
Potential Therapeutic Targets and Promising Agents for Combating NAFLD


 , , , and
, , , and
Abstract
:1. Introduction
2. Factors Associated with NAFLD Pathophysiology
3. Pathophysiology of NAFLD
4. Drugs
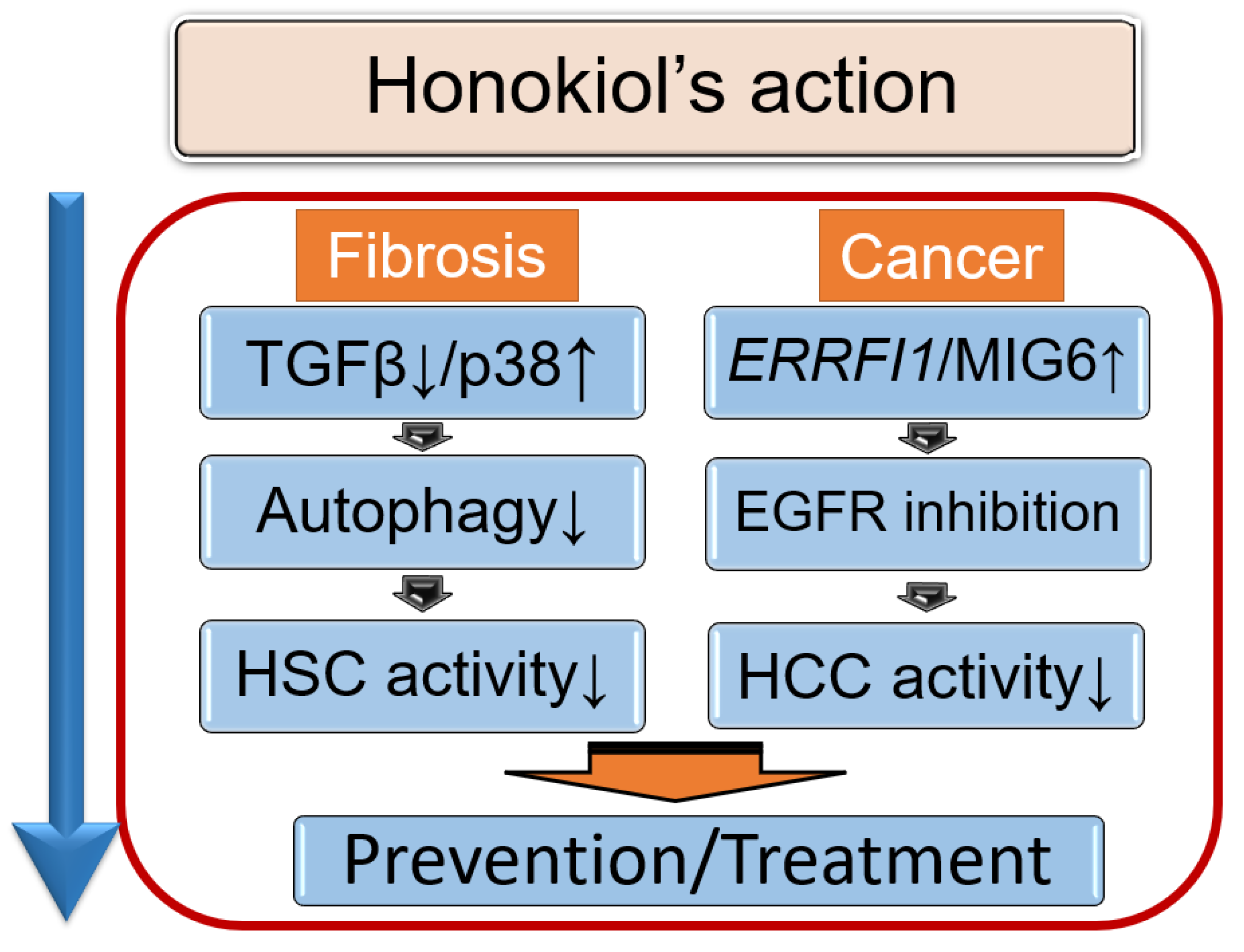
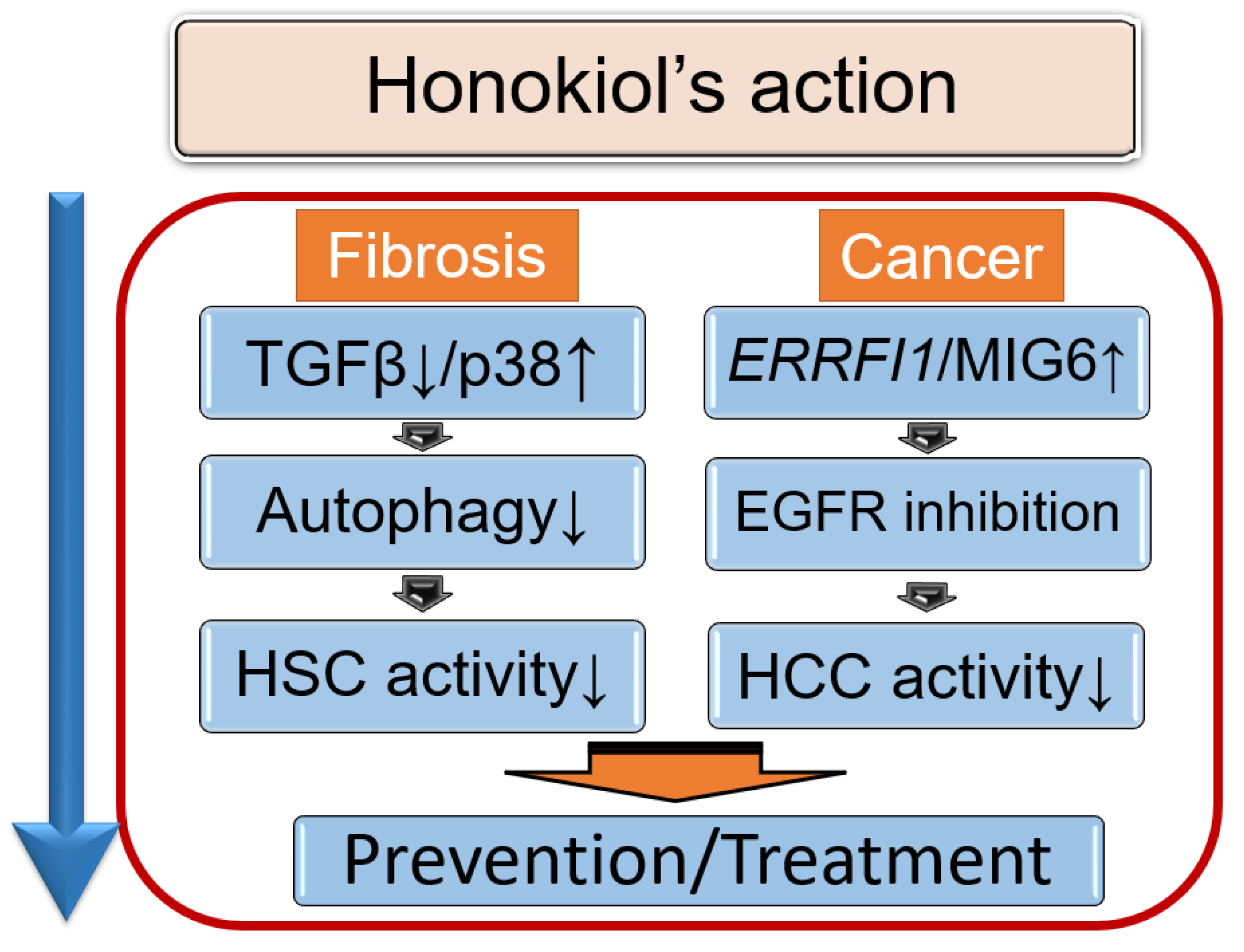
5. Therapeutic Potential of Honokiol in NAFLD
5.1. Honokiol, an Anti-Fibrotic Agent That Modulates Autophagy
5.2. Honokiol and Its Anti-Tumor Effects
6. Future Therapeutic Drugs in Development
6.1. Pharmacological Agents in Clinical Trials

{kind=link}
| Pharmacological Agent | Target | Trial Phase | |
|---|---|---|---|
| 1 | Obeticholic acid | FXR agonist | 3 |
| 2 | Semaglutide | GLP-1 receptor agonist | 3 |
| 3 | Resmetirom (MGL-3196) | THR-β agonist | 3 |
| 4 | Aramchol | SCD1 inhibitor | 3 |
| 5 | Lanifbranor | PPARα/δ/γ agonist | 3 |
| 6 | Belapectin (GR-MD-02) | Galectin-3 inhibitor | 3 |
| 7 | Pegbelfermin (BMS-986036) | FGF21 analogue | 3 |
| 8 | MK-3655 | Monoclonal antibody agonist of the b-Klotho/FGFR1c receptor complex | 2 |
| 9 | TVB-2640 | FASN inhibitor | 2 |
| 10 | Tirzepatide | Dual GIP and GLP-1 receptor agonist | 2 |
| 11 | BI456906 | Dual GIP and GLP-1 receptor agonist | 2 |
| 12 | BMS-986263 | HSP47 siRNA | 2 |
| 13 | CC-90001 | JNK inhibitor | 2 |
6.2. Drugs with Suspended or Discontinued Development
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Stepanova, M.; Ong, J.; Trimble, G.; AlQahtani, S.; Younossi, I.; Ahmed, A.; Racila, A.; Henry, L. Nonalcoholic Steatohepatitis Is the Most Rapidly Increasing Indication for Liver Transplantation in the United States. Clin. Gastroenterol. Hepatol. 2021, 19, 580–589.e585. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Van Natta, M.L.; Clark, J.; Neuschwander-Tetri, B.A.; Diehl, A.; Dasarathy, S.; Loomba, R.; Chalasani, N.; Kowdley, K.; Hameed, B.; et al. Prospective Study of Outcomes in Adults with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2021, 385, 1559–1569. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Koda, M.; Kawakami, M.; Murawaki, Y.; Senda, M. The impact of visceral fat in nonalcoholic fatty liver disease: Cross-sectional and longitudinal studies. J. Gastroenterol. 2007, 42, 897–903. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Raff, E.J.; Kakati, D.; Bloomer, J.R.; Shoreibah, M.; Rasheed, K.; Singal, A.K. Diabetes Mellitus Predicts Occurrence of Cirrhosis and Hepatocellular Cancer in Alcoholic Liver and Non-alcoholic Fatty Liver Diseases. J. Clin. Transl. Hepatol. 2015, 3, 9–16. [Google Scholar] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2019, 30, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Sumida, Y.; Umemura, A.; Matsuo, K.; Takahashi, M.; Takamura, T.; Yasui, K.; Saibara, T.; Hashimoto, E.; Kawanaka, M.; et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS ONE 2012, 7, e38322. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Shima, T.; Mizuno, M.; Mitsumoto, Y.; Umemura, A.; Kanbara, Y.; Tanaka, S.; Sumida, Y.; Yasui, K.; Takahashi, M.; et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE 2018, 13, e0185490. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Castano, G.O.; Scian, R.; Mallardi, P.; Fernandez Gianotti, T.; Burgueno, A.L.; San Martino, J.; Pirola, C.J. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Yamaguchi, K.; Tochiki, N.; Yano, K.; Takahashi, A.; Okishio, S.; Kataoka, S.; Okuda, K.; Umemura, A.; Moriguchi, M.; et al. Attenuated effect of PNPLA3 on hepatic fibrosis by HSD17B13 in Japanese patients with non-alcoholic fatty liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Yamaguchi, K.; Mizuno, N.; Okuda, K.; Takemura, M.; Taketani, H.; Hara, T.; Umemura, A.; Nishikawa, T.; Moriguchi, M.; et al. Combination of PNPLA3 and TLL1 polymorphism can predict advanced fibrosis in Japanese patients with nonalcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e1216. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.L.; Mohamed, R.; Mohamed, Z.; Zain, S.M. Phosphatidylethanolamine N-methyltransferase gene rs7946 polymorphism plays a role in risk of nonalcoholic fatty liver disease: Evidence from meta-analysis. Pharmacogenet. Genom. 2016, 26, 88–95. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Wang, S.; Friedman, S.L. Hepatic fibrosis: A convergent response to liver injury that is reversible. J. Hepatol. 2020, 73, 210–211. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar]
- Mehal, W.Z.; Schuppan, D. Antifibrotic therapies in the liver. Semin. Liver Dis. 2015, 35, 184–198. [Google Scholar]
- Amir, M.; Czaja, M.J. Autophagy in nonalcoholic steatohepatitis. Exp. Rev. Gastroenterol. Hepatol. 2011, 5, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature. 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Buse, J.B.; Taylor, K.; Kendall, D.M.; Trautmann, M.; Zhuang, D.; Porter, L.; Group, D.-S. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: A randomised, open-label, non-inferiority study. Lancet 2008, 372, 1240–1250. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Team, L.t.; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; Investigators, N.N. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Cui, J.; Philo, L.; Nguyen, P.; Hofflich, H.; Hernandez, C.; Bettencourt, R.; Richards, L.; Salotti, J.; Bhatt, A.; Hooker, J.; et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2016, 65, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Gosho, M.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Nakade, Y.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Vitamin E has a beneficial effect on nonalcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Nutrition 2015, 31, 923–930. [Google Scholar] [CrossRef]
- Li, B.; Wang, Y.; Ye, Z.; Yang, H.; Cui, X.; Wang, Z.; Liu, L. Effects of Canagliflozin on Fatty Liver Indexes in Patients with Type 2 Diabetes: A Meta-analysis of Randomized Controlled Trials. J. Pharm. Pharm. Sci. 2018, 21, 222–235. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S90–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e1515. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Ciampalini, P.; Alisi, A.; Devito, R.; Bugianesi, E.; Marcellini, M.; Marchesini, G. Metformin use in children with nonalcoholic fatty liver disease: An open-label, 24-month, observational pilot study. Clin. Ther. 2008, 30, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulten, H.J. Pleiotropic Effects of Metformin on Cancer. Int. J. Mol. Sci. 2018, 19, 2850. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- Loomba, R.; Sirlin, C.B.; Ang, B.; Bettencourt, R.; Jain, R.; Salotti, J.; Soaft, L.; Hooker, J.; Kono, Y.; Bhatt, A.; et al. Ezetimibe for the treatment of nonalcoholic steatohepatitis: Assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (MOZART trial). Hepatology 2015, 61, 1239–1250. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Chen, Y.P.; Ma, K.F.; Ye, Y.F.; Zheng, L.; Yang, Y.D.; Li, Y.M.; Jin, X. The role of ursodeoxycholic acid in non-alcoholic steatohepatitis: A systematic review. BMC Gastroenterol. 2013, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; de Ledinghen, V.; Oberti, F.; Mathurin, P.; Wartelle-Bladou, C.; Renou, C.; Sogni, P.; Maynard, M.; Larrey, D.; Serfaty, L.; et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J. Hepatol. 2011, 54, 1011–1019. [Google Scholar] [CrossRef]
- Torres, D.M.; Jones, F.J.; Shaw, J.C.; Williams, C.D.; Ward, J.A.; Harrison, S.A. Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: A 12-month randomized, prospective, open- label trial. Hepatology 2011, 54, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Kabir, J.; Mustafa, G.; Gupta, U.; Hasan, S.K.; Alam, A.K. Effect of telmisartan on histological activity and fibrosis of non-alcoholic steatohepatitis: A 1-year randomized control trial. Saudi J. Gastroenterol. 2016, 22, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Gianotti, T.F.; Rosselli, M.S.; Burgueno, A.L.; Castano, G.O.; Pirola, C.J. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis 2011, 218, 378–385. [Google Scholar] [CrossRef]
- Ong, C.P.; Lee, W.L.; Tang, Y.Q.; Yap, W.H. Honokiol: A Review of Its Anticancer Potential and Mechanisms. Cancers 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Cao, A.H.; Vo, L.T.; King, R.G. Honokiol protects against carbon tetrachloride induced liver damage in the rat. Phytother. Res. 2005, 19, 932–937. [Google Scholar] [CrossRef]
- Elfeky, M.G.; Mantawy, E.M.; Gad, A.M.; Fawzy, H.M.; El-Demerdash, E. Mechanistic aspects of antifibrotic effects of honokiol in Con A-induced liver fibrosis in rats: Emphasis on TGF-beta/SMAD/MAPK signaling pathways. Life Sci. 2020, 240, 117096. [Google Scholar] [CrossRef]
- Chiang, C.K.; Sheu, M.L.; Lin, Y.W.; Wu, C.T.; Yang, C.C.; Chen, M.W.; Hung, K.Y.; Wu, K.D.; Liu, S.H. Honokiol ameliorates renal fibrosis by inhibiting extracellular matrix and pro-inflammatory factors in vivo and in vitro. Br. J. Pharmacol. 2011, 163, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.C.; Schwabe, R.F. Autophagy and hepatic stellate cell activation—Partners in crime? J. Hepatol. 2011, 55, 1176–1177. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, S.; Umemura, A.; Okuda, K.; Taketani, H.; Seko, Y.; Nishikawa, T.; Yamaguchi, K.; Moriguchi, M.; Kanbara, Y.; Arbiser, J.L.; et al. Honokiol Acts as a Potent Anti-Fibrotic Agent in the Liver through Inhibition of TGF-beta1/SMAD Signaling and Autophagy in Hepatic Stellate Cells. Int. J. Mol. Sci. 2021, 22, 13354. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Singh, S.; Piazza, G.A.; Contreras, C.M.; Panyam, J.; Singh, A.P. Honokiol: A novel natural agent for cancer prevention and therapy. Curr. Mol. Med. 2012, 12, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Henry, L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. 2021, 3, 100305. [Google Scholar] [CrossRef] [PubMed]
- Tokushige, K.; Hyogo, H.; Nakajima, T.; Ono, M.; Kawaguchi, T.; Honda, K.; Eguchi, Y.; Nozaki, Y.; Kawanaka, M.; Tanaka, S.; et al. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease and alcoholic liver disease: Multicenter survey. J. Gastroenterol. 2016, 51, 586–596. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e1342. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e1822. [Google Scholar] [CrossRef] [Green Version]
- Komposch, K.; Sibilia, M. EGFR Signaling in Liver Diseases. Int. J. Mol. Sci. 2015, 17, 30. [Google Scholar] [CrossRef]
- Natarajan, A.; Wagner, B.; Sibilia, M. The EGF receptor is required for efficient liver regeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 17081–17086. [Google Scholar] [CrossRef] [Green Version]
- Paranjpe, S.; Bowen, W.C.; Mars, W.M.; Orr, A.; Haynes, M.M.; DeFrances, M.C.; Liu, S.; Tseng, G.C.; Tsagianni, A.; Michalopoulos, G.K. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016, 64, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Bhushan, B.; Banerjee, S.; Paranjpe, S.; Koral, K.; Mars, W.M.; Stoops, J.W.; Orr, A.; Bowen, W.C.; Locker, J.; Michalopoulos, G.K. Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Non-alcoholic Fatty Liver Disease in Murine Fast-food Diet Model. Hepatology 2019, 70, 1546–1563. [Google Scholar] [CrossRef] [PubMed]
- Choung, S.; Kim, J.M.; Joung, K.H.; Lee, E.S.; Kim, H.J.; Ku, B.J. Epidermal growth factor receptor inhibition attenuates non-alcoholic fatty liver disease in diet-induced obese mice. PLoS ONE 2019, 14, e0210828. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Umemura, A.; Umemura, S.; Kataoka, S.; Taketani, H.; Seko, Y.; Nishikawa, T.; Yamaguchi, K.; Moriguchi, M.; Kanbara, Y.; et al. Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor-MIG6 Axis. Cancers 2021, 13, 1515. [Google Scholar] [CrossRef] [PubMed]
- Ferby, I.; Reschke, M.; Kudlacek, O.; Knyazev, P.; Pante, G.; Amann, K.; Sommergruber, W.; Kraut, N.; Ullrich, A.; Fassler, R.; et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006, 12, 568–573. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Staal, B.; Su, Y.; Swiatek, P.; Zhao, P.; Cao, B.; Resau, J.; Sigler, R.; Bronson, R.; Vande Woude, G.F. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene 2007, 26, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Amatschek, S.; Koenig, U.; Auer, H.; Steinlein, P.; Pacher, M.; Gruenfelder, A.; Dekan, G.; Vogl, S.; Kubista, E.; Heider, K.H.; et al. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res. 2004, 64, 844–856. [Google Scholar] [CrossRef] [Green Version]
- Reschke, M.; Ferby, I.; Stepniak, E.; Seitzer, N.; Horst, D.; Wagner, E.F.; Ullrich, A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology 2010, 51, 1383–1390. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), versus placebo in patients with non-alcoholic fatty liver disease. Aliment Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Idowu, M.O.; Parmar, D.; Borg, B.B.; Denham, D.; Loo, N.M.; Lazas, D.; Younes, Z.; Sanyal, A.J. A Phase 2 Double Blinded, Randomized Controlled Trial of Saroglitazar in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2021, 19, 2670–2672. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Pablo Frias, J.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients With NASH. Hepatol. Commun. 2021, 5, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 2021, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Charles, E.D.; Sanyal, A.J.; Harrison, S.A.; Neuschwander-Tetri, B.A.; Goodman, Z.; Ehman, R.A.; Karsdal, M.; Nakajima, A.; Du, S.; et al. The FALCON program: Two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemp. Clin Trials. 2021, 104, 106335. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A. Fatty liver disease and liver cancer. J. Kyoto Pref. Univ. Med. 2022, 131, 209–218. [Google Scholar]
- Lawitz, E.J.; Shevell, D.E.; Tirucherai, G.S.; Du, S.; Chen, W.; Kavita, U.; Coste, A.; Poordad, F.; Karsdal, M.; Nielsen, M.; et al. BMS-986263 in patients with advanced hepatic fibrosis: 36-week results from a randomized, placebo-controlled phase 2 trial. Hepatology 2021, 75, 912–923. [Google Scholar] [CrossRef]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e1335. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Goodman, Z.; Jabbar, A.; Vemulapalli, R.; Younes, Z.H.; Freilich, B.; Sheikh, M.Y.; Schattenberg, J.M.; Kayali, Z.; Zivony, A.; et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 2020, 72, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Okanoue, T.; Sakamoto, M.; Harada, K.; Inagaki, M.; Totsuka, N.; Hashimoto, G.; Kumada, H. Efficacy and safety of apararenone (MT-3995) in patients with nonalcoholic steatohepatitis: A randomized controlled study. Hepatol. Res. Off. J. Japan Soc. Hepatol. 2021, 51, 943–956. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e211. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Aguilar Schall, R.; Jia, C.; McColgan, B.; et al. The Natural History of Advanced Fibrosis Due to Nonalcoholic Steatohepatitis: Data From the Simtuzumab Trials. Hepatology 2019, 70, 1913–1927. [Google Scholar] [CrossRef]
| Pharmacological Agent | Target | Trial Phase | |
|---|---|---|---|
| 1 | Elafibranor | PPARα/δ agonist | 3 |
| 2 | Cenicriviroc | CCR2/5 antagonist | 3 |
| 3 | Selonsertib | ASK1 inhibitor | 3 |
| 4 | Seladelpar (MBX-8025) | PPARδ agonist | 2 |
| 5 | Emricasan | Caspase inhibitor | 2 |
| 6 | Apararenone (MT-3995) | Non-steroidal MR antagonist | 2 |
| 7 | Aldafermin (NGM-282) | FGF19 analogue | 2 |
| 8 | Simtuzumab | Anti-LOXL2 antibody | 2 |
| Reasons for discontinuation | |||
| 1 Elafibranor failed to reach the primary endpoint in a phase 3 trial. | |||
| 2 A phase 3 trial of cenicriviroc was terminated early due to lack of efficacy. | |||
| 3 Selonsertib failed to reach the primary endpoint in a phase 3 trial. | |||
| 4 Seladelpar showed signs of significant adverse effects in a phase 2 trial. | |||
| 5 Emricasan failed to reach the primary endpoints in a phase 2 trial. | |||
| 6 There are no plans for a phase 3 trial of apararenone at this time. | |||
| 7 Aldafermin (NGM282) failed to reach the primary endpoint in a phase 2 trial. | |||
| 8 Simtuzumab failed to reach the primary endpoints in a phase 2 trial. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umemura, A.; Kataoka, S.; Okuda, K.; Seko, Y.; Yamaguchi, K.; Moriguchi, M.; Okanoue, T.; Itoh, Y. Potential Therapeutic Targets and Promising Agents for Combating NAFLD. Biomedicines 2022, 10, 901. https://doi.org/10.3390/biomedicines10040901
Umemura A, Kataoka S, Okuda K, Seko Y, Yamaguchi K, Moriguchi M, Okanoue T, Itoh Y. Potential Therapeutic Targets and Promising Agents for Combating NAFLD. Biomedicines. 2022; 10(4):901. https://doi.org/10.3390/biomedicines10040901
Chicago/Turabian StyleUmemura, Atsushi, Seita Kataoka, Keiichiro Okuda, Yuya Seko, Kanji Yamaguchi, Michihisa Moriguchi, Takeshi Okanoue, and Yoshito Itoh. 2022. "Potential Therapeutic Targets and Promising Agents for Combating NAFLD" Biomedicines 10, no. 4: 901. https://doi.org/10.3390/biomedicines10040901
APA StyleUmemura, A., Kataoka, S., Okuda, K., Seko, Y., Yamaguchi, K., Moriguchi, M., Okanoue, T., & Itoh, Y. (2022). Potential Therapeutic Targets and Promising Agents for Combating NAFLD. Biomedicines, 10(4), 901. https://doi.org/10.3390/biomedicines10040901