otal, P.; Bennet, A.; Parmentier, F.; Plantavid, M.; Lacroix, A.; Lefebvre, H.; Caron, P. Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. Eur. J. Endocrinol. 2007, 156, 21–31. [Google Scholar] [CrossRef]
- Gagliardi, L.; Hotu, C.; Casey, G.; Braund, W.J.; Ling, K.H.; Dodd, T.; Manavis, J.; Devitt, P.G.; Cutfield, R.; Rudzki, Z.; et al. Familial vasopressin-sensitive ACTH-independent macronodular adrenal hyperplasia (VPs-AIMAH): Clinical studies of three kindreds. Clin. Endocrinol. 2009, 70, 883–891. [Google Scholar] [CrossRef]
- Lampron, A.; Bourdeau, I.; Hamet, P.; Tremblay, J.; Lacroix, A. Whole genome expression profiling of glucose-dependent insulinotropic peptide (GIP)- and adrenocorticotropin-dependent adrenal hyperplasias reveals novel targets for the study of GIP-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 3611–3618. [Google Scholar] [CrossRef][Green Version]
- Swords, F.M.; Noon, L.A.; King, P.J.; Clark, A.J. Constitutive activation of the human ACTH receptor resulting from a synergistic interaction between two naturally occurring missense mutations in the MC2R gene. Mol. Cell Endocrinol. 2004, 213, 149–154. [Google Scholar] [CrossRef]
- Bourdeau, I.; Matyakhina, L.; Stergiopoulos, S.G.; Sandrini, F.; Boikos, S.; Stratakis, C.A. 17q22-24 chromosomal losses and alterations of protein kinase a subunit expression and activity in adrenocorticotropin-independent macronodular adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2006, 91, 3626–3632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fragoso, M.C.; Domenice, S.; Latronico, A.C.; Martin, R.M.; Pereira, M.A.; Zerbini, M.C.; Lucon, A.M.; Mendonca, B.B. Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J. Clin. Endocrinol. Metab. 2003, 88, 2147–2151. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, L.; Brady, R. McCune Albright Syndrome; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lumbroso, S.; Paris, F.; Sultan, C. McCune-Albright syndrome: Molecular genetics. J. Pediatr. Endocrinol. Metab. 2002, 15 (Suppl. 3), 875–882. [Google Scholar] [PubMed]
- Alam, N.A.; Bevan, S.; Churchman, M.; Barclay, E.; Barker, K.; Jaeger, E.E.; Nelson, H.M.; Healy, E.; Pembroke, A.C.; Friedmann, P.S.; et al. Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3-q43. Am. J. Hum. Genet. 2001, 68, 1264–1269. [Google Scholar] [CrossRef]
- Shuch, B.; Ricketts, C.J.; Vocke, C.D.; Valera, V.A.; Chen, C.C.; Gautam, R.; Gupta, G.N.; Gomez Macias, G.S.; Merino, M.J.; Bratslavsky, G.; et al. Adrenal nodular hyperplasia in hereditary leiomyomatosis and renal cell cancer. J. Urol. 2013, 189, 430–435. [Google Scholar] [CrossRef]
- Gaujoux, S.; Pinson, S.; Gimenez-Roqueplo, A.P.; Amar, L.; Ragazzon, B.; Launay, P.; Meatchi, T.; Libe, R.; Bertagna, X.; Audebourg, A.; et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin. Cancer Res. 2010, 16, 5133–5141. [Google Scholar] [CrossRef]
- Gatta-Cherifi, B.; Chabre, O.; Murat, A.; Niccoli, P.; Cardot-Bauters, C.; Rohmer, V.; Young, J.; Delemer, B.; Du Boullay, H.; Verger, M.F.; et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’etude des Tumeurs Endocrines database. Eur. J. Endocrinol. 2012, 166, 269–279. [Google Scholar] [CrossRef]
- Shiroky, J.S.; Lerner-Ellis, J.P.; Govindarajan, A.; Urbach, D.R.; Devon, K.M. Characteristics of Adrenal Masses in Familial Adenomatous Polyposis. Dis. Colon Rectum 2018, 61, 679–685. [Google Scholar] [CrossRef]
- Matyakhina, L.; Freedman, R.J.; Bourdeau, I.; Wei, M.H.; Stergiopoulos, S.G.; Chidakel, A.; Walther, M.; Abu-Asab, M.; Tsokos, M.; Keil, M.; et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: A clinical and molecular genetic investigation. J. Clin. Endocrinol. Metab. 2005, 90, 3773–3779. [Google Scholar] [CrossRef]
- Faucz, F.R.; Zilbermint, M.; Lodish, M.B.; Szarek, E.; Trivellin, G.; Sinaii, N.; Berthon, A.; Libe, R.; Assie, G.; Espiard, S.; et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: A clinical and genetic investigation. J. Clin. Endocrinol. Metab. 2014, 99, E1113–E1119. [Google Scholar] [CrossRef] [PubMed]
- Espiard, S.; Drougat, L.; Libe, R.; Assie, G.; Perlemoine, K.; Guignat, L.; Barrande, G.; Brucker-Davis, F.; Doullay, F.; Lopez, S.; et al. ARMC5 Mutations in a Large Cohort of Primary Macronodular Adrenal Hyperplasia: Clinical and Functional Consequences. J. Clin. Endocrinol. Metab. 2015, 100, E926–E935. [Google Scholar] [CrossRef] [PubMed]
- Elbelt, U.; Trovato, A.; Kloth, M.; Gentz, E.; Finke, R.; Spranger, J.; Galas, D.; Weber, S.; Wolf, C.; Konig, K.; et al. Molecular and clinical evidence for an ARMC5 tumor syndrome: Concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J. Clin. Endocrinol. Metab. 2015, 100, E119–E128. [Google Scholar] [CrossRef] [PubMed]
- Zilbermint, M.; Xekouki, P.; Faucz, F.R.; Berthon, A.; Gkourogianni, A.; Schernthaner-Reiter, M.H.; Batsis, M.; Sinaii, N.; Quezado, M.M.; Merino, M.; et al. Primary Aldosteronism and ARMC5 Variants. J. Clin. Endocrinol. Metab. 2015, 100, E900–E909. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lao, L.; Mao, J.; Jin, W.; Luo, H.; Charpentier, T.; Qi, S.; Peng, J.; Hu, B.; Marcinkiewicz, M.M.; et al. Armc5 deletion causes developmental defects and compromises T-cell immune responses. Nat. Commun. 2017, 8, 13834. [Google Scholar] [CrossRef]
- Berthon, A.; Faucz, F.R.; Espiard, S.; Drougat, L.; Bertherat, J.; Stratakis, C.A. Age-dependent effects of Armc5 haploinsufficiency on adrenocortical function. Hum. Mol. Genet. 2017, 26, 3495–3507. [Google Scholar] [CrossRef]
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagnere, A.M.; Rene-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627. [Google Scholar] [CrossRef]
- Cao, Y.; He, M.; Gao, Z.; Peng, Y.; Li, Y.; Li, L.; Zhou, W.; Li, X.; Zhong, X.; Lei, Y.; et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science 2014, 344, 913–917. [Google Scholar] [CrossRef]
- Zhu, J.; Cui, L.; Wang, W.; Hang, X.Y.; Xu, A.X.; Yang, S.X.; Dou, J.T.; Mu, Y.M.; Zhang, X.; Gao, J.P. Whole exome sequencing identifies mutation of EDNRA involved in ACTH-independent macronodular adrenal hyperplasia. Fam. Cancer 2013, 12, 657–667. [Google Scholar] [CrossRef]
- Stratakis, C.A. Cyclic AMP-dependent protein kinase catalytic subunit A (PRKACA): The expected, the unexpected, and what might be next. J. Pathol. 2018, 244, 257–259. [Google Scholar] [CrossRef]
- Sato, Y.; Maekawa, S.; Ishii, R.; Sanada, M.; Morikawa, T.; Shiraishi, Y.; Yoshida, K.; Nagata, Y.; Sato-Otsubo, A.; Yoshizato, T.; et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014, 344, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Scholl, U.I.; Healy, J.M.; Choi, M.; Prasad, M.L.; Nelson-Williams, C.; Kunstman, J.W.; Korah, R.; Suttorp, A.C.; Dietrich, D.; et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 2014, 46, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Di Dalmazi, G.; Kisker, C.; Calebiro, D.; Mannelli, M.; Canu, L.; Arnaldi, G.; Quinkler, M.; Rayes, N.; Tabarin, A.; Laure Jullie, M.; et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: A European multicentric study. J. Clin. Endocrinol. Metab. 2014, 99, E2093–E2100. [Google Scholar] [CrossRef] [PubMed]
- Bathon, K.; Weigand, I.; Vanselow, J.T.; Ronchi, C.L.; Sbiera, S.; Schlosser, A.; Fassnacht, M.; Calebiro, D. Alterations in Protein Kinase A Substrate Specificity as a Potential Cause of Cushing Syndrome. Endocrinology 2019, 160, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Espiard, S.; Knape, M.J.; Bathon, K.; Assie, G.; Rizk-Rabin, M.; Faillot, S.; Luscap-Rondof, W.; Abid, D.; Guignat, L.; Calebiro, D.; et al. Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 2018, 3, e98296. [Google Scholar] [CrossRef]
- Libe, R.; Bertherat, J. Molecular genetics of adrenocortical tumours, from familial to sporadic diseases. Eur. J. Endocrinol. 2005, 153, 477–487. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Di Dalmazi, G.; Faillot, S.; Sbiera, S.; Assie, G.; Weigand, I.; Calebiro, D.; Schwarzmayr, T.; Appenzeller, S.; Rubin, B.; et al. Genetic Landscape of Sporadic Unilateral Adrenocortical Adenomas without PRKACA p.Leu206Arg Mutation. J. Clin. Endocrinol. Metab. 2016, 101, 3526–3538. [Google Scholar] [CrossRef]
- Kobayashi, H.; Usui, T.; Fukata, J.; Yoshimasa, T.; Oki, Y.; Nakao, K. Mutation analysis of Gsalpha, adrenocorticotropin receptor and p53 genes in Japanese patients with adrenocortical neoplasms: Including a case of Gsalpha mutation. Endocr. J. 2000, 47, 461–466. [Google Scholar] [CrossRef][Green Version]
- Almeida, M.Q.; Azevedo, M.F.; Xekouki, P.; Bimpaki, E.I.; Horvath, A.; Collins, M.T.; Karaviti, L.P.; Jeha, G.S.; Bhattacharyya, N.; Cheadle, C.; et al. Activation of cyclic AMP signaling leads to different pathway alterations in lesions of the adrenal cortex caused by germline PRKAR1A defects versus those due to somatic GNAS mutations. J. Clin. Endocrinol. Metab. 2012, 97, E687–E693. [Google Scholar] [CrossRef]
- Monticone, S.; Burrello, J.; Tizzani, D.; Bertello, C.; Viola, A.; Buffolo, F.; Gabetti, L.; Mengozzi, G.; Williams, T.A.; Rabbia, F.; et al. Prevalence and Clinical Manifestations of Primary Aldosteronism Encountered in Primary Care Practice. J. Am. Coll. Cardiol. 2017, 69, 1811–1820. [Google Scholar] [CrossRef]
- Hannemann, A.; Wallaschofski, H. Prevalence of primary aldosteronism in patient’s cohorts and in population-based studies—A review of the current literature. Horm. Metab. Res. 2012, 44, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F., Jr. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Douma, S.; Petidis, K.; Doumas, M.; Papaefthimiou, P.; Triantafyllou, A.; Kartali, N.; Papadopoulos, N.; Vogiatzis, K.; Zamboulis, C. Prevalence of primary hyperaldosteronism in resistant hypertension: A retrospective observational study. Lancet 2008, 371, 1921–1926. [Google Scholar] [CrossRef]
- Calhoun, D.A.; Nishizaka, M.K.; Zaman, M.A.; Thakkar, R.B.; Weissmann, P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002, 40, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Itcho, K.; Oki, K.; Ohno, H.; Yoneda, M. Update on Genetics of Primary Aldosteronism. Biomedicines 2021, 9, 409. [Google Scholar] [CrossRef] [PubMed]
- Young, W.F. Primary aldosteronism: Renaissance of a syndrome. Clin. Endocrinol. 2007, 66, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Mulatero, P.; Baudrand, R.; Adler, G.K. The Expanding Spectrum of Primary Aldosteronism: Implications for Diagnosis, Pathogenesis, and Treatment. Endocr. Rev. 2018, 39, 1057–1088. [Google Scholar] [CrossRef]
- Byrd, J.B.; Turcu, A.F.; Auchus, R.J. Primary Aldosteronism: Practical Approach to Diagnosis and Management. Circulation 2018, 138, 823–835. [Google Scholar] [CrossRef]
- Spat, A.; Hunyady, L. Control of aldosterone secretion: A model for convergence in cellular signaling pathways. Physiol. Rev. 2004, 84, 489–539. [Google Scholar] [CrossRef]
- Sutherland, D.J.; Ruse, J.L.; Laidlaw, J.C. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can. Med. Assoc. J. 1966, 95, 1109–1119. [Google Scholar]
- Lifton, R.P.; Dluhy, R.G.; Powers, M.; Rich, G.M.; Cook, S.; Ulick, S.; Lalouel, J.M. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992, 355, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Stowasser, M.; Bachmann, A.W.; Huggard, P.R.; Rossetti, T.R.; Gordon, R.D. Treatment of familial hyperaldosteronism type I: Only partial suppression of adrenocorticotropin required to correct hypertension. J. Clin. Endocrinol. Metab. 2000, 85, 3313–3318. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.D.; Stowasser, M.; Tunny, T.J.; Klemm, S.A.; Finn, W.L.; Krek, A.L. Clinical and pathological diversity of primary aldosteronism, including a new familial variety. Clin. Exp. Pharmacol. Physiol. 1991, 18, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Stowasser, M.; Gordon, R.D.; Tunny, T.J.; Klemm, S.A.; Finn, W.L.; Krek, A.L. Familial hyperaldosteronism type II: Five families with a new variety of primary aldosteronism. Clin. Exp. Pharmacol. Physiol. 1992, 19, 319–322. [Google Scholar] [CrossRef]
- Scholl, U.I.; Stolting, G.; Schewe, J.; Thiel, A.; Tan, H.; Nelson-Williams, C.; Vichot, A.A.; Jin, S.C.; Loring, E.; Untiet, V.; et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 2018, 50, 349–354. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Daniil, G.; Orozco, I.J.; Goppner, C.; El Zein, R.; Jain, V.; Boulkroun, S.; Jeunemaitre, X.; Amar, L.; Lefebvre, H.; et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 2018, 50, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Soderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41. [Google Scholar] [CrossRef]
- Rege, J.; Nanba, K.; Blinder, A.R.; Plaska, S.; Udager, A.M.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; Rainey, W.E.; Else, T. Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J. Endocr. Soc. 2020, 4, bvaa123. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Yue, P.; Bjorklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef]
- Oki, K.; Plonczynski, M.W.; Luis Lam, M.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology 2012, 153, 1774–1782. [Google Scholar] [CrossRef]
- Kuppusamy, M.; Caroccia, B.; Stindl, J.; Bandulik, S.; Lenzini, L.; Gioco, F.; Fishman, V.; Zanotti, G.; Gomez-Sanchez, C.; Bader, M.; et al. A novel KCNJ5-insT149 somatic mutation close to, but outside, the selectivity filter causes resistant hypertension by loss of selectivity for potassium. J. Clin. Endocrinol. Metab. 2014, 99, E1765–E1773. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.; Penton, D.; Stindl, J.; Humberg, E.; Tegtmeier, I.; Sterner, C.; Beuschlein, F.; Reincke, M.; Barhanin, J.; Bandulik, S.; et al. Pharmacology and pathophysiology of mutated KCNJ5 found in adrenal aldosterone-producing adenomas. Endocrinology 2014, 155, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Hattangady, N.G.; Karashima, S.; Yuan, L.; Ponce-Balbuena, D.; Jalife, J.; Gomez-Sanchez, C.E.; Auchus, R.J.; Rainey, W.E.; Else, T. Mutated KCNJ5 activates the acute and chronic regulatory steps in aldosterone production. J. Mol. Endocrinol. 2016, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Healy, J.M.; Thiel, A.; Fonseca, A.L.; Brown, T.C.; Kunstman, J.W.; Horne, M.J.; Dietrich, D.; Riemer, J.; Kucukkoylu, S.; et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. 2015, 83, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Omata, K.; Else, T.; Beck, P.C.C.; Nanba, A.T.; Turcu, A.F.; Miller, B.S.; Giordano, T.J.; Tomlins, S.A.; Rainey, W.E. Targeted Molecular Characterization of Aldosterone-Producing Adenomas in White Americans. J. Clin. Endocrinol. Metab. 2018, 103, 3869–3876. [Google Scholar] [CrossRef] [PubMed]
- Hardege, I.; Xu, S.; Gordon, R.D.; Thompson, A.J.; Figg, N.; Stowasser, M.; Murrell-Lagnado, R.; O’Shaughnessy, K.M. Novel Insertion Mutation in KCNJ5 Channel Produces Constitutive Aldosterone Release From H295R Cells. Mol. Endocrinol. 2015, 29, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 2014, 63, 188–195. [Google Scholar] [CrossRef]
- Cheng, C.J.; Sung, C.C.; Wu, S.T.; Lin, Y.C.; Sytwu, H.K.; Huang, C.L.; Lin, S.H. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J. Clin. Endocrinol. Metab. 2015, 100, E155–E163. [Google Scholar] [CrossRef]
- Akerstrom, T.; Crona, J.; Delgado Verdugo, A.; Starker, L.F.; Cupisti, K.; Willenberg, H.S.; Knoefel, W.T.; Saeger, W.; Feller, A.; Ip, J.; et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS ONE 2012, 7, e41926. [Google Scholar] [CrossRef]
- Nanba, K.; Omata, K.; Tomlins, S.A.; Giordano, T.J.; Hammer, G.D.; Rainey, W.E.; Else, T. Double adrenocortical adenomas harboring independent KCNJ5 and PRKACA somatic mutations. Eur. J. Endocrinol. 2016, 175, K1–K6. [Google Scholar] [CrossRef]
- Zheng, F.F.; Zhu, L.M.; Nie, A.F.; Li, X.Y.; Lin, J.R.; Zhang, K.; Chen, J.; Zhou, W.L.; Shen, Z.J.; Zhu, Y.C.; et al. Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension 2015, 65, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Omata, K.; Gomez-Sanchez, C.E.; Stratakis, C.A.; Demidowich, A.P.; Suzuki, M.; Thompson, L.D.R.; Cohen, D.L.; Luther, J.M.; Gellert, L.; et al. Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension 2019, 73, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, T.; Omura, M.; Suematsu, S.; Saito, J.; Nishikawa, T. KCNJ5 mutation as a predictor for resolution of hypertension after surgical treatment of aldosterone-producing adenoma. J. Hypertens. 2018, 36, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Azizan, E.A.; Murthy, M.; Stowasser, M.; Gordon, R.; Kowalski, B.; Xu, S.; Brown, M.J.; O’Shaughnessy, K.M. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension 2012, 59, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Azizan, E.A.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Lenzini, L.; Rossitto, G.; Maiolino, G.; Letizia, C.; Funder, J.W.; Rossi, G.P. A Meta-Analysis of Somatic KCNJ5 K+ Channel Mutations in 1636 Patients with an Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metab. 2015, 100, E1089–E1095. [Google Scholar] [CrossRef]
- Geller, D.S.; Zhang, J.; Wisgerhof, M.V.; Shackleton, C.; Kashgarian, M.; Lifton, R.P. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J. Clin. Endocrinol. Metab. 2008, 93, 3117–3123. [Google Scholar] [CrossRef]
- Scholl, U.I.; Nelson-Williams, C.; Yue, P.; Grekin, R.; Wyatt, R.J.; Dillon, M.J.; Couch, R.; Hammer, L.K.; Harley, F.L.; Farhi, A.; et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl. Acad. Sci. USA 2012, 109, 2533–2538. [Google Scholar] [CrossRef]
- Monticone, S.; Bandulik, S.; Stindl, J.; Zilbermint, M.; Dedov, I.; Mulatero, P.; Allgaeuer, M.; Lee, C.C.; Stratakis, C.A.; Williams, T.A.; et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J. Clin. Endocrinol. Metab. 2015, 100, E114–E118. [Google Scholar] [CrossRef]
- Charmandari, E.; Sertedaki, A.; Kino, T.; Merakou, C.; Hoffman, D.A.; Hatch, M.M.; Hurt, D.E.; Lin, L.; Xekouki, P.; Stratakis, C.A.; et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J. Clin. Endocrinol. Metab. 2012, 97, E1532–E1539. [Google Scholar] [CrossRef]
- Mulatero, P.; Tauber, P.; Zennaro, M.C.; Monticone, S.; Lang, K.; Beuschlein, F.; Fischer, E.; Tizzani, D.; Pallauf, A.; Viola, A.; et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 2012, 59, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Hattangady, N.G.; Penton, D.; Isales, C.M.; Edwards, M.A.; Williams, T.A.; Sterner, C.; Warth, R.; Mulatero, P.; Rainey, W.E. A Novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J. Clin. Endocrinol. Metab. 2013, 98, E1861–E1865. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.G.; Suzuki, M.; Berthon, A.; Kamilaris, C.; Demidowich, A.; Lack, J.; Zilbermint, M.; Hannah-Shmouni, F.; Faucz, F.R.; Stratakis, C.A. Mosaicism for KCNJ5 Causing Early-Onset Primary Aldosteronism due to Bilateral Adrenocortical Hyperplasia. Am. J. Hypertens. 2020, 33, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Nishimoto, K.; Seki, T.; Matsuzawa, Y.; Saito, J.; Omura, M.; Gomez-Sanchez, C.E.; Makita, K.; Matsui, S.; Moriya, N.; et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol. Cell Endocrinol. 2017, 441, 134–139. [Google Scholar] [CrossRef]
- Scholl, U.I.; Stolting, G.; Nelson-Williams, C.; Vichot, A.A.; Choi, M.; Loring, E.; Prasad, M.L.; Goh, G.; Carling, T.; Juhlin, C.C.; et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015, 4, e06315. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Rassi-Cruz, M.; Maria, A.G.; Faucz, F.R.; London, E.; Vilela, L.A.P.; Santana, L.S.; Benedetti, A.F.F.; Goldbaum, T.S.; Tanno, F.Y.; Srougi, V.; et al. Phosphodiesterase 2A and 3B variants are associated with primary aldosteronism. Endocr. Relat. Cancer 2021, 28, 1–13. [Google Scholar] [CrossRef]
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444. [Google Scholar] [CrossRef]
- Hong, A.R.; Kim, J.H.; Song, Y.S.; Lee, K.E.; Seo, S.H.; Seong, M.W.; Shin, C.S.; Kim, S.W.; Kim, S.Y. Genetics of Aldosterone-Producing Adenoma in Korean Patients. PLoS ONE 2016, 11, e0147590. [Google Scholar] [CrossRef]
- Wu, V.C.; Huang, K.H.; Peng, K.Y.; Tsai, Y.C.; Wu, C.H.; Wang, S.M.; Yang, S.Y.; Lin, L.Y.; Chang, C.C.; Lin, Y.H.; et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci. Rep. 2015, 5, 11396. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F. Genetic Causes of Functional Adrenocortical Adenomas. Endocr. Rev. 2017, 38, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Seidel, E.; Schewe, J.; Scholl, U.I. Genetic causes of primary aldosteronism. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Nanba, K.; Yamazaki, Y.; Bick, N.; Onodera, K.; Tezuka, Y.; Omata, K.; Ono, Y.; Blinder, A.R.; Tomlins, S.A.; Rainey, W.E.; et al. Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J. Clin. Endocrinol. Metab. 2020, 105, e4066–e4073. [Google Scholar] [CrossRef]
- De Sousa, K.; Boulkroun, S.; Baron, S.; Nanba, K.; Wack, M.; Rainey, W.E.; Rocha, A.; Giscos-Douriez, I.; Meatchi, T.; Amar, L.; et al. Genetic, Cellular, and Molecular Heterogeneity in Adrenals with Aldosterone-Producing Adenoma. Hypertension 2020, 75, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.; Aichinger, B.; Christ, C.; Stindl, J.; Rhayem, Y.; Beuschlein, F.; Warth, R.; Bandulik, S. Cellular Pathophysiology of an Adrenal Adenoma-Associated Mutant of the Plasma Membrane Ca2+-ATPase ATP2B3. Endocrinology 2016, 157, 2489–2499. [Google Scholar] [CrossRef]
- Shimada, H.; Yamazaki, Y.; Sugawara, A.; Sasano, H.; Nakamura, Y. Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway. Biomedicines 2021, 9, 892. [Google Scholar] [CrossRef]
- Tadjine, M.; Lampron, A.; Ouadi, L.; Bourdeau, I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin. Endocrinol. 2008, 68, 264–270. [Google Scholar] [CrossRef]
- Berthon, A.; Drelon, C.; Ragazzon, B.; Boulkroun, S.; Tissier, F.; Amar, L.; Samson-Couterie, B.; Zennaro, M.C.; Plouin, P.F.; Skah, S.; et al. WNT/beta-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum. Mol. Genet. 2014, 23, 889–905. [Google Scholar] [CrossRef]
- Akerstrom, T.; Maharjan, R.; Sven Willenberg, H.; Cupisti, K.; Ip, J.; Moser, A.; Stalberg, P.; Robinson, B.; Alexander Iwen, K.; Dralle, H.; et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci. Rep. 2016, 6, 19546. [Google Scholar] [CrossRef]
- Teo, A.E.; Garg, S.; Shaikh, L.H.; Zhou, J.; Karet Frankl, F.E.; Gurnell, M.; Happerfield, L.; Marker, A.; Bienz, M.; Azizan, E.A.; et al. Pregnancy, Primary Aldosteronism, and Adrenal CTNNB1 Mutations. N. Engl. J. Med. 2015, 373, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, N.; Caceres-Gorriti, K.Y.; Corbeil, G.; El Ghoyareb, N.; Ludwig, N.; Latour, M.; Lacroix, A.; Bourdeau, I. Genetic Characterization of GnRH/LH-Responsive Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2018, 103, 2926–2935. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Familial Hyperaldosteronism | Gene | Clinical Characteristics |
|---|---|---|
| Type I | CYP11B1/CYP11B2 chimeric gene | Glucocorticoid-suppressive hyperaldosteronism |
| Type II | CLCN2 | Early onset PA |
| Type III | KCNJ5 | Severe early-onset PA (T158A, I157S, E145Q, G151R) Mild PA (G151E, Y152C) |
| Type IV | CACNA1H | Early onset PA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitsava, G.; Stratakis, C.A. Genetic Alterations in Benign Adrenal Tumors. Biomedicines 2022, 10, 1041. https://doi.org/10.3390/biomedicines10051041
Pitsava G, Stratakis CA. Genetic Alterations in Benign Adrenal Tumors. Biomedicines. 2022; 10(5):1041. https://doi.org/10.3390/biomedicines10051041
Chicago/Turabian StylePitsava, Georgia, and Constantine A. Stratakis. 2022. "Genetic Alterations in Benign Adrenal Tumors" Biomedicines 10, no. 5: 1041. https://doi.org/10.3390/biomedicines10051041
APA StylePitsava, G., & Stratakis, C. A. (2022). Genetic Alterations in Benign Adrenal Tumors. Biomedicines, 10(5), 1041. https://doi.org/10.3390/biomedicines10051041