
Autophagy Modulation in Aggresome Formation: Emerging Implications and Treatments of Alzheimer’s Disease

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
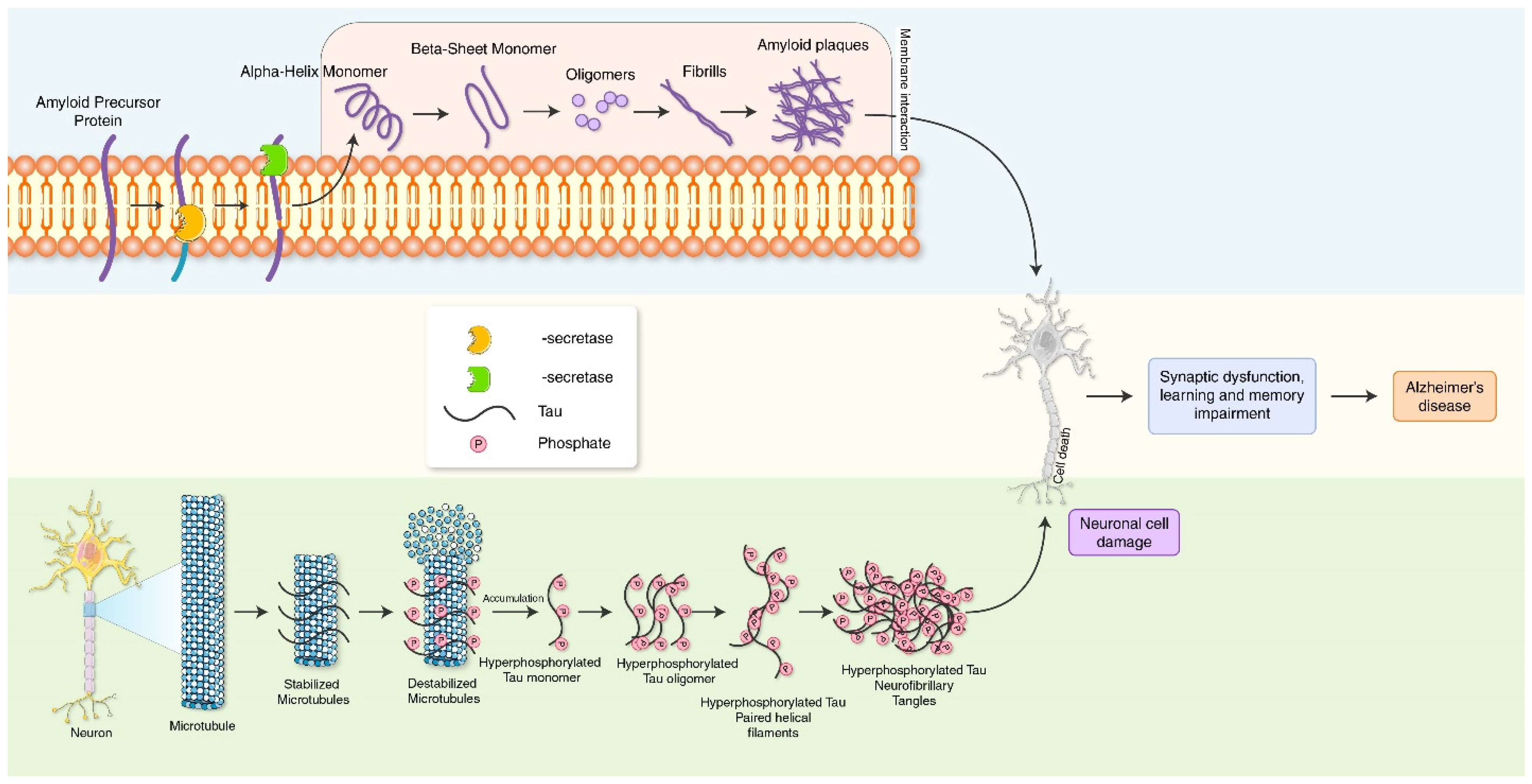
2. Molecular Mechanism of Alzheimer’s Disease
3. Molecular Pathway of Autophagy Process
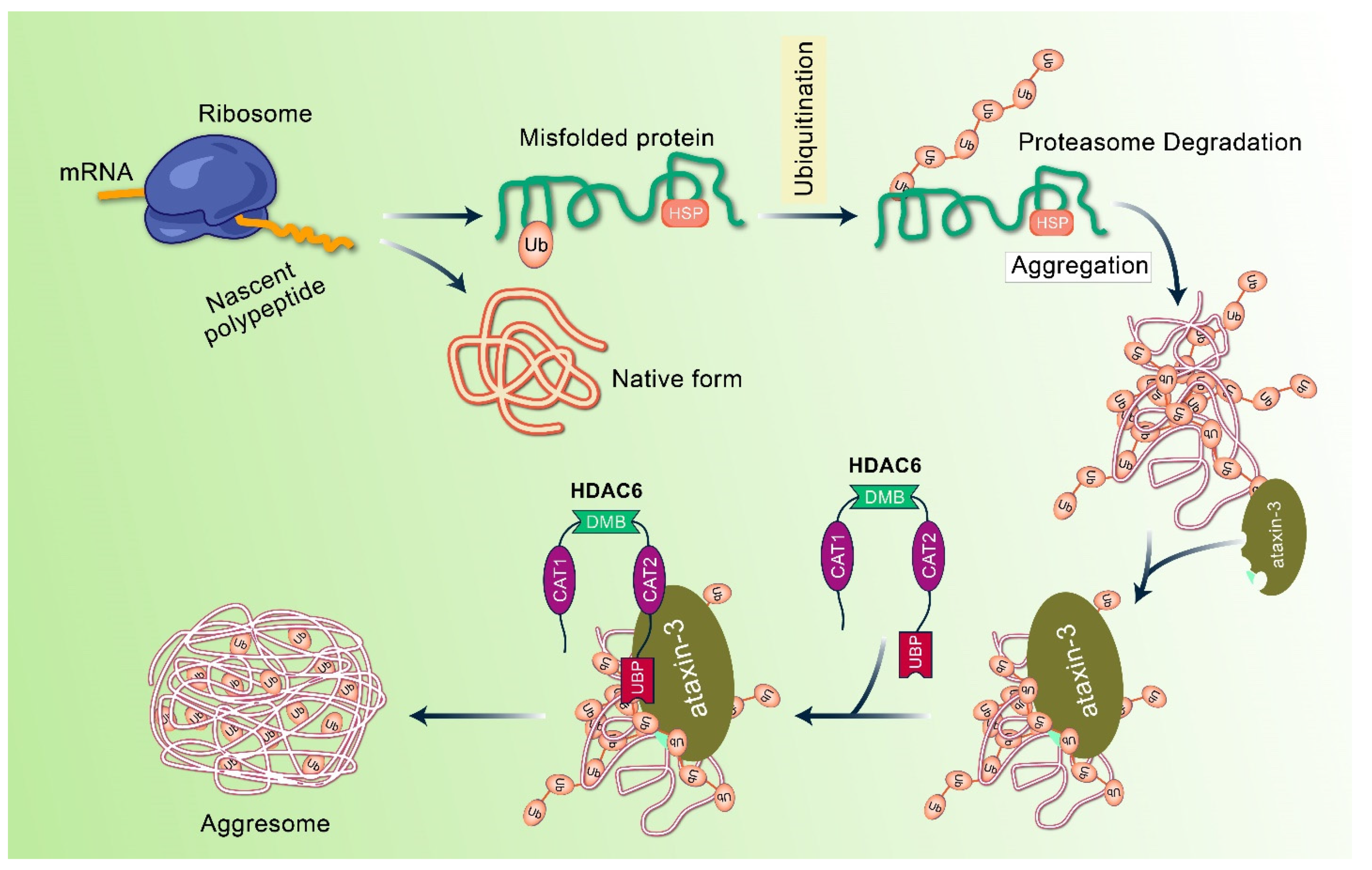
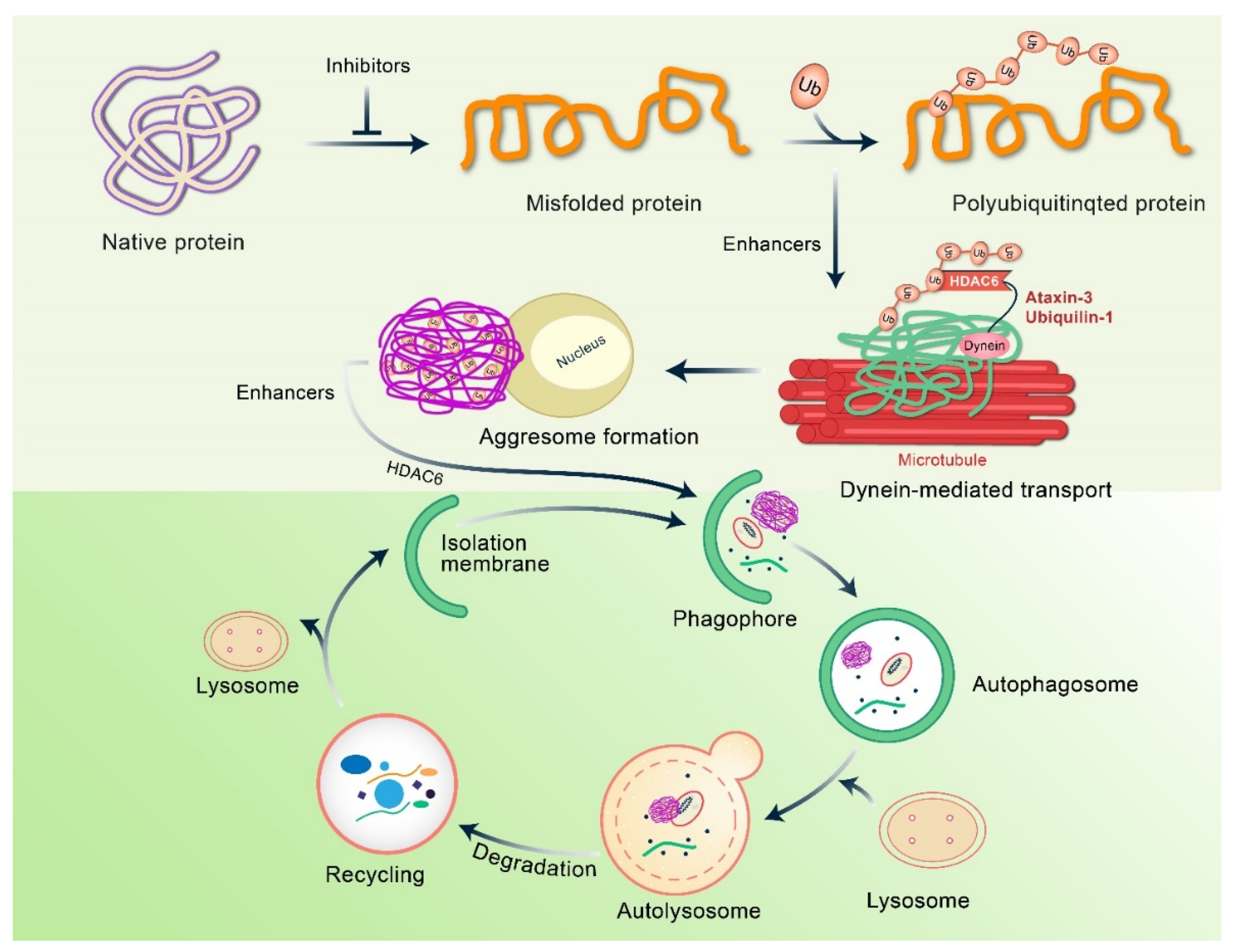
4. Mechanism of Aggresome Formation
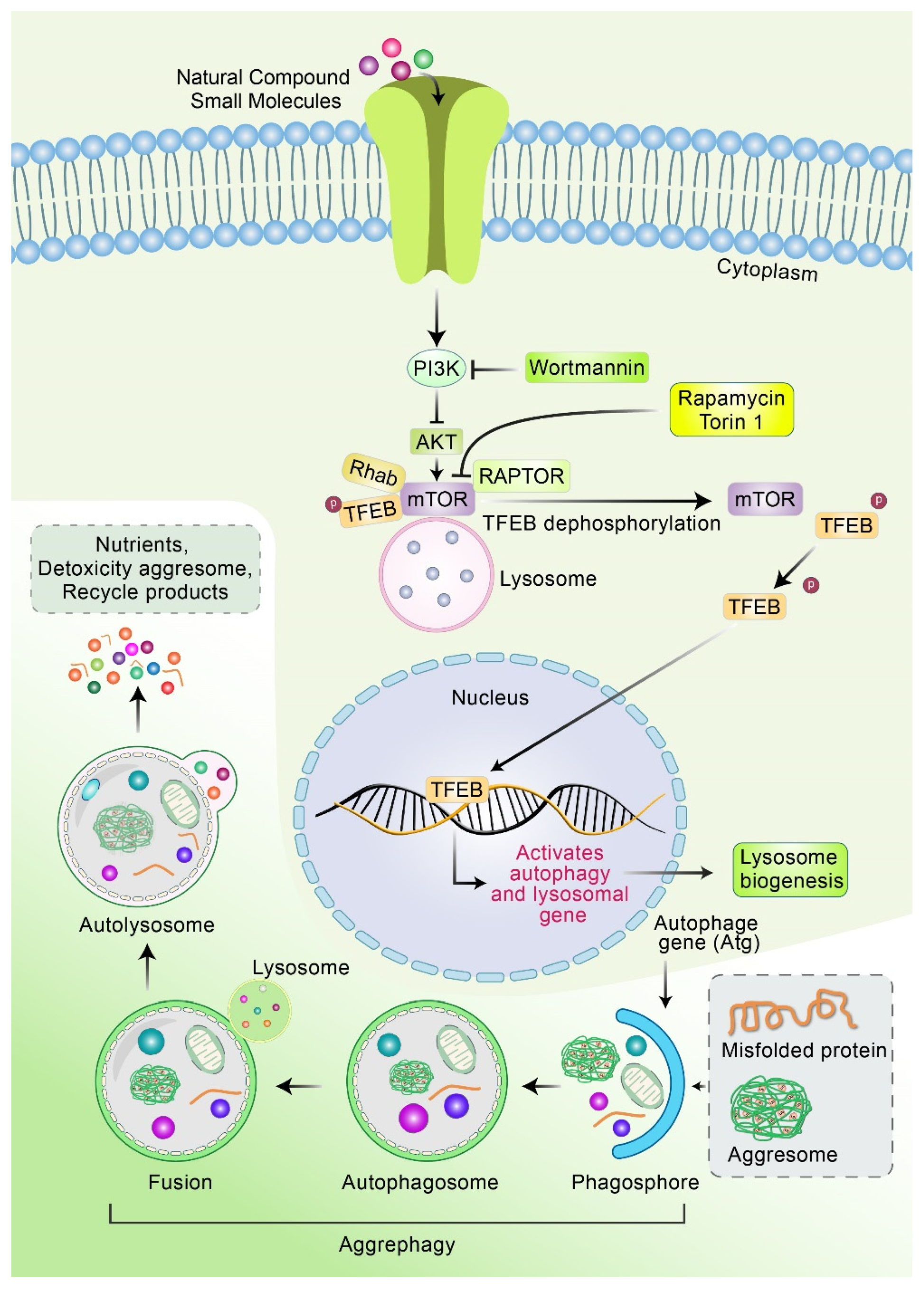
5. Clearance of Aggresomes through Autophagy
6. Molecular Mechanism of the Fusion of Aggresome and Lysosome
7. The Role of Autophagy in Modulating Aggregation in AD
8. Potential Therapeutic Action of Autophagy to Control Aggresome Formation in AD Pathogenesis
9. Future Prospective of Inhibiting Aggresome Formation as a Treatment for AD
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar] [PubMed]
- Hyttinen, J.M.T.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Malampati, S.; Song, J.X.; Tong, B.C.K.; Nalluri, A.; Yang, C.B.; Wang, Z.Y.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.F.; et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells 2020, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, M.N. Alzheimer’s Disease Drug Development Pipeline 2020. J. Prev. Alzheimer’s Dis. 2020, 7, 66–67. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes, R.S.; Delorme-Axford, E.; Klionsky, D.J.; Reggiori, F. Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae. Methods 2015, 75, 141–150. [Google Scholar] [CrossRef]
- Kraft, C.; Reggiori, F.; Peter, M. Selective types of autophagy in yeast. Biochim. Biophys. Acta 2009, 1793, 1404–1412. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Rubinsztein, D.C. Can autophagy protect against neurodegeneration caused by aggregate-prone proteins? Neuroreport 2004, 15, 2443–2445. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chin, L.S. Aggresome formation and neurodegenerative diseases: Therapeutic implications. Curr. Med. Chem. 2008, 15, 47–60. [Google Scholar] [PubMed] [Green Version]
- Gregersen, N. Protein misfolding disorders: Pathogenesis and intervention. J. Inherit. Metab. Dis. 2006, 29, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Sorgdrager, F.J.H.; Vermeiren, Y.; Van Faassen, H.J.R.; Van Der Ley, C.P.; Nollen, E.A.A.; Kema, I.P.; De Deyn, P.P. Age- and Disease-Specific Changes of the Kynurenine Pathway in Parkinson’s and Alzheimer’s Disease. J. Neurochem. 2019, 151, 656–668. [Google Scholar] [CrossRef] [Green Version]
- Castro-Chavira, S.A.; Fernandez, T.; Nicolini, H.; Diaz-Cintra, S.; Prado-Alcala, R.A. Genetic markers in biological fluids for aging-related major neurocognitive disorder. Curr. Alzheimer Res. 2015, 12, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Rhim, H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Moya-Alvarado, G.; Gershoni-Emek, N.; Perlson, E.; Bronfman, F.C. Neurodegeneration and Alzheimer’s disease (AD). What Can Proteomics Tell Us About the Alzheimer’s Brain? Mol. Cell Proteom. 2016, 15, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Rahman, M.S.; Uddin, M.J.; Mamum-Or-Rashid, A.N.M.; Pang, M.G.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. R. 2020, 27, 44659–44672. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Rahman, M.S.; Rahman, M.H.; Rasheduzzaman, M.; Mamun-Or-Rashid, A.N.M.; Uddin, M.J.; Rahman, M.R.; Hwang, H.; Pang, M.G.; Rhim, H. Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines 2021, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prevot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabaninia, M.; Tourchi, A.; Di Carlo, H.; Gearhart, J.P. Autophagy, apoptosis, and cell proliferation in exstrophy-epispadias complex. Urology 2018, 111, 157–161. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, C.; Li, R.; Zhang, L.; Tian, J. TEOA, a triterpenoid from Actinidia eriantha, induces autophagy in SW620 cells via endoplasmic reticulum stress and ROS-dependent mitophagy. Arch. Pharmacal Res. 2017, 40, 579–591. [Google Scholar] [CrossRef]
- Cordani, M.; Sánchez-Álvarez, M.; Strippoli, R.; Bazhin, A.V.; Donadelli, M. Sestrins at the interface of ROS control and autophagy regulation in health and disease. Oxidative Med. Cell. Longev. 2019, 2019, 1283075. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.-S.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Jia, J.; Zhang, X.; Dai, H. Selective binding of mitophagy receptor protein Bcl-rambo to LC3/GABARAP family proteins. Biochem. Biophys. Res. Commun. 2020, 530, 292–300. [Google Scholar] [CrossRef]
- Chmurska, A.; Matczak, K.; Marczak, A. Two Faces of Autophagy in the Struggle against Cancer. Int. J. Mol. Sci. 2021, 22, 2981. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Cho, Y.; Nam, G.; Rhim, H. Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes. Antioxidants 2021, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Hannan, M.A.; Dash, R.; Rahman, M.H.; Islam, R.; Uddin, M.J.; Sohag, A.A.; Rahman, M.H.; Rhim, H. Phytochemicals as a Complement to Cancer Chemotherapy: Pharmacological Modulation of the Autophagy-Apoptosis Pathway. Front. Pharmacol. 2021, 12, 639628. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, L.; Yan, G.; Yan, X. PFKP facilitates ATG4B phosphorylation during amino acid deprivation-induced autophagy. Cell. Signal. 2021, 82, 109956. [Google Scholar] [CrossRef]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Akter, S.; Rahman, M.A.; Hasan, M.N.; Akhter, H.; Noor, P.; Islam, R.; Shin, Y.; Rahman, M.D.H.; Gazi, M.S.; Huda, M.N.; et al. Recent Advances in Ovarian Cancer: Therapeutic Strategies, Potential Biomarkers, and Technological Improvements. Cells 2022, 11, 650. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2020, 124, 1–12. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [Green Version]
- Suresh, S.N.; Chakravorty, A.; Giridharan, M.; Garimella, L.; Manjithaya, R. Pharmacological Tools to Modulate Autophagy in Neurodegenerative Diseases. J. Mol. Bio. 2020, 432, 2822–2842. [Google Scholar] [CrossRef]
- Fortun, J.; Dunn, W.A.L., Jr.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef] [Green Version]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Markossian, K.A.; Kurganov, B.I. Protein folding, misfolding, and aggregation. Formation of inclusion bodies and aggresomes. Biochem. Mosc. 2004, 69, 971–984. [Google Scholar] [CrossRef]
- Ventura, S. Sequence determinants of protein aggregation: Tools to increase protein solubility. Microb. Cell Fact. 2005, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012, 287, 2317–2327. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.C.; Tan, M.S.; Wang, H.; Xie, A.M.; Yu, J.T.; Tan, L. Heat Shock Protein 70 in Alzheimer’s Disease. Biomed. Res. Int. 2014, 2014, 435203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Graham, K.; Foote, M.; Liang, F.S.; Rizkallah, R.; Hurt, M.; Wang, Y.C.; Wu, Y.Y.; Zhou, Y. 14-3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 2013, 126, 4173–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heir, R.; Ablasou, C.; Dumontier, E.; Elliott, M.; Fagotto-Kaufmann, C.; Bedford, F.K. The UBL domain of PLIC-1 regulates aggresome formation. EMBO Rep. 2006, 7, 1252–1258. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef]
- Ryhanen, T.; Viiri, J.; Hyttinen, J.M.T.; Uusitalo, H.; Salminen, A.; Kaarniranta, K. Influence of Hsp90 and HDAC Inhibition and Tubulin Acetylation on Perinuclear Protein Aggregation in Human Retinal Pigment Epithelial Cells. J. Biomed. Biotechnol. 2011, 2011, 798052. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Saha, S.K.; Rahman, M.S.; Uddin, M.J.; Uddin, M.S.; Pang, M.G.; Rhim, H.; Cho, S.G. Molecular Insights into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Front. Cell Dev. Biol. 2020, 8, 283. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.D.; Feldman, J.L. Microtubule-organizing centers: From the centrosome to non-centrosomal sites. Curr. Opin. Cell Biol. 2017, 44, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Rahman, M.R.; Zaman, T.; Uddin, M.S.; Islam, R.; Abdel-Daim, M.M.; Rhim, H. Emerging Potential of Naturally Occurring Autophagy Modulators Against Neurodegeneration. Curr. Pharm. Des. 2020, 26, 772–779. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hwang, H.; Nah, S.Y.; Rhim, H. Gintonin stimulates autophagic flux in primary cortical astrocytes. J. Ginseng Res. 2020, 44, 67–78. [Google Scholar] [CrossRef]
- Yim, W.W.Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, X.H.; He, S.K.; Ma, B.Y. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Rahman, M.A.; Bishayee, K.; Habib, K.; Sadra, A.; Huh, S.O. 18alpha-Glycyrrhetinic acid lethality for neuroblastoma cells via de-regulating the Beclin-1/Bcl-2 complex and inducing apoptosis. Biochem. Pharmacol. 2016, 117, 97–112. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Abdullah, A.; Rahman, M.A.; Kabir, M.T.; Alkahtani, S.; Alanazi, I.S.; Perveen, A.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Promise of Flavonoids to Combat Neuropathic Pain: From Molecular Mechanisms to Therapeutic Implications. Front. Neurosci. 2020, 14, 478. [Google Scholar] [CrossRef] [PubMed]
- Peker, N.; Gozuacik, D. Autophagy as a Cellular Stress Response Mechanism in the Nervous System. J. Mol. Biol. 2020, 432, 2560–2588. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, E.K.; Gardner, R.G. Selective destruction of abnormal proteins by ubiquitin-mediated protein quality control degradation. Semin. Cell Dev. Biol. 2012, 23, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Rahman, M.A.; Kabir, M.T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 2020, 72, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Bishayee, K.; Sadra, A.; Huh, S.O. Oxyresveratrol activates parallel apoptotic and autophagic cell death pathways in neuroblastoma cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 23–36. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Tang, Z.; Chen, D.; Moughon, D.; Ding, X.; Chen, S.; Zhu, M.; Zhong, Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010, 6, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Hwang, H.; Cho, Y.; Rhim, H. Modulation of O-GlcNAcylation Regulates Autophagy in Cortical Astrocytes. Oxid. Med. Cell Longev. 2019, 2019, 6279313. [Google Scholar] [CrossRef]
- Islam, M.A.; Sooro, M.A.; Zhang, P.H. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Ocak, U.; Gao, L.; Tu, S.; Lenahan, C.J.; Zhang, J.; Shao, A. Selective autophagy as a therapeutic target for neurological diseases. Cell. Mol. Life Sci. 2021, 78, 1369–1392. [Google Scholar] [CrossRef]
- You, Z.; Jiang, W.X.; Qin, L.Y.; Gong, Z.; Wan, W.; Li, J.; Wang, Y.; Zhang, H.; Peng, C.; Zhou, T.; et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat. Commun. 2019, 10, 5792. [Google Scholar] [CrossRef] [PubMed]
- Wu, S. Biochemical and Structural Studies of p62/SQSTM1 and the Beclin1-UVRAG Interaction in Autophagy and Endosomal Trafficking. 2016. Page XVI. 167. Available online: https://theses.lib.polyu.edu.hk/handle/200/9877.
- Tang, J.; Li, Y.; Xia, S.; Li, J.; Yang, Q.; Ding, K.; Zhang, H. Sequestosome 1/p62: A multitasker in the regulation of malignant tumor aggression. Int. J. Oncol. 2021, 59, 1–20. [Google Scholar] [CrossRef]
- Pellegrini, P.; Hervera, A.; Varea, O.; Brewer, M.K.; López-Soldado, I.; Guitart, A.; Aguilera, M.; Prats, N.; Del Río, J.A.; Guinovart, J.J. Lack of p62 impairs glycogen aggregation and exacerbates pathology in a mouse model of myoclonic epilepsy of Lafora. Mol. Neurobiol. 2022, 59, 1214–1229. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Ürmösi, A.; Jipa, A.; Kovács, L.; Deák, P.; Szabó, Á.; Juhász, G. Loss of ubiquitinated protein autophagy is compensated by persistent cnc/NFE2L2/Nrf2 antioxidant responses. Autophagy 2022, 20, 1–12. [Google Scholar] [CrossRef]
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, I.; Waster, P.; Ollinger, K. Restoration of lysosomal function after damage is accompanied by recycling of lysosomal membrane proteins. Cell Death Dis. 2020, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Song, H.K.; Orr, A.; Duan, M.T.; Merz, A.J.; Wickner, W. Sec17/Sec18 act twice, enhancing membrane fusion and then disassembling cis-SNARE complexes. Elife 2017, 6, e26646. [Google Scholar] [CrossRef]
- Steinauer, A.; LaRochelle, J.R.; Knox, S.L.; Wissner, R.F.; Berry, S.; Schepartz, A. HOPS-dependent endosomal fusion required for efficient cytosolic delivery of therapeutic peptides and small proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Nair, U.; Jotwani, A.; Geng, J.F.; Gammoh, N.; Richerson, D.; Yen, W.L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE Proteins Are Required for Macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, S.; Manjithaya, R. A reversible autophagy inhibitor blocks autophagosome-lysosome fusion by preventing Stx17 loading onto autophagosomes. Mol. Biol. Cell 2019, 30, 2283–2295. [Google Scholar] [CrossRef] [PubMed]
- Aivazidis, S.; Jain, A.; Rauniyar, A.K.; Anderson, C.C.; Marentette, J.O.; Orlicky, D.J.; Fritz, K.S.; Harris, P.S.; Siegel, D.; Maclean, K.N.; et al. SNARE proteins rescue impaired autophagic flux in Down syndrome. PLoS ONE 2019, 14, e0223254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arasaki, K.; Nagashima, H.; Kurosawa, Y.; Kimura, H.; Nishida, N.; Dohmae, N.; Yamamoto, A.; Yanagi, S.; Wakana, Y.; Inoue, H.; et al. MAP1B-LC1 prevents autophagosome formation by linking syntaxin 17 to microtubules. EMBO Rep. 2018, 19, e45584. [Google Scholar] [CrossRef] [PubMed]
- Danieli, A.; Martens, S. p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J. Cell Sci. 2018, 131, jcs214304. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2008, 1782, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-Beta Protein Clearance and Degradation (ABCD) Pathways and their Role in Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.S.; Komatsu, M. p62/SQSTM1–steering the cell through health and disease. J. Cell Sci. 2018, 131, 222836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 2017, 22, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.F.; Wooten, M.C.; Wooten, M.W. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol. Dis. 2009, 35, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef]
- Yang, Y.; Willis, T.L.; Button, R.W.; Strang, C.J.; Fu, Y.H.; Wen, X.; Grayson, P.R.C.; Evans, T.; Sipthorpe, R.J.; Roberts, S.L.; et al. Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response. Nat. Commun. 2019, 10, 3759. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.F.; Attarwala, I.Y.; Xie, X.Q. SQSTM1/p62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef]
- Babu, J.R.; Seibenhener, M.L.; Peng, J.M.; Strom, A.L.; Kemppainen, R.; Cox, N.; Zhu, H.N.; Wooten, M.C.; Diaz-Meco, M.T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448. [Google Scholar] [CrossRef] [Green Version]
- McClean, P.L.; Holscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.J.; Wan, L.P.; Wang, Y.F.; Zhang, H.; Zhang, W. Liraglutide Attenuates A beta 42 Generation in APPswe/SH-SY5Y Cells Through the Regulation of Autophagy. Neuropsych. Dis. Treat. 2020, 16, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Zheng, W.; Halloran, J.J.; Burbank, R.R.; Hussong, S.A.; Hart, M.J.; Javors, M.; Shih, Y.Y.I.; Muir, E.; Fonseca, R.S.; et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin Attenuates the Progression of Tau Pathology in P301S Tau Transgenic Mice. PLoS ONE 2013, 8, e62459. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, X.; Teng, Z.P.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Luccarini, I.; Grossi, C.; Rigacci, S.; Coppi, E.; Pugliese, A.M.; Pantano, D.; la Marca, G.; Ed Dami, T.; Berti, A.; Stefani, M.; et al. Oleuropein aglycone protects against pyroglutamylated-3 amyloid-ss toxicity: Biochemical, epigenetic and functional correlates. Neurobiol. Aging 2015, 36, 648–663. [Google Scholar] [CrossRef]
- Wu, X.L.; Kosaraju, J.; Zhou, W.; Tam, K.Y. Neuroprotective Effect of SLM, a Novel Carbazole-Based Fluorophore, on SH-SY5Y Cell Model and 3xTg-AD Mouse Model of Alzheimer’s Disease. ACS Chem. Neurosci. 2017, 8, 676–685. [Google Scholar] [CrossRef]
- Holubova, M.; Hruba, L.; Popelova, A.; Bencze, M.; Prazienkova, V.; Gengler, S.; Kratochvilova, H.; Haluzik, M.; Zelezna, B.; Kunes, J.; et al. Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of beta-amyloid pathology. Neuropharmacology 2019, 144, 377–387. [Google Scholar] [CrossRef]
- Kim, S.; Choi, K.J.; Cho, S.J.; Yun, S.M.; Jeon, J.P.; Koh, Y.H.; Song, J.; Johnson, G.V.W.; Jo, C. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Song, H.L.; Demirev, A.V.; Kim, N.Y.; Kim, D.H.; Yoon, S.Y. Ouabain activates transcription factor EB and exerts neuroprotection in models of Alzheimer’s disease. Mol. Cell Neurosci. 2019, 95, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Jana, M.; Pahan, K. Aspirin Induces Lysosomal Biogenesis and Attenuates Amyloid Plaque Pathology in a Mouse Model of Alzheimer’s Disease via PPAR alpha. J. Neurosci. 2018, 38, 6682–6699. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Roy, A.; Jana, M.; Pahan, K. Cinnamic acid activates PPARalpha to stimulate Lysosomal biogenesis and lower Amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol. Dis. 2019, 124, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Kruger, U.; Wang, Y.P.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Portbury, S.D.; Hare, D.J.; Sgambelloni, C.; Perronnes, K.; Portbury, A.J.; Finkelstein, D.I.; Adlard, P.A. Trehalose Improves Cognition in the Transgenic Tg2576 Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.Q.; Chen, L.W.; Huang, X.H.; Wang, X.; Jian, Y.L.; Tang, G.H.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Wang, H.F.; Tan, M.S.; Gao, Q.; Qin, H.; Zhang, Y.D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef]
- Lesire, L.; Chaput, L.; De Casas, P.C.; Rousseau, F.; Piveteau, C.; Dumont, J.; Pointu, D.; Deprez, B.; Leroux, F. High-Throughput Image-Based Aggresome Quantification. SLAS Discov. 2020, 25, 783–791. [Google Scholar] [CrossRef]
- Schmidt, J.; Marques, M.R.G.; Botti, S.; Marques, M.A.L. Recent advances and applications of machine learning in solid-state materials science. NPJ Comput. Mater. 2019, 5, 83. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, P.; Wang, Y.; Shi, C.; Jing, N.; Sun, H.; Yang, J.; Liu, Y.; Wen, X.; Zhang, J. Establishment of induced pluripotent stem cell line (ZZUi010-A) from an Alzheimer’s disease patient carrying an APP gene mutation. Stem Cell Res. 2017, 25, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, W.; Pan, G.; Zhou, J. Advances in understanding the cell types and approaches used for generating induced pluripotent stem cells. J. Hematol. Oncol. 2014, 7, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Guerra, E.; Oria-Muriel, M.A.; Moreno-Jiménez, E.P.; de Rojasb, I.; Rodríguez, C.; Rodríguez-Traver, E.; Orera, M.; Hernándezb, I.; Ruizb, A.; Vicario, C. Generation of an integration-free iPSC line, ICCSICi006-A, derived from a male Alzheimer’s disease patient carrying the PSEN1-G206D mutation. Stem Cell Res. 2019, 40, 101574. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, V.D.; Qiu, L.; Tan, E.K.; Zeng, L.; Zhang, Y. Modelling Alzheimer’s disease: Insights from in vivo to in vitro three-dimensional culture platforms. J. Tissue Eng. Regen. Med. 2018, 12, 1944–1958. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Rahman, M.A.; Behl, T.; Perveen, A.; Hafeez, A.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Emerging Proof of Protein Misfolding and Interaction in Multifactorial Alzheimer’s Disease. Curr. Top. Med. Chem. 2020, 20, 2380–2390. [Google Scholar] [CrossRef]
- Ding, L.; Nan, W.H.; Zhu, X.B.; Li, X.M.; Zhou, L.Y.; Chen, H.J.; Yu, L.; Khan, F.U.; Zhong, H.B.; Shi, X.J. Rapamycin and FK506 derivative TH2849 could ameliorate neurodegenerative diseases through autophagy with low immunosuppressive effect. CNS Neurosci. Ther. 2019, 25, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Zhao, B.; Li, K.S.; Zhang, L.Q.; Li, C.H.; Quazi, S.H.; Tan, Y. Mammalian target of rapamycin: A valid therapeutic target through the autophagy pathway for alzheimer’s disease? J. Neurosci. Res. 2012, 90, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.C.; Kang, R.; Sun, X.F.; Zhong, M.Z.; Huang, J.; Klionsky, D.J.; Tang, D.L. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–784. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Compounds/Small Molecules | AD Model | Molecular Mechanism | Research Outcomes | References |
|---|---|---|---|---|
| Fisetin | Mouse and rat primary cortical neurons | mTOR inhibition, TFEB and Nrf2 activation | Autophagy induction, decreases sarkosyl-insoluble tau phosphorylation | [111] |
| Ouabain | Tau transgenic fly, P301L mice | Inactivation of mTOR, activation of TFEB | Increases autophagy, decreases toxic tau, increases memory function | [112] |
| SLM, a carbazole-based fluorophore | 3xTg-AD | Activation of GSK-3β, reduces neuroinflammation | Decreases Aβ40 and Aβ42 levels, reduces phosphorylation of tau | [109] |
| Aspirin | 5xFAD | Activation of PPARα and TFEB | Increases lysosomal biogenesis, decreases Aβ | [113] |
| Liraglutide | APP/PS1, APPswe/SH-SY5Y cells | Increase in IDE levels, mTOR-independent, JNK activation | Improves cognitive function, reduces Aβ plaque deposition and inflammation, enhances LTP and autophagy activation | [102,103] |
| Rapamycin | Transgenic (h)APP mice | mTOR inactivation | Improves memory, decreases sarkosyl-insoluble tau | [104,105] |
| Cinnamic acid | 5 × FAD | Activation of PPARα, upregulation of TFEB | Reduces cerebral Aβ plaque burden, improves memory function, stimulates lysosomal biogenesis. | [114] |
| Trehalose | APP/PS1, Tg2576 | Increase in synaptophysin, doublecortin, and progranulin | Inhibits tau, improves cognitive and learning ability | [115,116] |
| Curcumin | APP/PS1 | mTOR inactivation | Reduces Aβ plaque, increases memory function | [106] |
| Oleuropein aglycone | TgCRND8 mice | Inhibition of mTOR and Ca2+ liberating | Reduces Aβ plaque, increases synaptic plasticity | [108] |
| Hep-14 | APP/PS1 | Upregulation of TFEB | Reduces Aβ plaque | [117] |
| Palm11-PrRP31 | APP/PS1 | Activation of pre-synaptic marker synaptophysin | Decreases Tau phosphorylation, reduces Aβ plaque and microgliosis | [110] |
| Methylene blue | JNPL3 | mTOR inactivation | reduces insoluble tau, increases memory function | [107] |
| Temsirolimus | P301S mice | Inhibition of mTOR | Improves motor and memory function, reduces sarkosyl-insoluble tau | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Rahman, M.H.; Mamun-Or-Rashid, A.N.M.; Hwang, H.; Chung, S.; Kim, B.; Rhim, H. Autophagy Modulation in Aggresome Formation: Emerging Implications and Treatments of Alzheimer’s Disease. Biomedicines 2022, 10, 1027. https://doi.org/10.3390/biomedicines10051027
Rahman MA, Rahman MH, Mamun-Or-Rashid ANM, Hwang H, Chung S, Kim B, Rhim H. Autophagy Modulation in Aggresome Formation: Emerging Implications and Treatments of Alzheimer’s Disease. Biomedicines. 2022; 10(5):1027. https://doi.org/10.3390/biomedicines10051027
Chicago/Turabian StyleRahman, Md. Ataur, MD. Hasanur Rahman, A. N. M. Mamun-Or-Rashid, Hongik Hwang, Sooyoung Chung, Bonglee Kim, and Hyewhon Rhim. 2022. "Autophagy Modulation in Aggresome Formation: Emerging Implications and Treatments of Alzheimer’s Disease" Biomedicines 10, no. 5: 1027. https://doi.org/10.3390/biomedicines10051027
APA StyleRahman, M. A., Rahman, M. H., Mamun-Or-Rashid, A. N. M., Hwang, H., Chung, S., Kim, B., & Rhim, H. (2022). Autophagy Modulation in Aggresome Formation: Emerging Implications and Treatments of Alzheimer’s Disease. Biomedicines, 10(5), 1027. https://doi.org/10.3390/biomedicines10051027