
Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
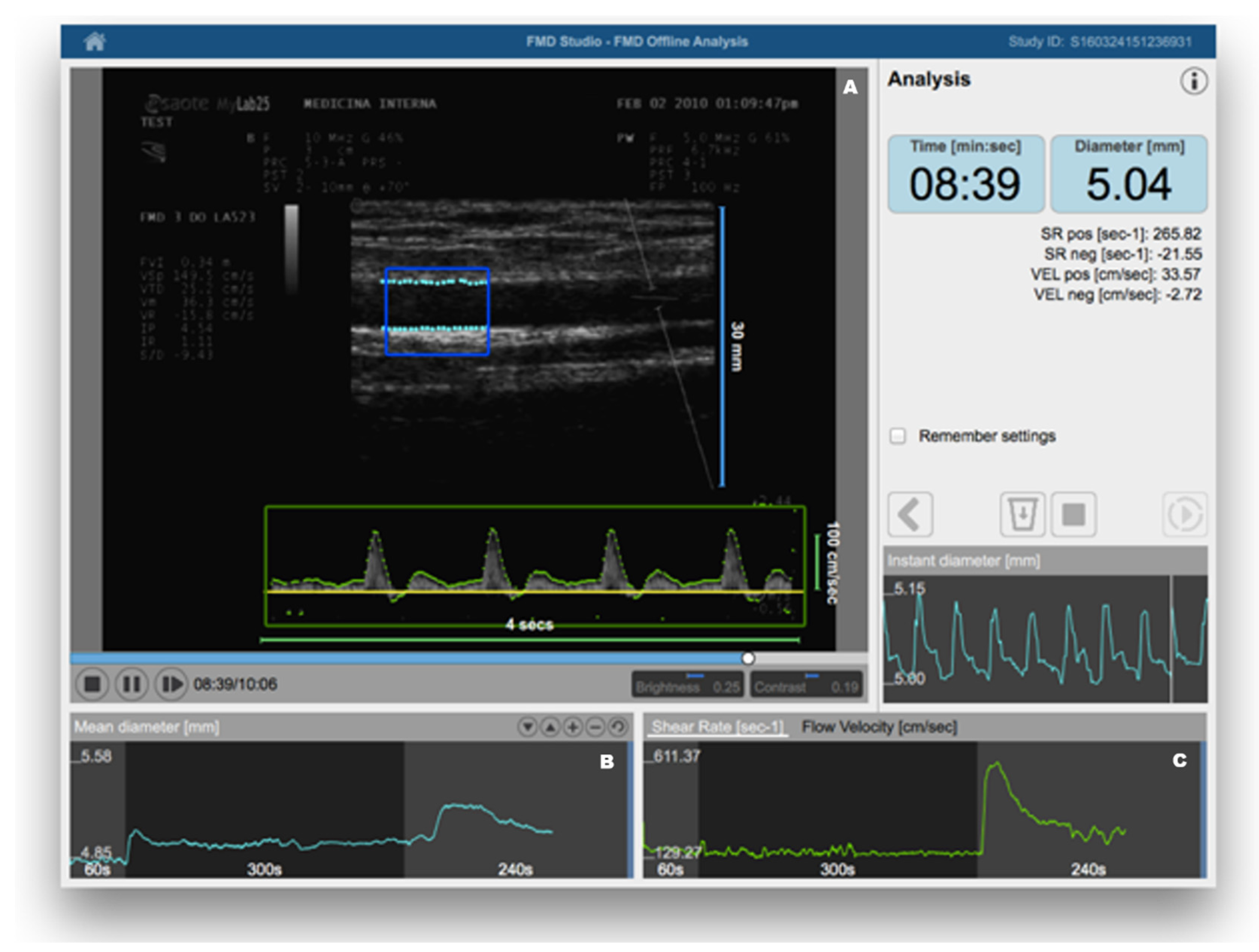
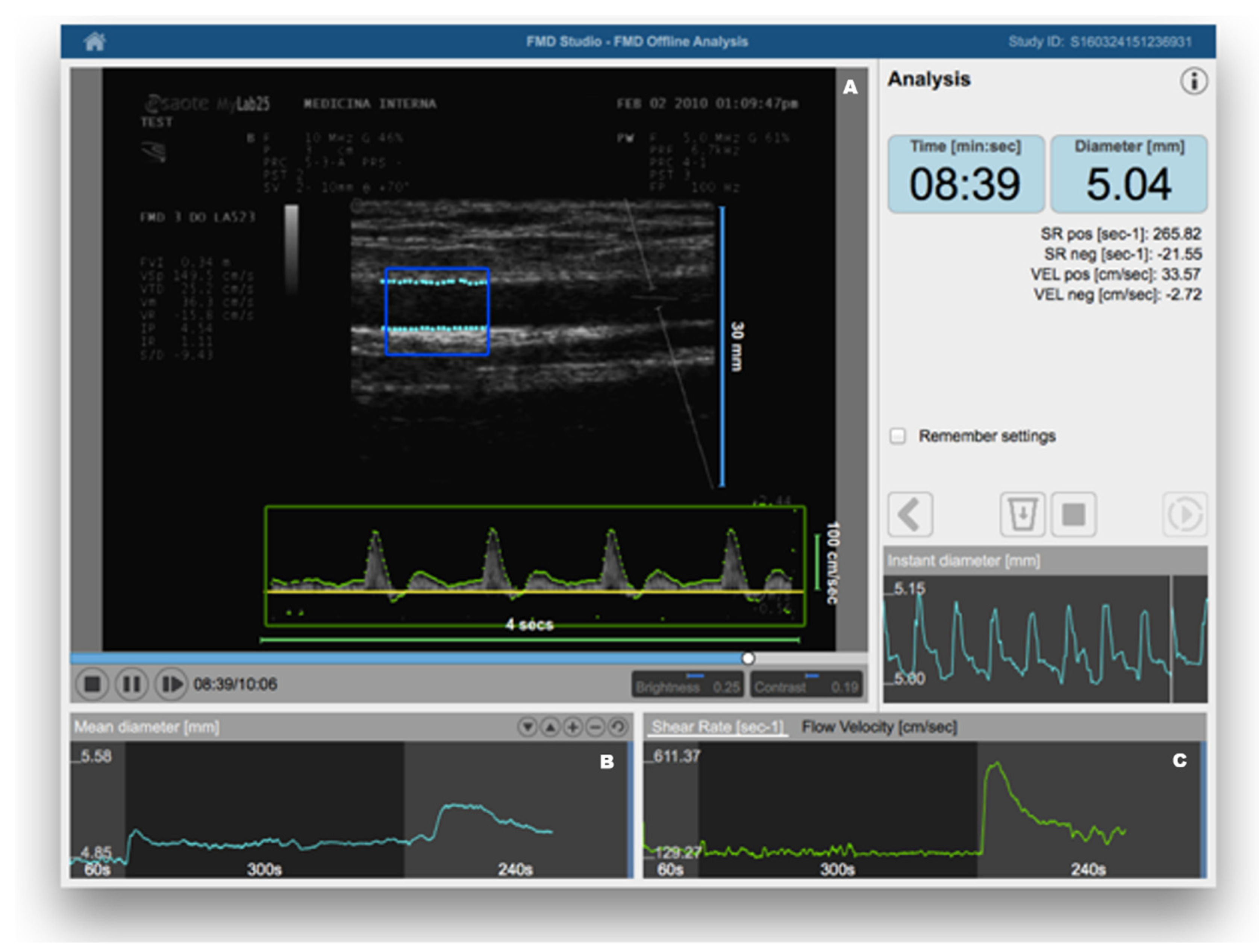{kind=link}
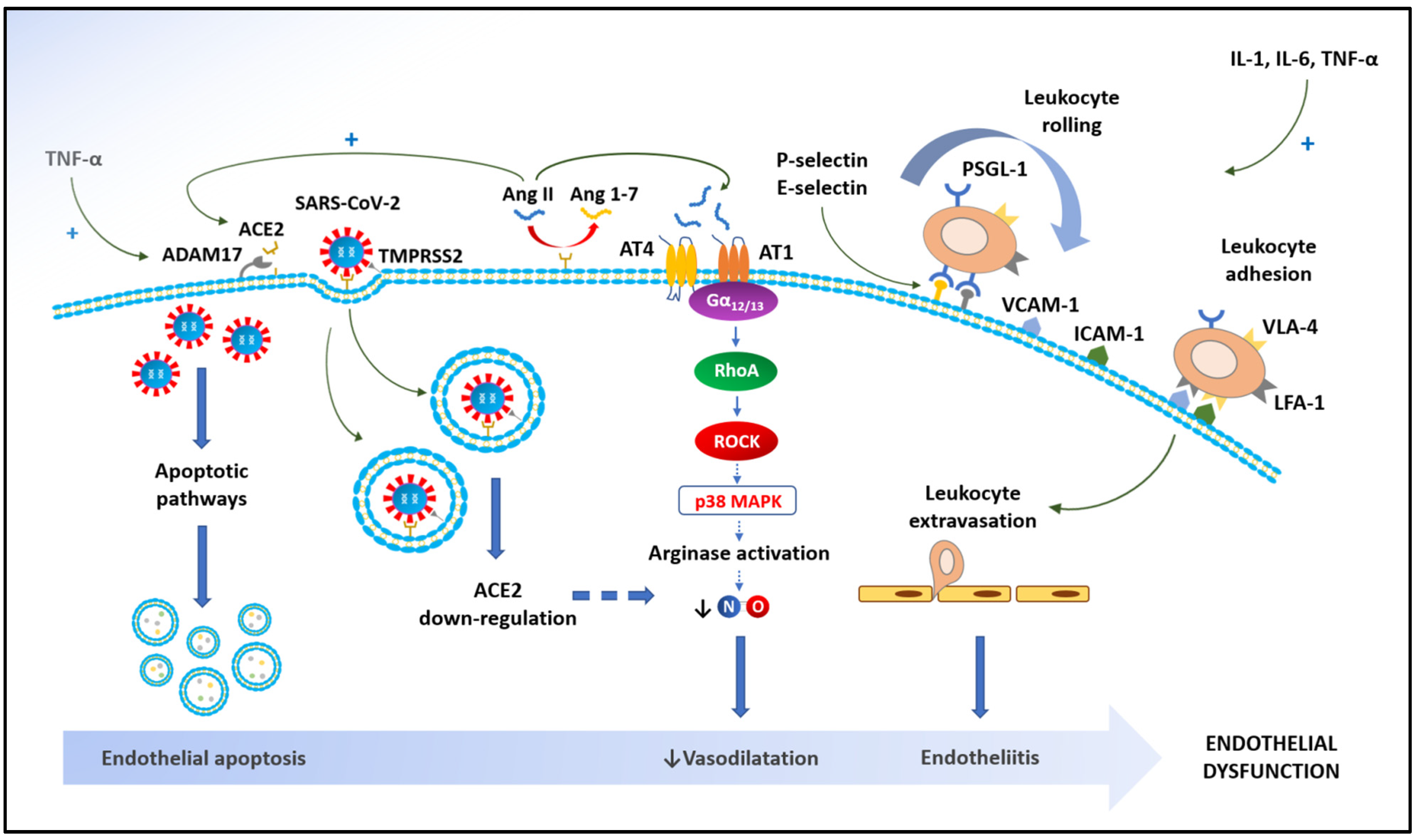
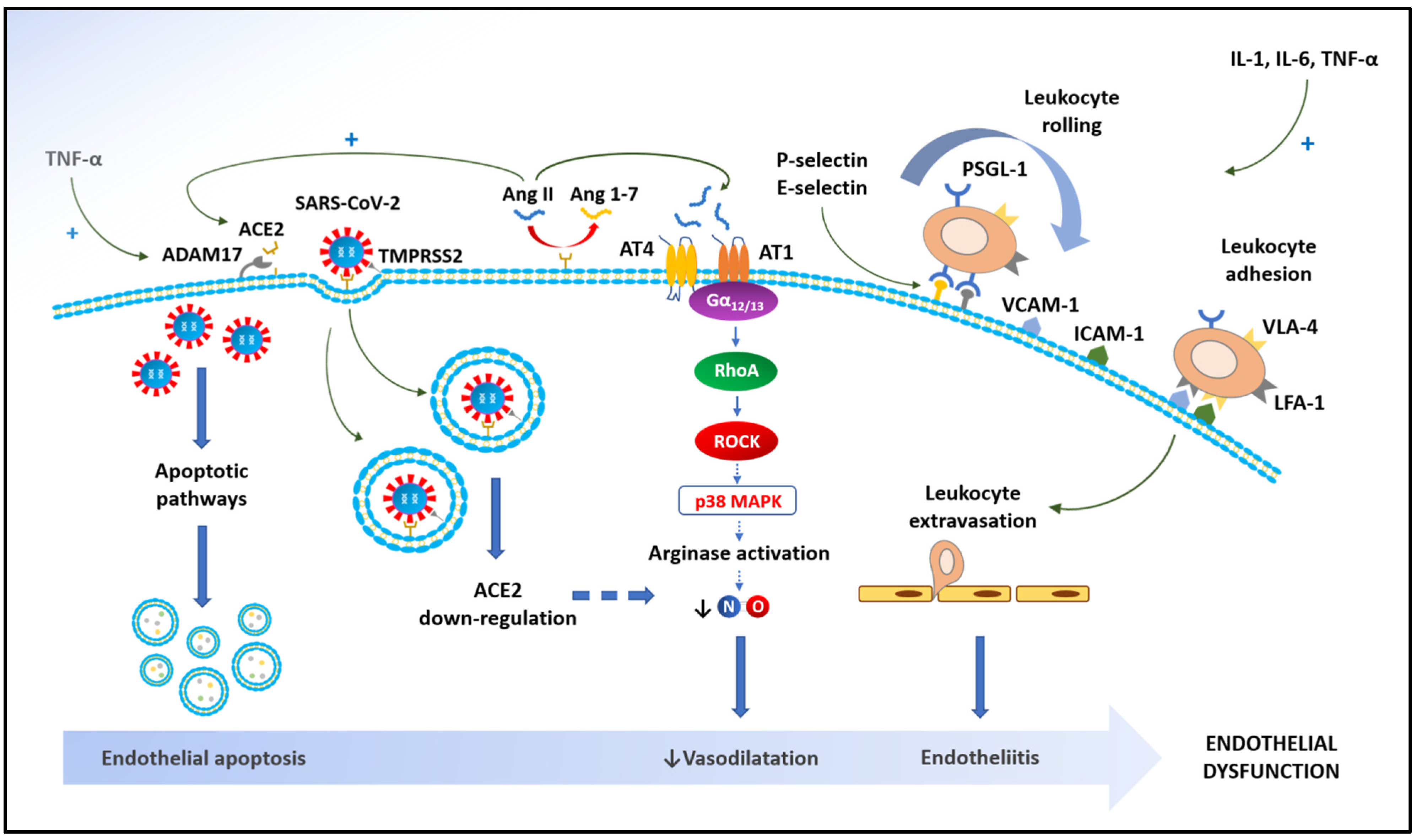{kind=link}
Abstract
:1. Introduction
2. Endothelial Cell Homeostasis
3. Endothelial Function Assessment
3.1. Clinical Methods
3.2. Laboratory Methods
4. Evidence of Endothelial Dysfunction in COVID-19
5. Pathophysiology of Endothelial Dysfunction in COVID-19
5.1. Direct Viral Action
5.2. Indirect Viral Action
5.3. Potential Inflammatory Mechanisms from Other Respiratory Diseases
6. Targeting Endothelial Dysfunction in COVID-19
6.1. RAS Inhibitors
6.2. Statins
6.3. Antioxidants and Other Pharmacological Strategies
6.4. Rehabilitation and Exercise-Based Approaches
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosino, P.; Papa, A.; Maniscalco, M.; Di Minno, M.N.D. COVID-19 and functional disability: Current insights and rehabilitation strategies. Postgrad. Med. J. 2021, 97, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.C.; Rainger, G.E.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef]
- Fodor, A.; Tiperciuc, B.; Login, C.; Orasan, O.H.; Lazar, A.L.; Buchman, C.; Hanghicel, P.; Sitar-Taut, A.; Suharoschi, R.; Vulturar, R.; et al. Endothelial Dysfunction, Inflammation, and Oxidative Stress in COVID-19-Mechanisms and Therapeutic Targets. Oxid. Med. Cell. Longev. 2021, 2021, 8671713. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ambrosino, P.; Calcaterra, I.; Molino, A.; Moretta, P.; Lupoli, R.; Spedicato, G.A.; Papa, A.; Motta, A.; Maniscalco, M.; Di Minno, M.N.D. Persistent Endothelial Dysfunction in Post-Acute COVID-19 Syndrome: A Case-Control Study. Biomedicines 2021, 9, 957. [Google Scholar] [CrossRef]
- Cuker, A.; Tseng, E.K.; Nieuwlaat, R.; Angchaisuksiri, P.; Blair, C.; Dane, K.; Davila, J.; DeSancho, M.T.; Diuguid, D.; Griffin, D.O.; et al. American Society of Hematology 2021 guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19. Blood Adv. 2021, 5, 872–888. [Google Scholar] [CrossRef]
- Di Minno, A.; Ambrosino, P.; Calcaterra, I.; Di Minno, M.N.D. COVID-19 and Venous Thromboembolism: A Meta-analysis of Literature Studies. Semin. Thromb. Hemost. 2020, 46, 763–771. [Google Scholar] [CrossRef]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential Effects of Coronaviruses on the Cardiovascular System: A Review. JAMA Cardiol. 2020, 5, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Corretti, M.C.; Anderson, T.J.; Benjamin, E.J.; Celermajer, D.; Charbonneau, F.; Creager, M.A.; Deanfield, J.; Drexler, H.; Gerhard-Herman, M.; Herrington, D.; et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: A report of the International Brachial Artery Reactivity Task Force. J. Am. Coll. Cardiol. 2002, 39, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Holder, S.M.; Bruno, R.M.; Shkredova, D.A.; Dawson, E.A.; Jones, H.; Hopkins, N.D.; Hopman, M.T.E.; Bailey, T.G.; Coombes, J.S.; Askew, C.D.; et al. Reference Intervals for Brachial Artery Flow-Mediated Dilation and the Relation With Cardiovascular Risk Factors. Hypertension 2021, 77, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Chen, J.A.; Bergmann, S.R. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: A meta-analysis. Int. J. Cardiovasc. Imaging 2010, 26, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Ergul, E.; Yilmaz, A.S.; Ogutveren, M.M.; Emlek, N.; Kostakoglu, U.; Cetin, M. COVID 19 disease independently predicted endothelial dysfunction measured by flow-mediated dilatation. Int. J. Cardiovasc. Imaging 2022, 38, 25–32. [Google Scholar] [CrossRef]
- Tu, T.M.; Seet, C.Y.H.; Koh, J.S.; Tham, C.H.; Chiew, H.J.; De Leon, J.A.; Chua, C.Y.K.; Hui, A.C.; Tan, S.S.Y.; Vasoo, S.S.; et al. Acute Ischemic Stroke During the Convalescent Phase of Asymptomatic COVID-2019 Infection in Men. JAMA Netw. Open 2021, 4, e217498. [Google Scholar] [CrossRef]
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; Ten Cate-Hoek, A.J.; Ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F. Endothelium-Derived Relaxing Factor: Discovery, Early Studies, and Identifcation as Nitric Oxide (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1999, 38, 1870–1880. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Sawdey, M.S.; Loskutoff, D.J. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J. Clin. Investig. 1991, 88, 1346–1353. [Google Scholar] [CrossRef] [Green Version]
- Levin, E.G.; Loskutoff, D.J. Cultured bovine endothelial cells produce both urokinase and tissue-type plasminogen activators. J. Cell Biol. 1982, 94, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Nachman, R.L.; Rafii, S. Platelets, petechiae, and preservation of the vascular wall. N. Engl. J. Med. 2008, 359, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, D.H.; Black, M.A.; Pyke, K.E.; Padilla, J.; Atkinson, G.; Harris, R.A.; Parker, B.; Widlansky, M.E.; Tschakovsky, M.E.; Green, D.J. Assessment of flow-mediated dilation in humans: A methodological and physiological guideline. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2–H12. [Google Scholar] [CrossRef] [Green Version]
- Celermajer, D.S.; Sorensen, K.E.; Bull, C.; Robinson, J.; Deanfield, J.E. Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J. Am. Coll. Cardiol. 1994, 24, 1468–1474. [Google Scholar] [CrossRef]
- Anderson, T.J. Prognostic significance of brachial flow-mediated vasodilation. Circulation 2007, 115, 2373–2375. [Google Scholar] [CrossRef] [Green Version]
- Bots, M.L.; Westerink, J.; Rabelink, T.J.; de Koning, E.J. Assessment of flow-mediated vasodilatation (FMD) of the brachial artery: Effects of technical aspects of the FMD measurement on the FMD response. Eur. Heart J. 2005, 26, 363–368. [Google Scholar] [CrossRef]
- Greyling, A.; van Mil, A.C.; Zock, P.L.; Green, D.J.; Ghiadoni, L.; Thijssen, D.H.; Dilation, T.I.W.G.o.F.M. Adherence to guidelines strongly improves reproducibility of brachial artery flow-mediated dilation. Atherosclerosis 2016, 248, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klonizakis, M.; Manning, G.; Donnelly, R. Assessment of lower limb microcirculation: Exploring the reproducibility and clinical application of laser Doppler techniques. Skin Pharmacol. Physiol. 2011, 24, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinshtein, R.; Kuvin, J.T.; Soffler, M.; Lennon, R.J.; Lavi, S.; Nelson, R.E.; Pumper, G.M.; Lerman, L.O.; Lerman, A. Assessment of endothelial function by non-invasive peripheral arterial tonometry predicts late cardiovascular adverse events. Eur. Heart J. 2010, 31, 1142–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell. Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef]
- Celik, T.; Balta, S.; Karaman, M.; Ahmet Ay, S.; Demirkol, S.; Ozturk, C.; Dinc, M.; Unal, H.U.; Yilmaz, M.I.; Kilic, S.; et al. Endocan, a novel marker of endothelial dysfunction in patients with essential hypertension: Comparative effects of amlodipine and valsartan. Blood Press. 2015, 24, 55–60. [Google Scholar] [CrossRef]
- Savoia, C.; Grassi, G. Exercise activity and endothelial function: The uprising role of endothelial progenitor cells in vascular protection. J. Hypertens. 2012, 30, 2083–2084. [Google Scholar] [CrossRef]
- Sabatier, F.; Camoin-Jau, L.; Anfosso, F.; Sampol, J.; Dignat-George, F. Circulating endothelial cells, microparticles and progenitors: Key players towards the definition of vascular competence. J. Cell. Mol. Med. 2009, 13, 454–471. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Kolivras, A.; Dehavay, F.; Delplace, D.; Feoli, F.; Meiers, I.; Milone, L.; Olemans, C.; Sass, U.; Theunis, A.; Thompson, C.T.; et al. Coronavirus (COVID-19) infection-induced chilblains: A case report with histopathologic findings. JAAD Case Rep. 2020, 6, 489–492. [Google Scholar] [CrossRef]
- Dou, Q.; Wei, X.; Zhou, K.; Yang, S.; Jia, P. Cardiovascular Manifestations and Mechanisms in Patients with COVID-19. Trends Endocrinol. Metab. 2020, 31, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, S.; Pourgholaminejad, A.; Saberi, A. Stroke Associated with SARS-CoV-2 Infection and its Pathogenesis: A Systematic Review. Basic Clin. Neurosci. 2021, 12, 569–586. [Google Scholar] [CrossRef]
- Pathirathna, M.L.; Samarasekara, B.P.P.; Dasanayake, T.S.; Saravanakumar, P.; Weerasekara, I. Adverse Perinatal Outcomes in COVID-19 Infected Pregnant Women: A Systematic Review and Meta-Analysis. Healthcare 2022, 10, 203. [Google Scholar] [CrossRef]
- Delli Muti, N.; Finocchi, F.; Tossetta, G.; Salvio, G.; Cutini, M.; Marzioni, D.; Balercia, G. Could SARS-CoV-2 infection affect male fertility and sexuality? APMIS, 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Andrianto; Al-Farabi, M.J.; Nugraha, R.A.; Marsudi, B.A.; Azmi, Y. Biomarkers of endothelial dysfunction and outcomes in coronavirus disease 2019 (COVID-19) patients: A systematic review and meta-analysis. Microvasc. Res. 2021, 138, 104224. [Google Scholar] [CrossRef] [PubMed]
- Lampsas, S.; Tsaplaris, P.; Pantelidis, P.; Oikonomou, E.; Marinos, G.; Charalambous, G.; Souvaliotis, N.; Mystakidi, V.C.; Goliopoulou, A.; Katsianos, E.; et al. The Role of Endothelial Related Circulating Biomarkers in COVID-19. A Systematic Review and Meta-analysis. Curr. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Mancuso, P.; Gidaro, A.; Gregato, G.; Raveane, A.; Cremonesi, P.; Quarna, J.; Caccia, S.; Gusso, L.; Rusconi, S.; Giacomelli, A.; et al. Circulating endothelial progenitors are increased in COVID-19 patients and correlate with SARS-CoV-2 RNA in severe cases. J. Thromb. Haemost. 2020, 18, 2744–2750. [Google Scholar] [CrossRef]
- Poyatos, P.; Luque, N.; Eizaguirre, S.; Sabater, G.; Sebastian, L.; Albesa, I.F.; Peracaula, M.; Boixade, M.; Orriols, R.; Tura-Ceide, O. Post-COVID-19 patients show an increased endothelial progenitor cell production. Transl. Res. 2022, 243, 14–20. [Google Scholar] [CrossRef]
- Paneroni, M.; Pasini, E.; Vitacca, M.; Scalvini, S.; Comini, L.; Pedrinolla, A.; Venturelli, M. Altered Vascular Endothelium-Dependent Responsiveness in Frail Elderly Patients Recovering from COVID-19 Pneumonia: Preliminary Evidence. J. Clin. Med. 2021, 10, 2558. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Cimino, G.; Vizzardi, E.; Calvi, E.; Pancaldi, E.; Pascariello, G.; Bernardi, N.; Cersosimo, A.; Amore, L.; Inciardi, R.M.; Raddino, R.; et al. Endothelial dysfunction in COVID-19 patients assessed with Endo-PAT2000. Monaldi Arch. Chest Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A vascular disease. Trends Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, K.; Gronski, P.A.; Kiyan, Y.; Seeliger, B.; Bertram, A.; Pape, T.; Welte, T.; Hoeper, M.M.; Haller, H.; David, S. Injury to the Endothelial Glycocalyx in Critically Ill Patients with COVID-19. Am. J. Respir. Crit. Care Med. 2020, 202, 1178–1181. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schroder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mae, M.; Muhl, L.; et al. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffre, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136. [Google Scholar] [CrossRef]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Berletch, J.B.; Yang, F.; Xu, J.; Carrel, L.; Disteche, C.M. Genes that escape from X inactivation. Hum. Genet. 2011, 130, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Viveiros, A.; Rasmuson, J.; Vu, J.; Mulvagh, S.L.; Yip, C.Y.Y.; Norris, C.M.; Oudit, G.Y. Sex differences in COVID-19: Candidate pathways, genetics of ACE2, and sex hormones. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H296–H304. [Google Scholar] [CrossRef]
- Bukowska, A.; Spiller, L.; Wolke, C.; Lendeckel, U.; Weinert, S.; Hoffmann, J.; Bornfleth, P.; Kutschka, I.; Gardemann, A.; Isermann, B.; et al. Protective regulation of the ACE2/ACE gene expression by estrogen in human atrial tissue from elderly men. Exp. Biol. Med. 2017, 242, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.C.; Tieri, P.; Ortona, E.; Ruggieri, A. ACE2 expression and sex disparity in COVID-19. Cell Death Discov. 2020, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Fujimoto, S.; Arakawa, S.; Yada, T.; Namikoshi, T.; Haruna, Y.; Horike, H.; Sasaki, T.; Kashihara, N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 3806–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Yu, M.; Jiang, J.; Luo, Y.; Zhang, Q.; Wang, S.; Yang, F.; Wang, A.; Wang, L.; Zhuang, M.; et al. Angiotensin II Decreases Endothelial Nitric Oxide Synthase Phosphorylation via AT1R Nox/ROS/PP2A Pathway. Front. Physiol. 2020, 11, 566410. [Google Scholar] [CrossRef]
- Wolf, G.; Wenzel, U.; Burns, K.D.; Harris, R.C.; Stahl, R.A.; Thaiss, F. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors. Kidney Int. 2002, 61, 1986–1995. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Sansico, F.; Miroballo, M.; Bianco, D.S.; Tamiro, F.; Colucci, M.; Santis, E.; Rossi, G.; Rosati, J.; Di Mauro, L.; Miscio, G.; et al. COVID-19 Specific Immune Markers Revealed by Single Cell Phenotypic Profiling. Biomedicines 2021, 9, 1794. [Google Scholar] [CrossRef]
- Wang, J.M.; Sica, A.; Peri, G.; Walter, S.; Padura, I.M.; Libby, P.; Ceska, M.; Lindley, I.; Colotta, F.; Mantovani, A. Expression of monocyte chemotactic protein and interleukin-8 by cytokine-activated human vascular smooth muscle cells. Arterioscler. Thromb. 1991, 11, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Kandere-Grzybowska, K.; Letourneau, R.; Kempuraj, D.; Donelan, J.; Poplawski, S.; Boucher, W.; Athanassiou, A.; Theoharides, T.C. IL-1 induces vesicular secretion of IL-6 without degranulation from human mast cells. J. Immunol. 2003, 171, 4830–4836. [Google Scholar] [CrossRef] [Green Version]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochemistry 2020, 85, 1543–1553. [Google Scholar] [CrossRef]
- Angelini, D.J.; Hyun, S.W.; Grigoryev, D.N.; Garg, P.; Gong, P.; Singh, I.S.; Passaniti, A.; Hasday, J.D.; Goldblum, S.E. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1232–L1245. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, J.M.; Niikura, M.; Yang, C.W.T.; Sin, D.D. COVID-19 and COPD. Eur. Respir. J. 2020, 56, 2002108. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Kheradmand, F. COVID-19, COPD, and AECOPD: Immunological, Epidemiological, and Clinical Aspects. Front. Med. 2020, 7, 627278. [Google Scholar] [CrossRef] [PubMed]
- Szucs, B.; Szucs, C.; Petrekanits, M.; Varga, J.T. Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. Int. J. Mol. Sci. 2019, 20, 4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesker, J.J.; Ten Hacken, N.H.; Zeinstra-Smith, M.; Rutgers, S.R.; Postma, D.S.; Timens, W. Reticular basement membrane in asthma and COPD: Similar thickness, yet different composition. Int. J. Chron. Obstruct. Pulmon. Dis. 2009, 4, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Arafah, M.A.; Raddaoui, E.; Kassimi, F.A.; Alhamad, E.H.; Alboukai, A.A.; Alshedoukhy, A.A.; Ouban, A. Endobronchial biopsy in the final diagnosis of chronic obstructive pulmonary disease and asthma: A clinicopathological study. Ann. Saudi Med. 2018, 38, 118–124. [Google Scholar] [CrossRef]
- Henson, P.M.; Vandivier, R.W.; Douglas, I.S. Cell death, remodeling, and repair in chronic obstructive pulmonary disease? Proc. Am. Thorac. Soc. 2006, 3, 713–717. [Google Scholar] [CrossRef]
- Kraen, M.; Frantz, S.; Nihlen, U.; Engstrom, G.; Lofdahl, C.G.; Wollmer, P.; Dencker, M. Matrix Metalloproteinases in COPD and atherosclerosis with emphasis on the effects of smoking. PLoS ONE 2019, 14, e0211987. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Luscher, T.F.; Camici, G.G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef]
- Ayres-Sander, C.E.; Lauridsen, H.; Maier, C.L.; Sava, P.; Pober, J.S.; Gonzalez, A.L. Transendothelial migration enables subsequent transmigration of neutrophils through underlying pericytes. PLoS ONE 2013, 8, e60025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosyreva, A.; Dzhalilova, D.; Lokhonina, A.; Vishnyakova, P.; Fatkhudinov, T. The Role of Macrophages in the Pathogenesis of SARS-CoV-2-Associated Acute Respiratory Distress Syndrome. Front. Immunol. 2021, 12, 682871. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xu, Z.; Chen, B.; He, W.; Hu, J.; Zhang, L.; Liu, X.; Chen, F. The Role of Vascular Endothelial Growth Factor in Small-airway Remodelling in a Rat Model of Chronic Obstructive Pulmonary Disease. Sci. Rep. 2017, 7, 41202. [Google Scholar] [CrossRef] [Green Version]
- Won, T.; Wood, M.K.; Hughes, D.M.; Talor, M.V.; Ma, Z.; Schneider, J.; Skinner, J.T.; Asady, B.; Goerlich, E.; Halushka, M.K.; et al. Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. EBioMedicine 2022, 75, 103812. [Google Scholar] [CrossRef]
- Ambrosino, P.; Grassi, G.; Maniscalco, M. Endothelial Dysfunction: From a Pathophysiological Mechanism to a Potential Therapeutic Target. Biomedicines 2021, 10, 78. [Google Scholar] [CrossRef]
- Ambrosino, P.; Molino, A.; Calcaterra, I.; Formisano, R.; Stufano, S.; Spedicato, G.A.; Motta, A.; Papa, A.; Di Minno, M.N.D.; Maniscalco, M. Clinical Assessment of Endothelial Function in Convalescent COVID-19 Patients Undergoing Multidisciplinary Pulmonary Rehabilitation. Biomedicines 2021, 9, 614. [Google Scholar] [CrossRef]
- Nagele, M.P.; Haubner, B.; Tanner, F.C.; Ruschitzka, F.; Flammer, A.J. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis 2020, 314, 58–62. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Shahin, Y.; Khan, J.A.; Samuel, N.; Chetter, I. Angiotensin converting enzyme inhibitors effect on endothelial dysfunction: A meta-analysis of randomised controlled trials. Atherosclerosis 2011, 216, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Napoleone, E.; Di Santo, A.; Camera, M.; Tremoli, E.; Lorenzet, R. Angiotensin-converting enzyme inhibitors downregulate tissue factor synthesis in monocytes. Circ. Res. 2000, 86, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hippisley-Cox, J.; Young, D.; Coupland, C.; Channon, K.M.; Tan, P.S.; Harrison, D.A.; Rowan, K.; Aveyard, P.; Pavord, I.D.; Watkinson, P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: Cohort study including 8.3 million people. Heart 2020, 106, 1503–1511. [Google Scholar] [CrossRef]
- Gao, C.; Cai, Y.; Zhang, K.; Zhou, L.; Zhang, Y.; Zhang, X.; Li, Q.; Li, W.; Yang, S.; Zhao, X.; et al. Association of hypertension and antihypertensive treatment with COVID-19 mortality: A retrospective observational study. Eur. Heart J. 2020, 41, 2058–2066. [Google Scholar] [CrossRef]
- Angeli, F.; Verdecchia, P.; Balestrino, A.; Bruschi, C.; Ceriana, P.; Chiovato, L.; Dalla Vecchia, L.A.; Fanfulla, F.; La Rovere, M.T.; Perego, F.; et al. Renin Angiotensin System Blockers and Risk of Mortality in Hypertensive Patients Hospitalized for COVID-19: An Italian Registry. J. Cardiovasc. Dev. Dis. 2022, 9, 15. [Google Scholar] [CrossRef]
- Hasan, S.S.; Kow, C.S.; Hadi, M.A.; Zaidi, S.T.R.; Merchant, H.A. Mortality and Disease Severity Among COVID-19 Patients Receiving Renin-Angiotensin System Inhibitors: A Systematic Review and Meta-analysis. Am. J. Cardiovasc. Drugs 2020, 20, 571–590. [Google Scholar] [CrossRef]
- Lopes, R.D.; Macedo, A.V.S.; de Barros, E.S.P.G.M.; Moll-Bernardes, R.J.; Dos Santos, T.M.; Mazza, L.; Feldman, A.; D’Andrea Saba Arruda, G.; de Albuquerque, D.C.; Camiletti, A.S.; et al. Effect of Discontinuing vs Continuing Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers on Days Alive and Out of the Hospital in Patients Admitted With COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 254–264. [Google Scholar] [CrossRef]
- Kow, C.S.; Ming, L.C.; Hasan, S.S. Renin-angiotensin system inhibitor use and the risk of mortality in hospitalized patients with COVID-19: A meta-analysis of randomized controlled trials. Hypertens. Res. 2021, 44, 1042–1045. [Google Scholar] [CrossRef]
- Iacobucci, G. COVID-19: People who take statins may be less likely to die, research suggests. BMJ 2021, 375, n2536. [Google Scholar] [CrossRef]
- Hermida, N.; Balligand, J.L. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: The role of statins. Antioxid. Redox Signal. 2014, 20, 1216–1237. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arocutipa, C.; Melgar-Talavera, B.; Alvarado-Yarasca, A.; Saravia-Bartra, M.M.; Cazorla, P.; Belzusarri, I.; Hernandez, A.V. Statins reduce mortality in patients with COVID-19: An updated meta-analysis of 147 824 patients. Int. J. Infect. Dis. 2021, 110, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.S.; Lin, P.C.; Chen, Y.S.; Pan, T.C.; Tang, P.L. The use of statins was associated with reduced COVID-19 mortality: A systematic review and meta-analysis. Ann. Med. 2021, 53, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Investigators, I.-S. Atorvastatin versus placebo in patients with COVID-19 in intensive care: Randomized controlled trial. BMJ 2022, 376, e068407. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Castaneda, S.; Ancochea, J. Biologic Therapy in COVID-19. Arch. Bronconeumol. 2021, 57, 1–2. [Google Scholar] [CrossRef]
- Deng, H.; Tang, T.X.; Chen, D.; Tang, L.S.; Yang, X.P.; Tang, Z.H. Endothelial Dysfunction and SARS-CoV-2 Infection: Association and Therapeutic Strategies. Pathogens 2021, 10, 582. [Google Scholar] [CrossRef]
- Alwazeer, D.; Liu, F.F.; Wu, X.Y.; LeBaron, T.W. Combating Oxidative Stress and Inflammation in COVID-19 by Molecular Hydrogen Therapy: Mechanisms and Perspectives. Oxid. Med. Cell. Longev. 2021, 2021, 5513868. [Google Scholar] [CrossRef]
- Daiber, A.; Chlopicki, S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free Radic. Biol. Med. 2020, 157, 15–37. [Google Scholar] [CrossRef]
- Carlstrom, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 2019, 10, 3081. [Google Scholar] [CrossRef] [Green Version]
- Timpani, C.A.; Rybalka, E. Calming the (Cytokine) Storm: Dimethyl Fumarate as a Therapeutic Candidate for COVID-19. Pharmaceuticals 2020, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, A.A.; Bullen, C.K.; Villabona-Rueda, A.F.; Thompson, E.A.; Turner, M.L.; Davis, S.L.; Komm, O.; Powell, J.D.; D’Alessio, F.R.; Yolken, R.H.; et al. Sulforaphane exhibits in vitro and in vivo antiviral activity against pandemic SARS-CoV-2 and seasonal HCoV-OC43 coronaviruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Alam, M.S.; Czajkowsky, D.M. SARS-CoV-2 infection and oxidative stress: Pathophysiological insight into thrombosis and therapeutic opportunities. Cytokine Growth Factor Rev. 2021, 63, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Schwalfenberg, G.K. N-Acetylcysteine: A Review of Clinical Usefulness (an Old Drug with New Tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, R.I.; Freeman, P.R.; Bruzzese, J. Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: A report of 2 cases. Respir. Med. Case Rep. 2020, 30, 101063. [Google Scholar] [CrossRef]
- Poe, F.L.; Corn, J. N-Acetylcysteine: A potential therapeutic agent for SARS-CoV-2. Med. Hypotheses. 2020, 143, 109862. [Google Scholar] [CrossRef]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.K.; Micinski, D.; Parsanathan, R. l-Cysteine Stimulates the Effect of Vitamin D on Inhibition of Oxidative Stress, IL-8, and MCP-1 Secretion in High Glucose Treated Monocytes. J. Am. Coll. Nutr. 2021, 40, 327–332. [Google Scholar] [CrossRef]
- Gambardella, J.; Khondkar, W.; Morelli, M.B.; Wang, X.; Santulli, G.; Trimarco, V. Arginine and Endothelial Function. Biomedicines 2020, 8, 277. [Google Scholar] [CrossRef]
- Fiorentino, G.; Coppola, A.; Izzo, R.; Annunziata, A.; Bernardo, M.; Lombardi, A.; Trimarco, V.; Santulli, G.; Trimarco, B. Effects of adding L-arginine orally to standard therapy in patients with COVID-19: A randomized, double-blind, placebo-controlled, parallel-group trial. Results of the first interim analysis. EClinicalMedicine 2021, 40, 101125. [Google Scholar] [CrossRef]
- Ambrosino, P.; Fuschillo, S.; Papa, A.; Di Minno, M.N.D.; Maniscalco, M. Exergaming as a Supportive Tool for Home-Based Rehabilitation in the COVID-19 Pandemic Era. Games Health J. 2020, 9, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Wang, J.; Xi, X.; He, M.; Zhao, C.; Feng, F.; Wang, H.; Sun, W.; Mao, L.; Hu, X.; et al. Efficacy and safety of short-wave diathermy treatment for moderate COVID-19 patients: A prospective, double-blind, randomized controlled clinical study. Eur. J. Phys. Rehabil. Med. 2021, 58, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Parizad, N.; Goli, R.; Faraji, N.; Mam-Qaderi, M.; Mirzaee, R.; Gharebaghi, N.; Baghaie, R.; Feizipour, H.; Haghighi, M.M. Effect of guided imagery on anxiety, muscle pain, and vital signs in patients with COVID-19: A randomized controlled trial. Complement. Ther. Clin. Pract. 2021, 43, 101335. [Google Scholar] [CrossRef] [PubMed]
- Divanoglou, A.; Samuelsson, A.P.K.; Sjodahl, P.E.R.; Andersson, C.; Levi, P.R. Rehabilitation needs and mortality associated with the COVID-19 pandemic: A population-based study of all hospitalised and home-healthcare individuals in a Swedish healthcare region. EClinicalMedicine 2021, 36, 100920. [Google Scholar] [CrossRef] [PubMed]
- Maniscalco, M.; Fuschillo, S.; Ambrosino, P.; Martucci, M.; Papa, A.; Matera, M.G.; Cazzola, M. Preexisting cardiorespiratory comorbidity does not preclude the success of multidisciplinary rehabilitation in post-COVID-19 patients. Respir. Med. 2021, 184, 106470. [Google Scholar] [CrossRef] [PubMed]
- Sinoway, L.I.; Musch, T.I.; Minotti, J.R.; Zelis, R. Enhanced maximal metabolic vasodilatation in the dominant forearms of tennis players. J. Appl. Physiol. 1986, 61, 673–678. [Google Scholar] [CrossRef]
- Merlo, C.; Bernardi, E.; Bellotti, F.; Pomidori, L.; Cogo, A. Supervised exercise training improves endothelial function in COPD patients: A method to reduce cardiovascular risk? ERJ Open Res. 2020, 6, 00304–02019. [Google Scholar] [CrossRef]
- Kitzman, D.W.; Brubaker, P.H.; Herrington, D.M.; Morgan, T.M.; Stewart, K.P.; Hundley, W.G.; Abdelhamed, A.; Haykowsky, M.J. Effect of endurance exercise training on endothelial function and arterial stiffness in older patients with heart failure and preserved ejection fraction: A randomized, controlled, single-blind trial. J. Am. Coll. Cardiol. 2013, 62, 584–592. [Google Scholar] [CrossRef]
- Dai, R.; Zhuo, H.; Chen, Y.; Zhang, K.; Dong, Y.; Chen, C.; Wang, W. Mechanism of Isosorbide Dinitrate Combined with Exercise Training Rehabilitation to Mobilize Endothelial Progenitor Cells in Patients with Coronary Heart Disease. Bioengineered 2021. [Google Scholar] [CrossRef]
- Lanza, G.A.; Golino, M.; Villano, A.; Lanza, O.; Lamendola, P.; Fusco, A.; Leggio, M. Cardiac Rehabilitation and Endothelial Function. J. Clin. Med. 2020, 9, 2487. [Google Scholar] [CrossRef]
- Montero, D.; Padilla, J.; Diaz-Canestro, C.; Muris, D.M.; Pyke, K.E.; Obert, P.; Walther, G. Flow-mediated dilation in athletes: Influence of aging. Med. Sci. Sports Exerc. 2014, 46, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.D.; Malone, E.; Florida-James, G. Vascular Ageing and Exercise: Focus on Cellular Reparative Processes. Oxid. Med. Cell. Longev. 2016, 2016, 3583956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrosino, P.; Calcaterra, I.L.; Mosella, M.; Formisano, R.; D’Anna, S.E.; Bachetti, T.; Marcuccio, G.; Galloway, B.; Mancini, F.P.; Papa, A.; et al. Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target. Biomedicines 2022, 10, 812. https://doi.org/10.3390/biomedicines10040812
Ambrosino P, Calcaterra IL, Mosella M, Formisano R, D’Anna SE, Bachetti T, Marcuccio G, Galloway B, Mancini FP, Papa A, et al. Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target. Biomedicines. 2022; 10(4):812. https://doi.org/10.3390/biomedicines10040812
Chicago/Turabian StyleAmbrosino, Pasquale, Ilenia Lorenza Calcaterra, Marco Mosella, Roberto Formisano, Silvestro Ennio D’Anna, Tiziana Bachetti, Giuseppina Marcuccio, Brurya Galloway, Francesco Paolo Mancini, Antimo Papa, and et al. 2022. "Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target" Biomedicines 10, no. 4: 812. https://doi.org/10.3390/biomedicines10040812
APA StyleAmbrosino, P., Calcaterra, I. L., Mosella, M., Formisano, R., D’Anna, S. E., Bachetti, T., Marcuccio, G., Galloway, B., Mancini, F. P., Papa, A., Motta, A., Di Minno, M. N. D., & Maniscalco, M. (2022). Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target. Biomedicines, 10(4), 812. https://doi.org/10.3390/biomedicines10040812