
Structural Insights from Molecular Modeling of Isoindolin-1-One Derivatives as PI3Kγ Inhibitors against Gastric Carcinoma



Abstract
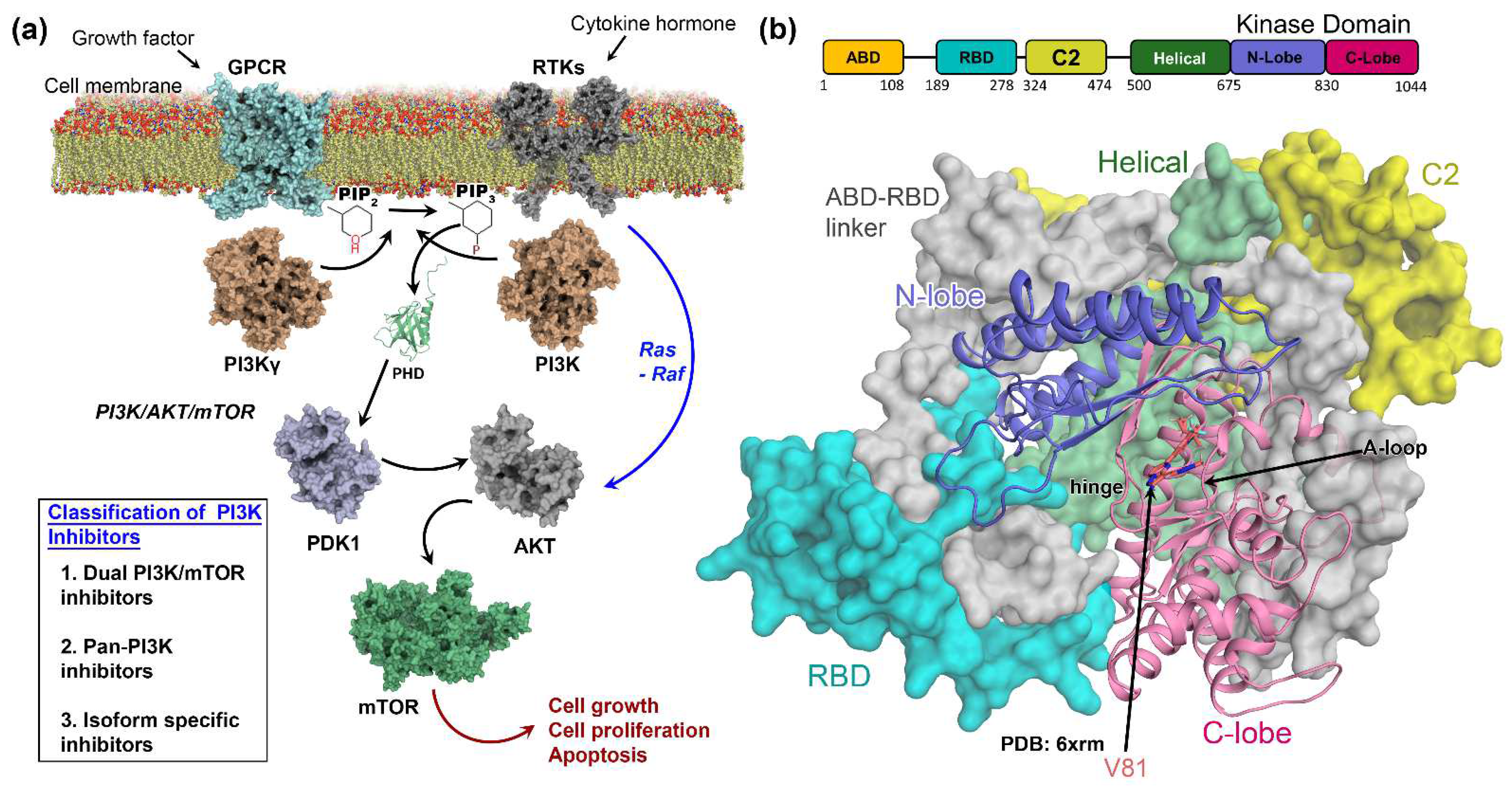
:1. Introduction
2. Materials and Methods
2.1. Protein Structure Preparation and Molecular Docking
2.2. Molecular Dynamics
2.3. Binding Free Energy Estimation
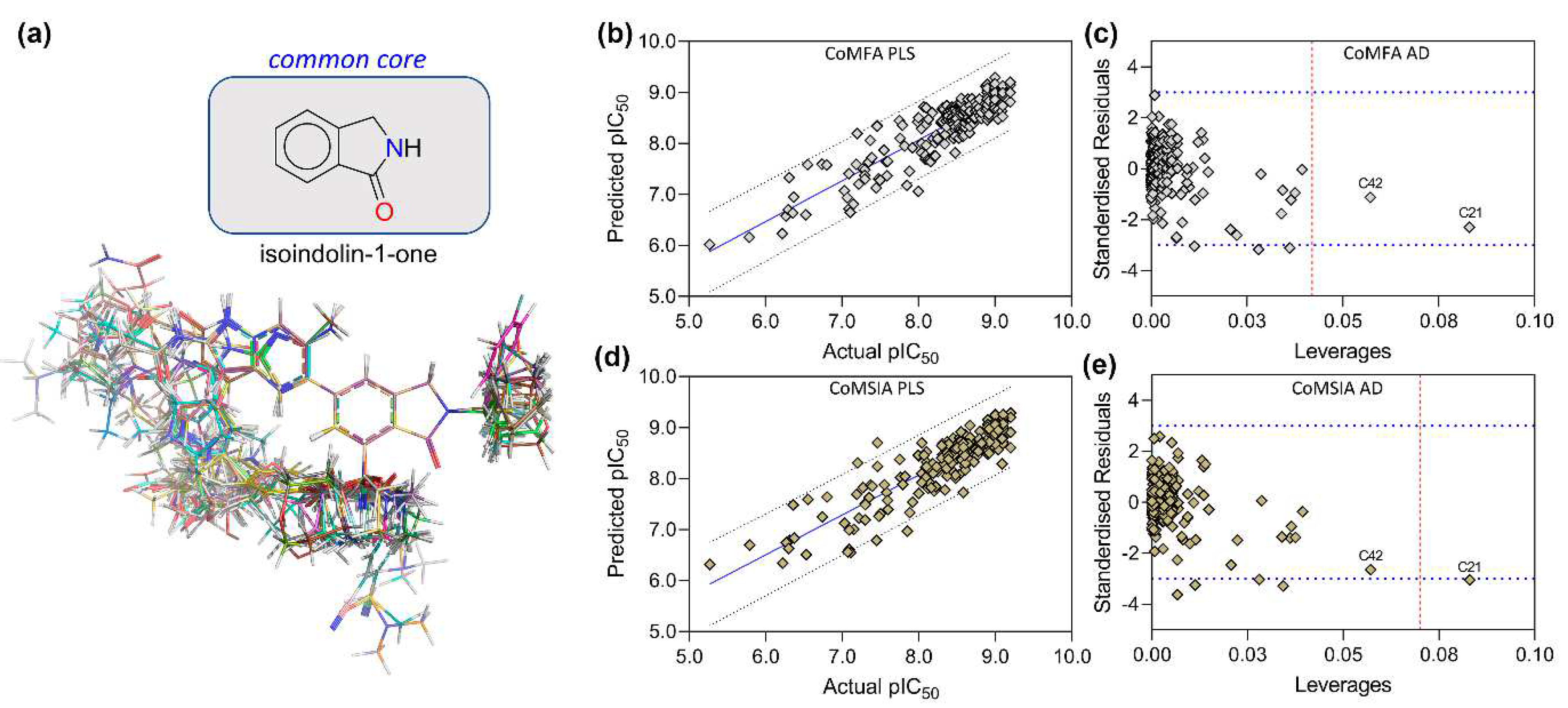
2.4. Molecular Alignment and Dataset Building
2.5. CoMFA and CoMSIA Model Building
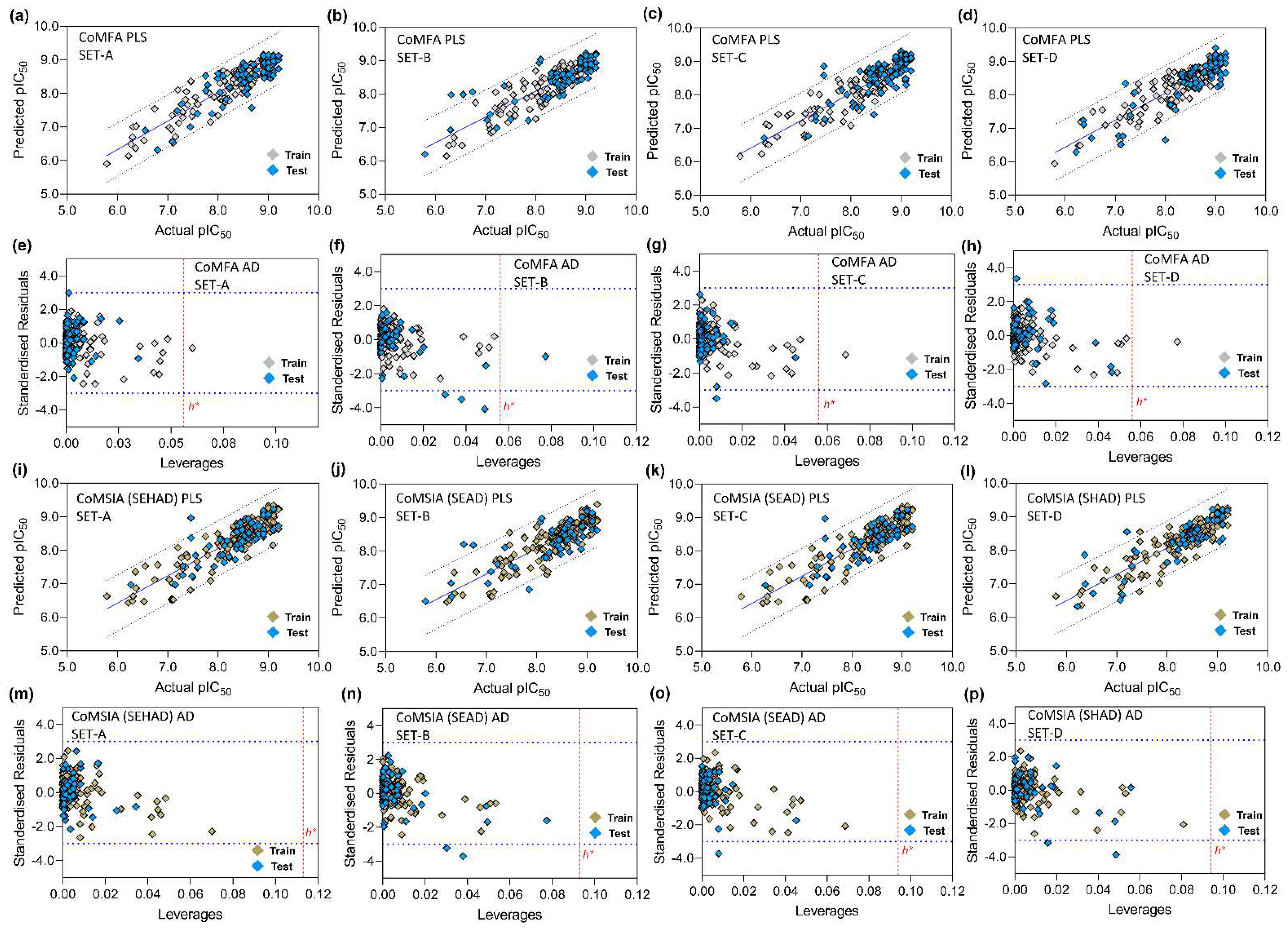
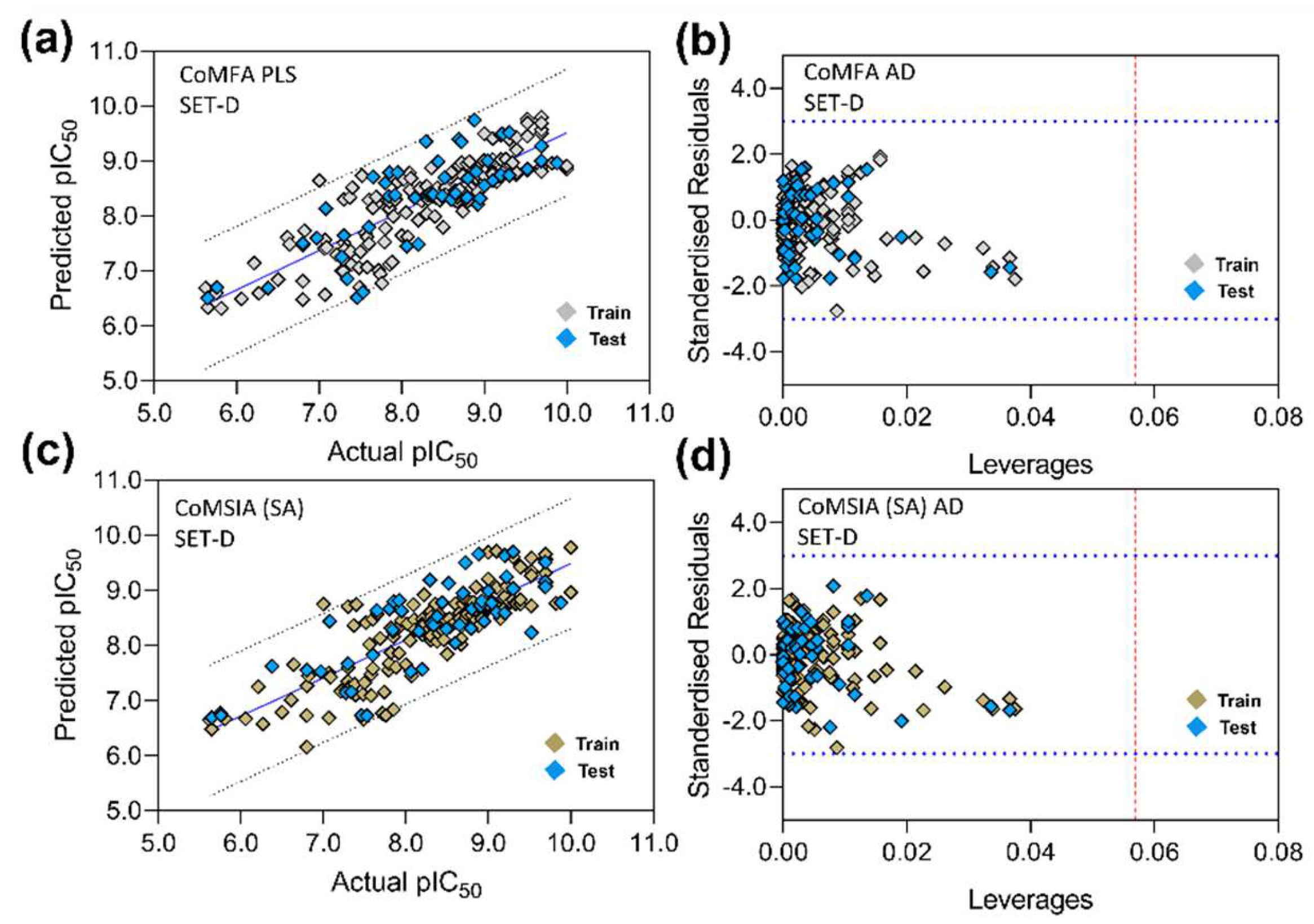
2.6. 3D-QSAR Model Validation
2.7. Applicability Domain Analysis
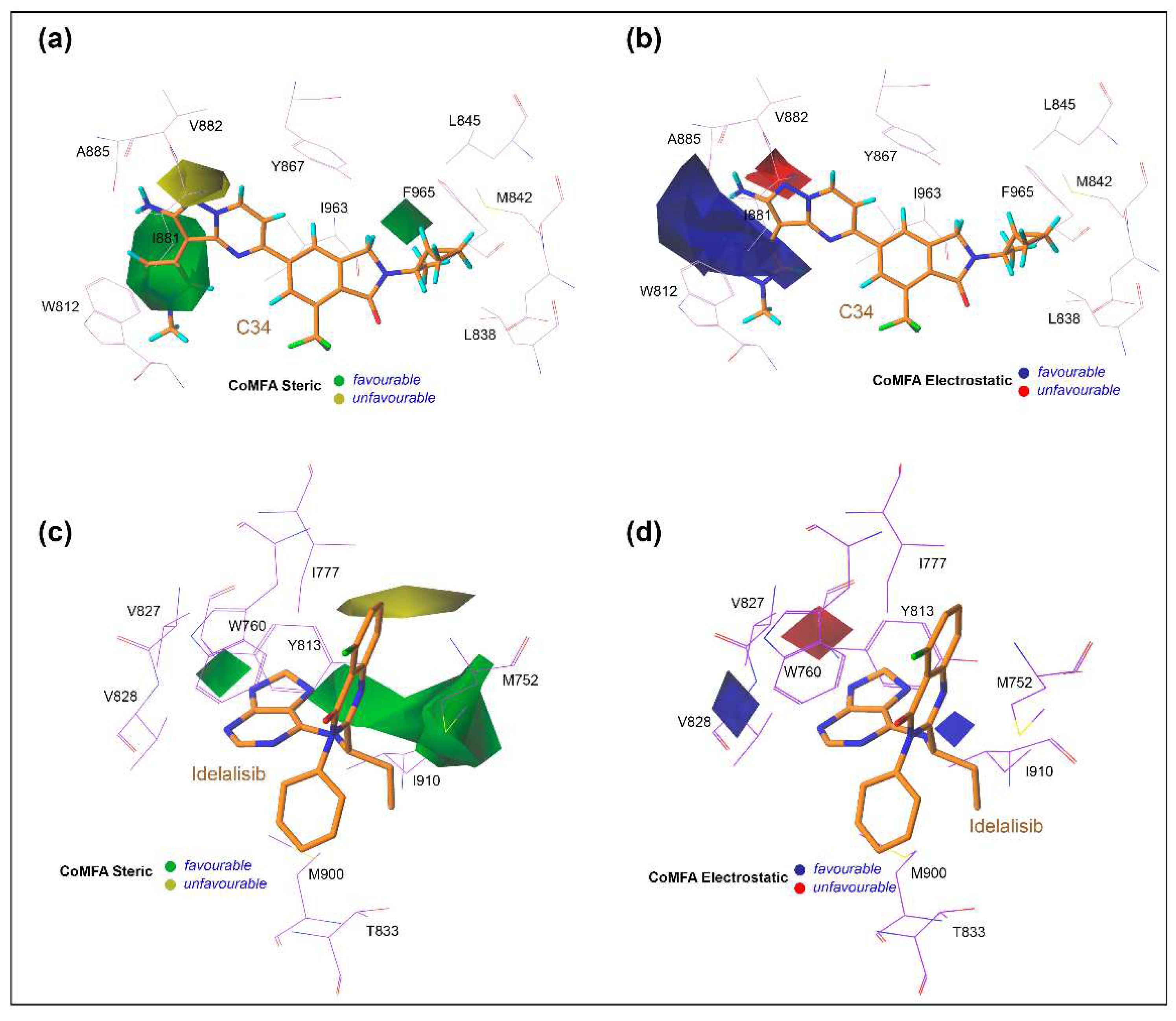
2.8. Contour Map Analysis and SAR Study
2.9. Designing of the New Compounds and Binding Affinity Calculation
3. Results
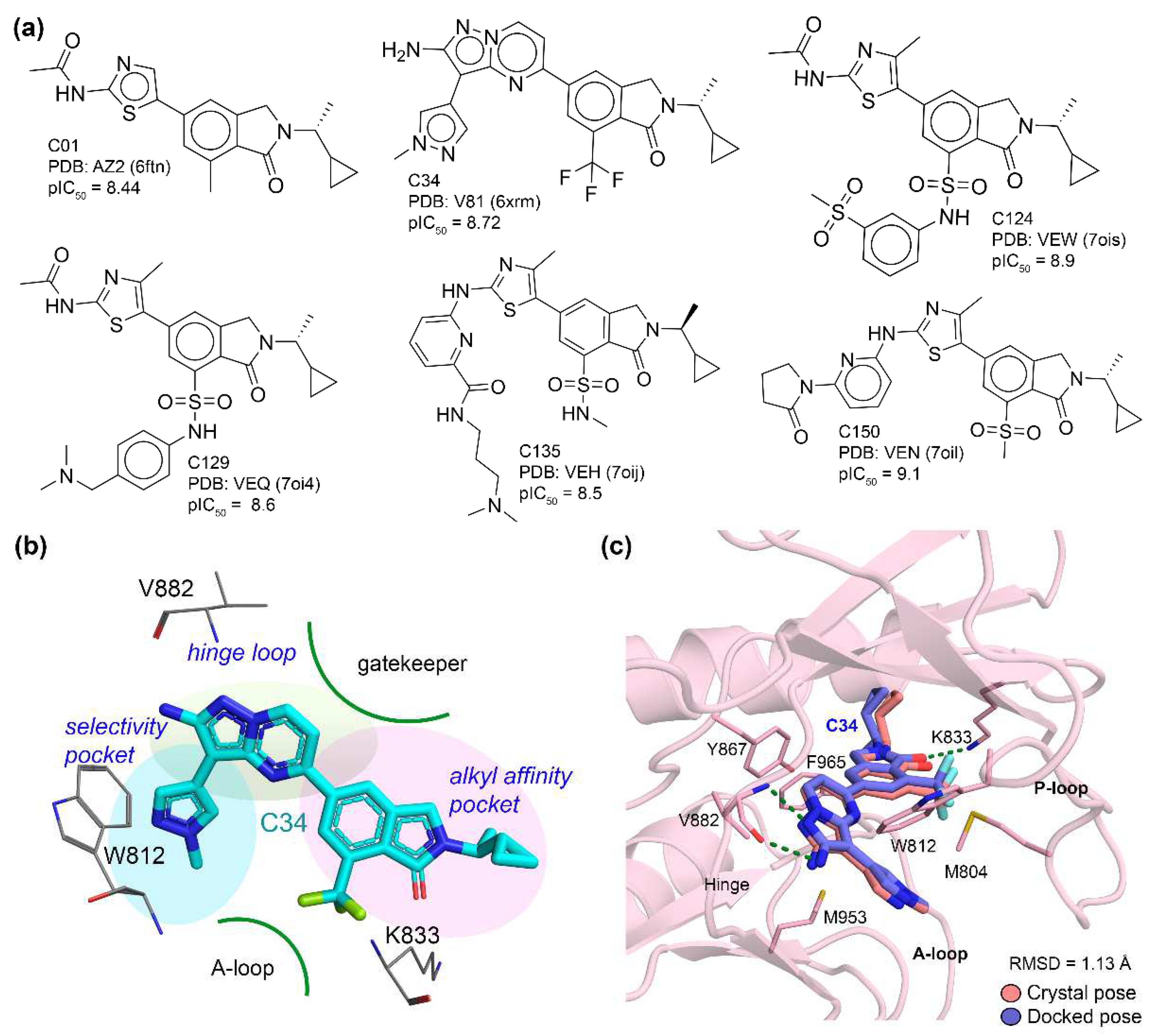
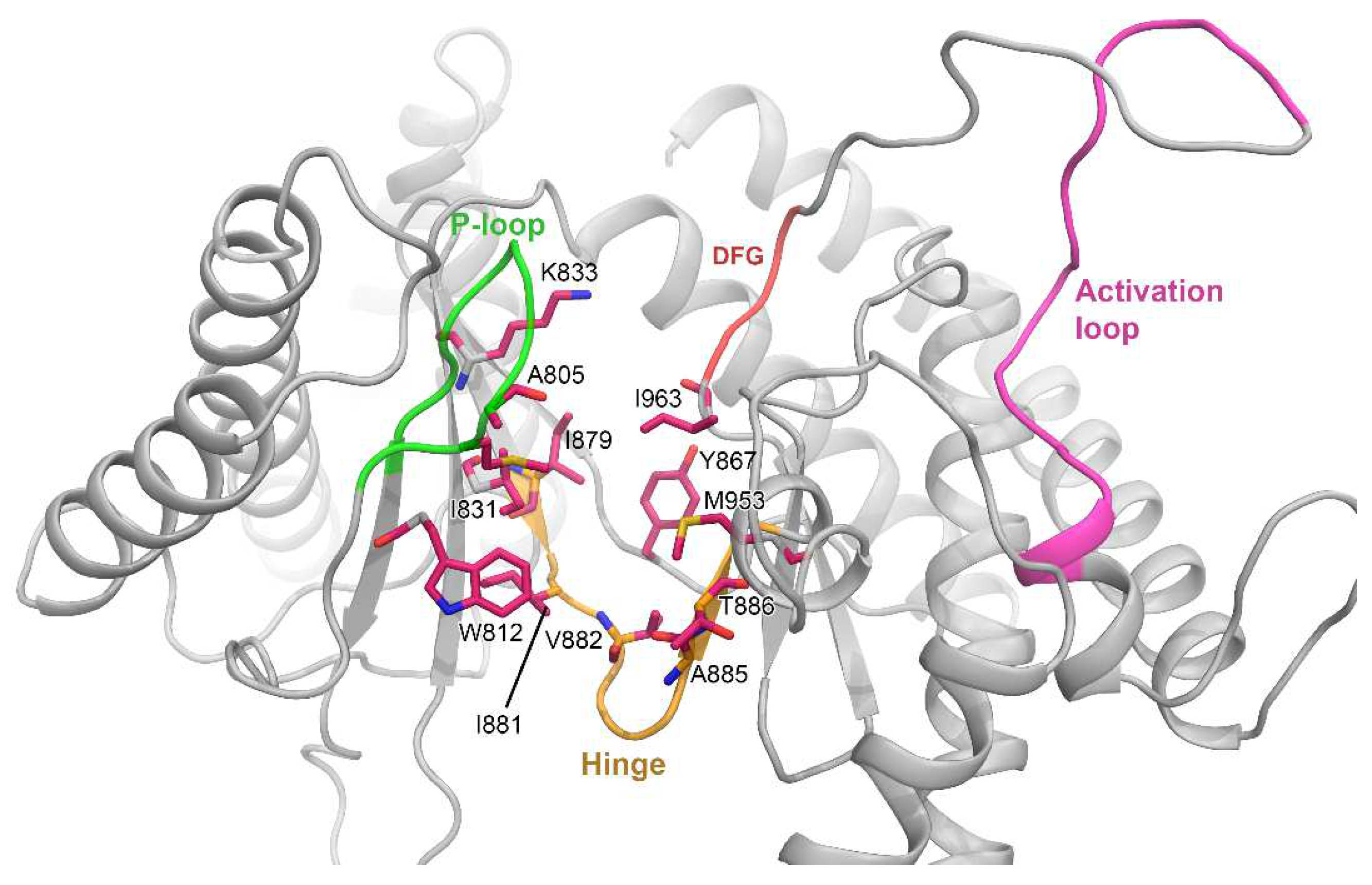
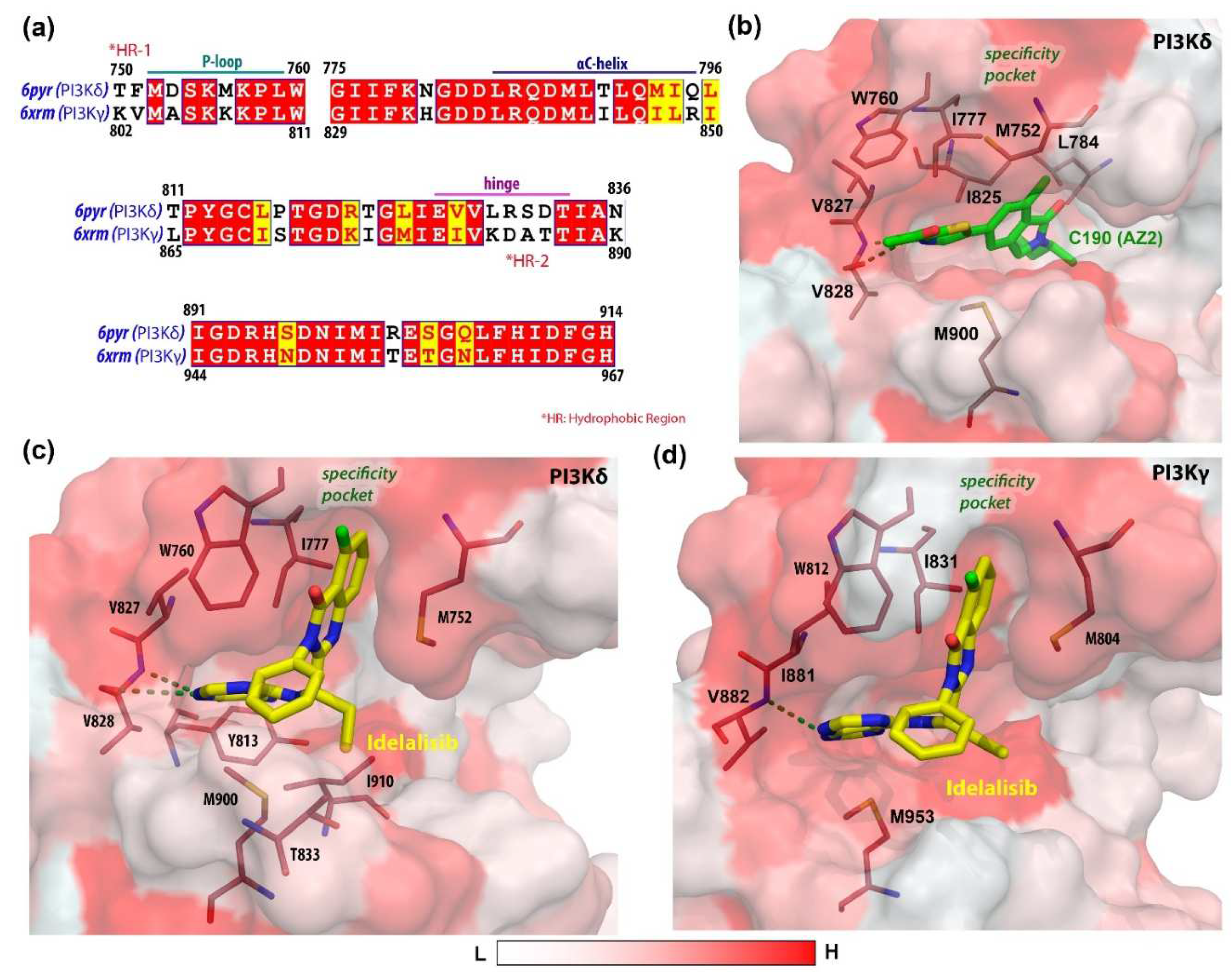
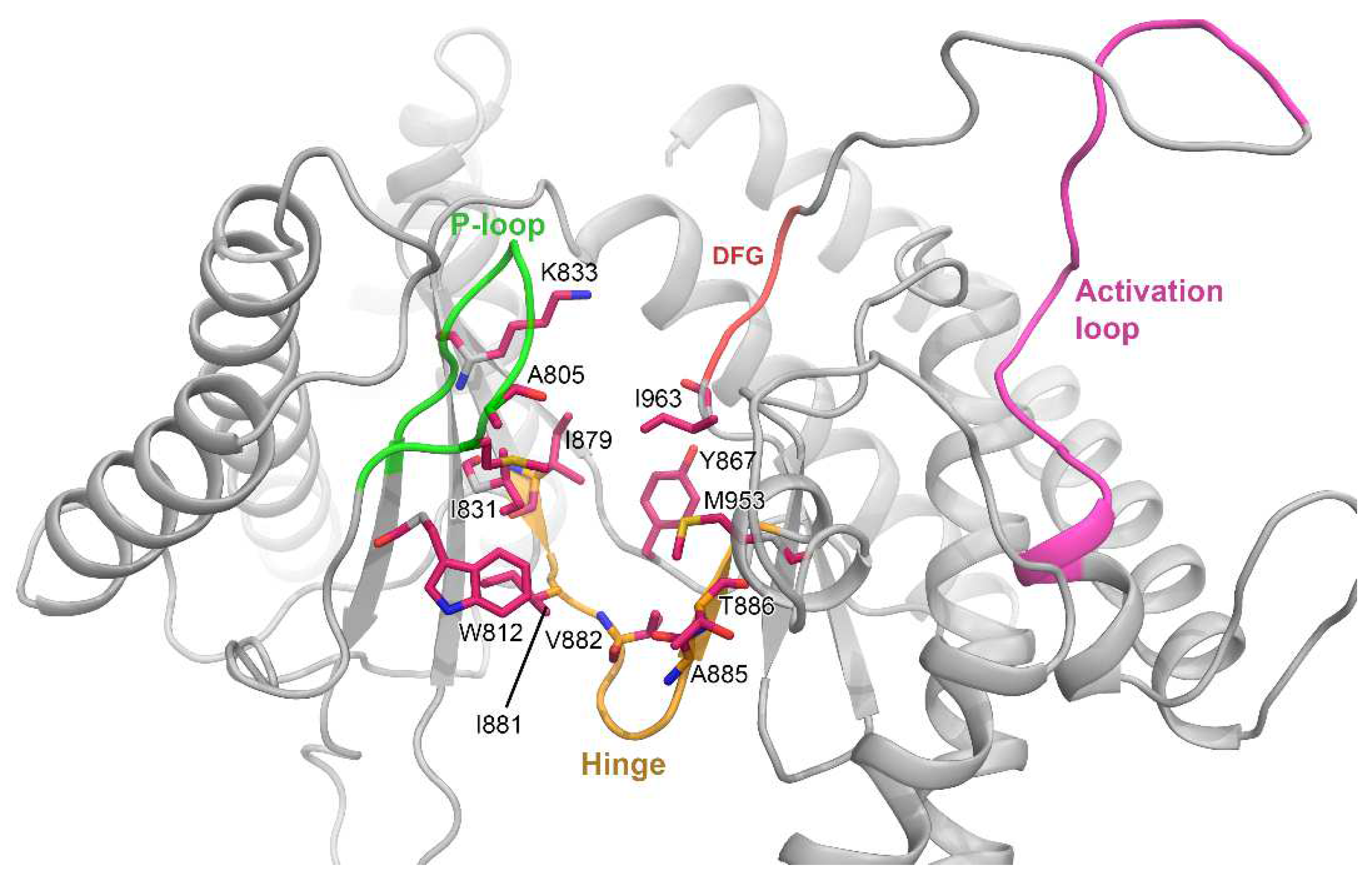
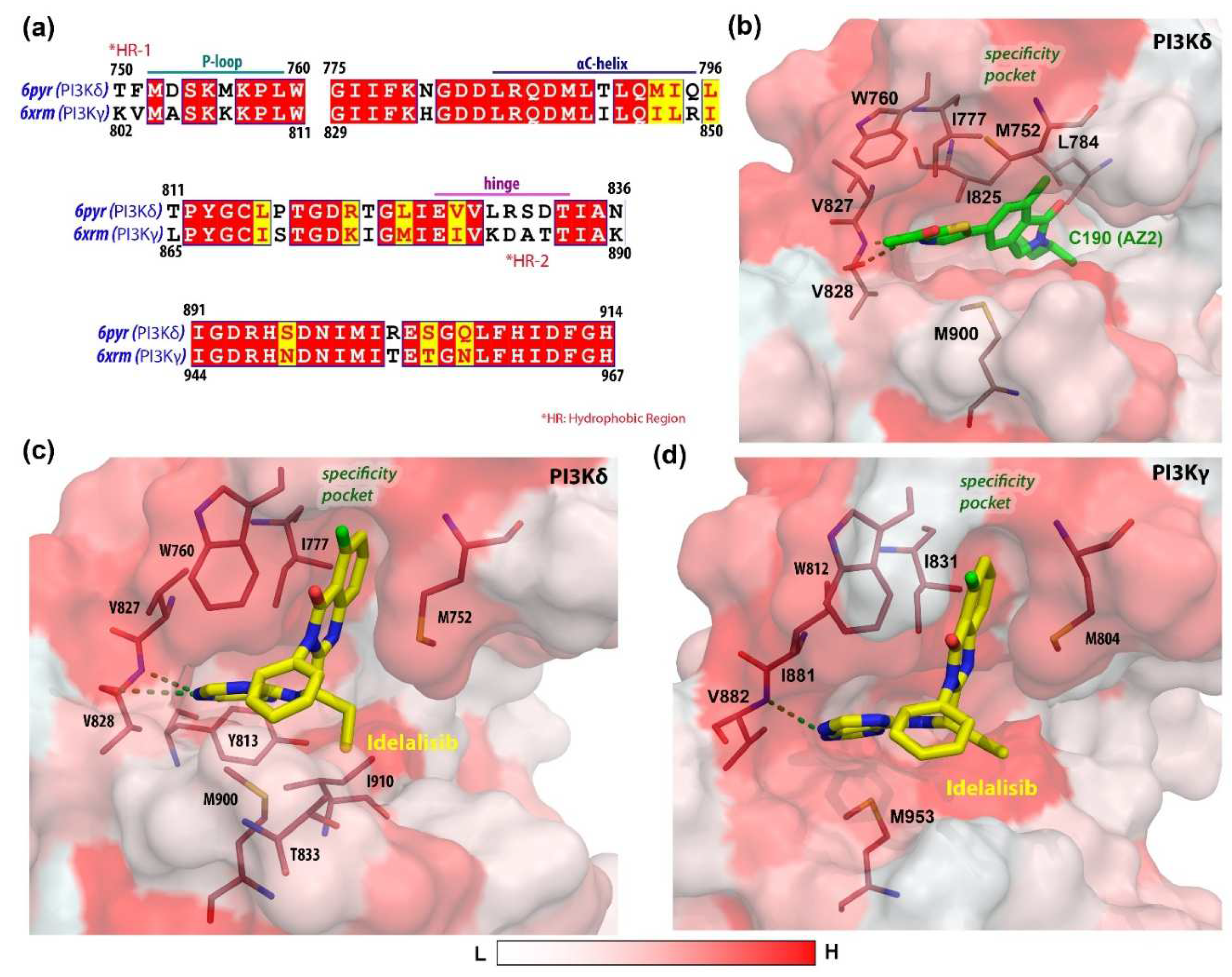
3.1. Molecular Docking Analysis
3.2. MD Simulation and Protein–Ligand Stability
3.3. Free Energy Calculation
3.4. Statistical Results from CoMFA and CoMSIA
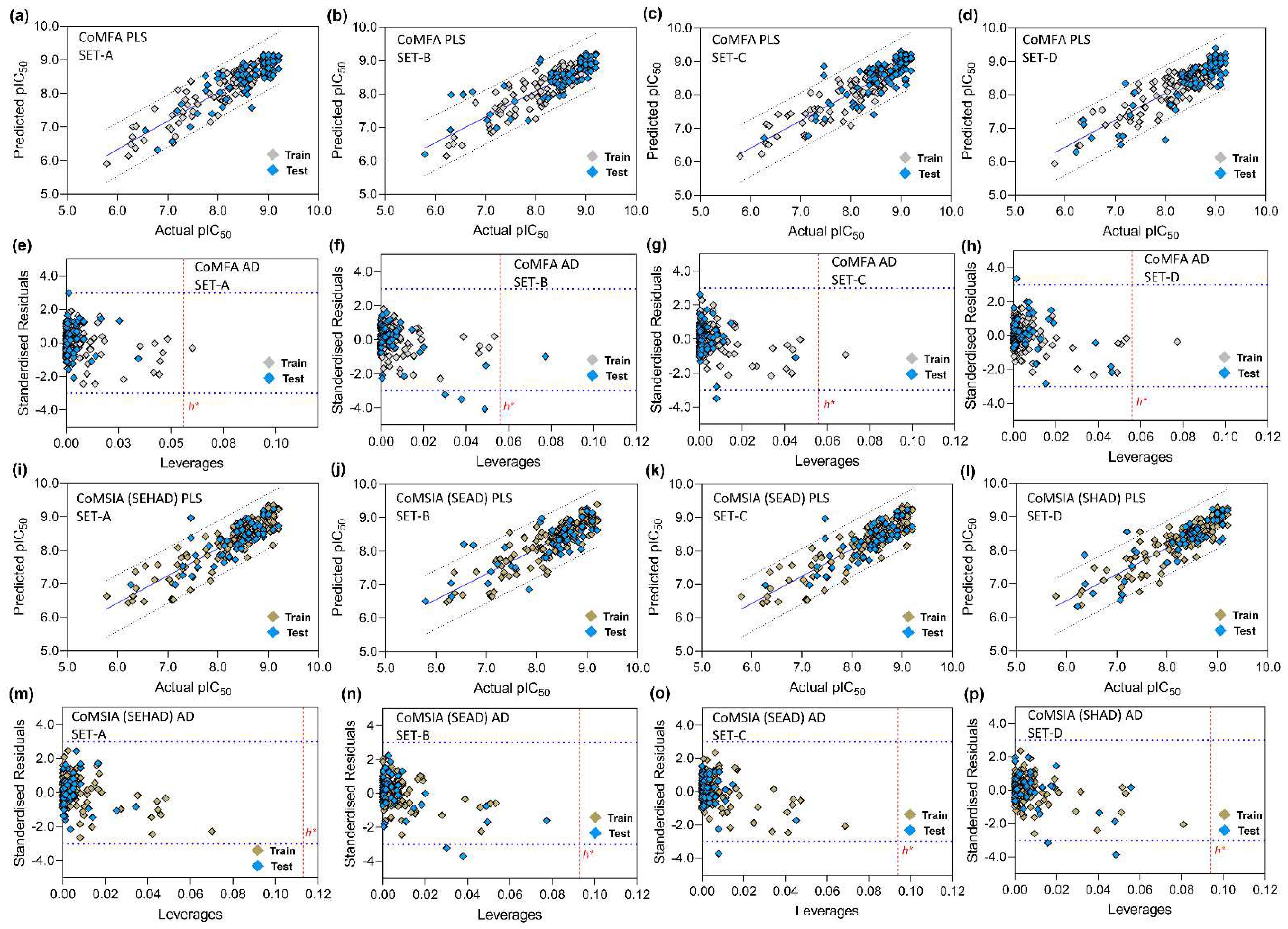
3.5. PLS Plots and Applicability Domain Analysis
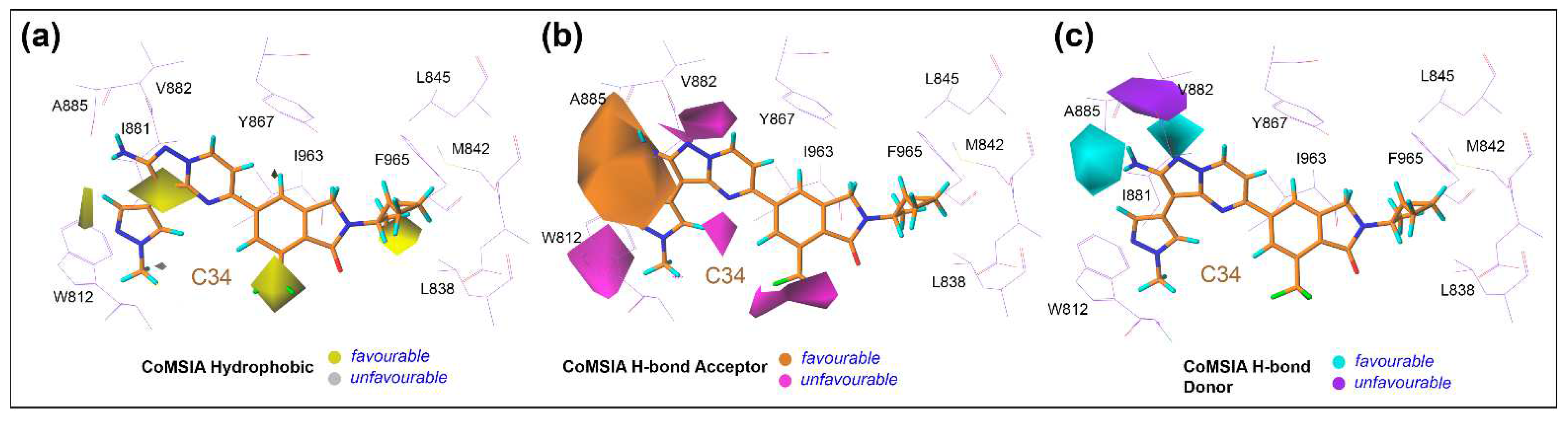
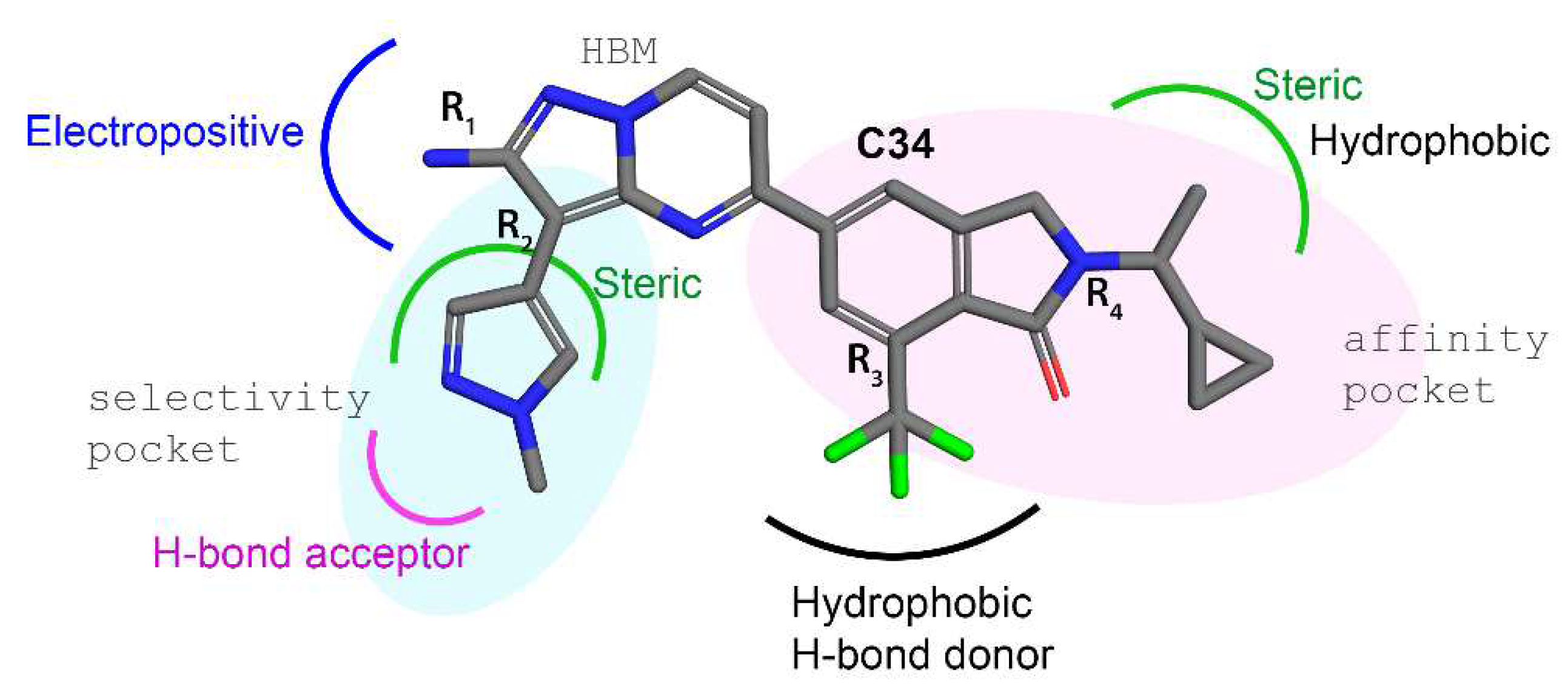
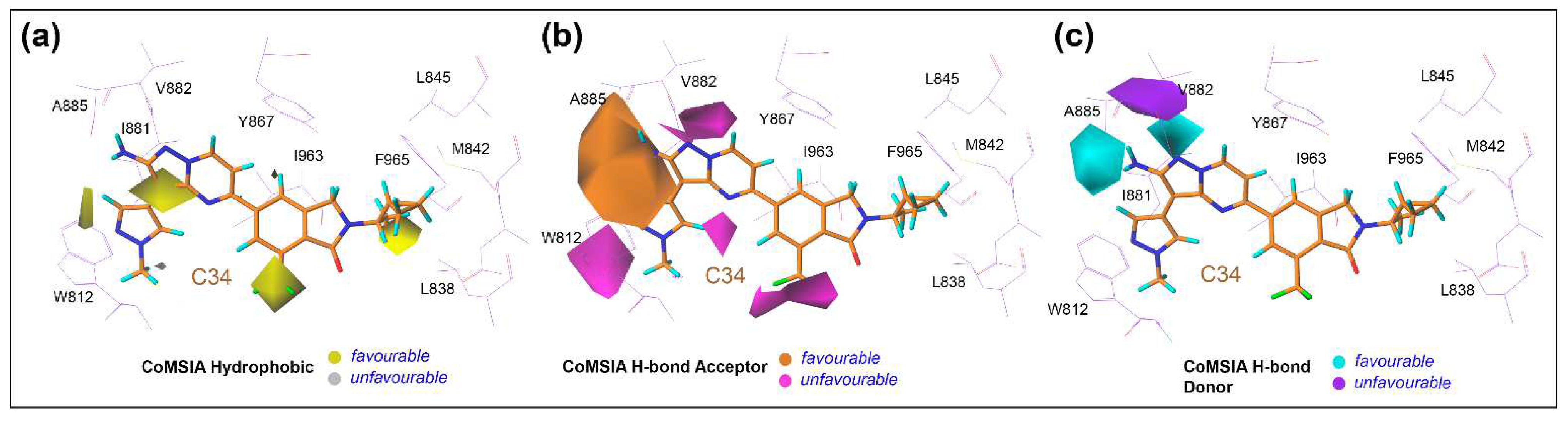
3.6. Contour Maps Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PI3K | Phosphoinositol-3-kinase. |
| GC | Gastric Carcinoma. |
| TAM | Tumor-associated macrophage. |
| MM-PB/GBSA | Molecular Mechanics-Poison–Boltzmann/generalized Born Surface Area. |
| LIE | Linear Interaction Energy. |
| BE | Binding Energy. |
| CoMFA | Comparative Molecular Field Analysis. |
| CoMSIA | Comparative Molecular Similarity Indices Analysis. |
| 3D-QSAR | 3-Dimensional Structure–Activity Relationship. |
References
- Valencia, G.A.; Rioja, P.; Morante, Z.; Araujo, J.M.; Vallejos, H.D.; Guerra, H.; Gomez, H.L. PIK3CA mutation in non-metastatic triple-negative breast cancer as a potential biomarker of early relapse: A case report. World J. Clin. Oncol. 2021, 12, 702. [Google Scholar] [CrossRef]
- Suppan, C.; Graf, R.; Jahn, S.; Zhou, Q.; Klocker, E.V.; Bartsch, R.; Terbuch, A.; Kashofer, K.; Regitnig, P.; Lindenmann, J. Sensitive and robust liquid biopsy-based detection of PIK3CA mutations in hormone-receptor-positive metastatic breast cancer patients. Br. J. Cancer 2021, 126, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, S.; Amjadi-Moheb, F.; Tabaripour, R.; Ashrafi, G.H.; Akhavan-Niaki, H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. 2020, 262, 118513. [Google Scholar] [CrossRef] [PubMed]
- Khorasani, A.B.S.; Pourbagheri-Sigaroodi, A.; Pirsalehi, A.; Safaroghli-Azar, A.; Zali, M.R.; Bashash, D. The PI3K/Akt/mTOR signaling pathway in gastric cancer; from oncogenic variations to the possibilities for pharmacologic interventions. Eur. J. Pharmacol. 2021, 898, 173983. [Google Scholar] [CrossRef] [PubMed]
- Berning, P.; Lenz, G. The role of PI3K inhibitors in the treatment of malignant lymphomas. Leuk. Lymphoma 2021, 62, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, K.; Yu, L.; Chen, Y.; Cai, Y.; Jin, J.; Hou, T. Targeting phosphatidylinositol 3-kinase gamma (PI3Kγ): Discovery and development of its selective inhibitors. Med. Res. Rev. 2021, 41, 1599–1621. [Google Scholar] [CrossRef]
- Roudsari, N.M.; Lashgari, N.-A.; Momtaz, S.; Abaft, S.; Jamali, F.; Safaiepour, P.; Narimisa, K.; Jackson, G.; Bishayee, A.; Rezaei, N. Inhibitors of the PI3K/Akt/mTOR pathway in prostate cancer chemoprevention and intervention. Pharmaceutics 2021, 13, 1195. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR signaling pathway in breast cancer: From molecular landscape to clinical aspects. Int. J. Mol. Sci. 2021, 22, 173. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR pathway as a pro-survival signaling and resistance-mediating mechanism to therapy of prostate cancer. Int. J. Mol. Sci. 2021, 22, 11088. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Taylor, M.A.; King, M.; Coenen-Stass, A.M.; Hughes, A.M.; Bell, S.; Proia, T.A.; Wang, Y.; Ramos-Montoya, A.; Wali, N. Macrophage Activation Status Rather than Repolarization Is Associated with Enhanced Checkpoint Activity in Combination with PI3Kγ Inhibition. Mol. Cancer Ther. 2021, 20, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Afify, S.M.; Du, J.; Liu, B.; Hassan, G.; Wang, Q.; Li, H.; Liu, Y.; Fu, X.; Zhu, Z.; et al. The efficacy of PI3Kγ and EGFR inhibitors on the suppression of the characteristics of cancer stem cells. Sci. Rep. 2022, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, 1–21. [Google Scholar] [CrossRef]
- Sapon-Cousineau, V.; Sapon-Cousineau, S.; Assouline, S. PI3K inhibitors and their role as novel agents for targeted therapy in lymphoma. Curr. Treat. Options Oncol. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Visentin, A.; Frezzato, F.; Severin, F.; Imbergamo, S.; Pravato, S.; Gargarella, L.R.; Manni, S.; Pizzo, S.; Ruggieri, E.; Facco, M. Lights and Shade of Next-Generation Pi3k Inhibitors in Chronic Lymphocytic Leukemia. OncoTargets Ther. 2020, 13, 9679. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hong, D.S.; Tolcher, A.W.; Patnaik, A.; Shapiro, G.; Chmielowski, B.; Ribas, A.; Brail, L.H.; Roberts, J.; Lee, L. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J. Clin. Oncol. 2018, 36, 3013. [Google Scholar] [CrossRef]
- Drew, S.L.; Thomas-Tran, R.; Beatty, J.W.; Fournier, J.; Lawson, K.V.; Miles, D.H.; Mata, G.; Sharif, E.U.; Yan, X.; Mailyan, A.K. Discovery of potent and selective PI3Kγ inhibitors. J. Med. Chem. 2020, 63, 11235–11257. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Nussinov, R. Structural Features that Distinguish Inactive and Active PI3K Lipid Kinases. J. Mol. Biol. 2020, 432, 5849–5859. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.W.; Bjorhall, K.; Bold, P.; Brulls, M.; Borjesson, U.; Carlsson, J.; Chang, H.-F.A.; Chen, Y.; Eriksson, A.; Fihn, B.-M. Discovery of AZD8154, a Dual PI3Kγδ Inhibitor for the Treatment of Asthma. J. Med. Chem. 2021, 64, 8053–8075. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.H.; Yan, X.; Thomas-Tran, R.; Fournier, J.; Sharif, E.U.; Drew, S.L.; Mata, G.; Lawson, K.V.; Ginn, E.; Wong, K.; et al. Discovery of Potent and Selective 7-Azaindole Isoindolinone-Based PI3Kγ Inhibitors. ACS Med. Chem. Lett. 2020, 11, 2244–2252. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, N.; Mogemark, M.; Arlbrandt, S.; Bold, P.; Cox, R.J.; Gardelli, C.; Holden, N.S.; Karabelas, K.; Karlsson, J.; Lever, S.; et al. Discovery of Highly Isoform Selective Orally Bioavailable Phosphoinositide 3-Kinase (PI3K)-γ Inhibitors. J. Med. Chem. 2018, 61, 5435–5441. [Google Scholar] [CrossRef] [PubMed]
- Mata, G.; Miles, D.H.; Drew, S.L.; Fournier, J.; Lawson, K.V.; Mailyan, A.K.; Sharif, E.U.; Yan, X.; Beatty, J.W.; Banuelos, J.; et al. Design, Synthesis, and Structure–Activity Relationship Optimization of Pyrazolopyrimidine Amide Inhibitors of Phosphoinositide 3-Kinase γ (PI3Kγ). J. Med. Chem. 2021, 65, 1418–1444. [Google Scholar] [CrossRef]
- Li, F.; Liang, X.; Jiang, Z.; Wang, A.; Wang, J.; Chen, C.; Wang, W.; Zou, F.; Qi, Z.; Liu, Q. Discovery of (S)-2-(1-(4-Amino-3-(3-fluoro-4-methoxyphenyl)-1 H-pyrazolo [3, 4-d] pyrimidin-1-yl) propyl)-3-cyclopropyl-5-fluoroquinazolin-4 (3 H)-one (IHMT-PI3Kδ-372) as a Potent and Selective PI3Kδ Inhibitor for the Treatment of Chronic Obstructive Pulmonary Disease. J. Med. Chem. 2020, 63, 13973–13993. [Google Scholar]
- Patel, L.; Chandrasekhar, J.; Evarts, J.; Haran, A.C.; Ip, C.; Kaplan, J.A.; Kim, M.; Koditek, D.; Lad, L.; Lepist, E.-I. 2, 4, 6-Triaminopyrimidine as a novel hinge binder in a series of PI3Kδ selective inhibitors. J. Med. Chem. 2016, 59, 3532–3548. [Google Scholar] [CrossRef]
- Feng, Y.; Duan, W.; Fan, S.; Zhang, H.; Zhang, S.-Q.; Xin, M. Synthesis and biological evaluation of 4-(piperid-3-yl) amino substituted 6-pyridylquinazolines as potent PI3Kδ inhibitors. Biorg. Med. Chem. 2019, 27, 115035. [Google Scholar] [CrossRef]
- Methot, J.L.; Zhou, H.; Kattar, S.D.; McGowan, M.A.; Wilson, K.; Garcia, Y.; Deng, Y.; Altman, M.; Fradera, X.; Lesburg, C. Structure Overhaul Affords a Potent Purine PI3Kδ Inhibitor with Improved Tolerability. J. Med. Chem. 2019, 62, 4370–4382. [Google Scholar] [CrossRef]
- Ma, C.-C.; Zhang, C.-M.; Tang, L.-Q.; Liu, Z.-P. Discovery of novel quinazolinone derivatives as high potent and selective PI3Kδ and PI3Kδ/γ inhibitors. Eur. J. Med. Chem. 2018, 151, 9–17. [Google Scholar] [CrossRef]
- Wang, N.-Y.; Zuo, W.-Q.; Hu, R.; Wang, W.-L.; Zhu, Y.-X.; Xu, Y.; Yu, L.-T.; Liu, Z.-H. Design, synthesis and structure-activity relationship study of piperazinone-containing thieno [3, 2-d] pyrimidine derivatives as new PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127479. [Google Scholar] [CrossRef] [PubMed]
- Erra, M.; Taltavull, J.; Bernal, F.J.; Caturla, J.F.; Carrascal, M.; Pagès, L.; Mir, M.; Espinosa, S.N.; Gràcia, J.; Domínguez, M. Discovery of a novel inhaled PI3Kδ inhibitor for the treatment of respiratory diseases. J. Med. Chem. 2018, 61, 9551–9567. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Keretsu, S.; Cho, S.J. 3D-QSAR, Docking and Molecular Dynamics Simulation Study of C-Glycosylflavones as GSK-3β Inhibitors. J. Chosun Nat. Sci. 2020, 13, 170–180. [Google Scholar]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Designing of the N-ethyl-4-(pyridin-4-yl)benzamide based potent ROCK1 inhibitors using docking, molecular dynamics, and 3D-QSAR. PeerJ 2021, 9, e11951. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller III, B.R.; McGee Jr, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- Bang, S.J.; Cho, S.J. Comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) study of mutagen X. Bull. Korean Chem. Soc. 2004, 25, 1525–1530. [Google Scholar]
- San Juan, A.A.; Cho, S.J. 3D-QSAR study of microsomal prostaglandin E 2 synthase (mPGES-1) inhibitors. J. Mol. Modeling 2007, 13, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “rm2” metrics for judging quality of quantitative structure–activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Sangion, A. A historical excursus on the statistical validation parameters for QSAR models: A clarification concerning metrics and terminology. J. Chem. Inf. Modeling 2016, 56, 1127–1131. [Google Scholar] [CrossRef]
- Todeschini, R.; Ballabio, D.; Grisoni, F. Beware of unreliable Q 2! A comparative study of regression metrics for predictivity assessment of QSAR models. J. Chem. Inf. Modeling 2016, 56, 1905–1913. [Google Scholar] [CrossRef]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Molecular Modeling Studies of N-phenylpyrimidine-4-amine Derivatives for Inhibiting FMS-like Tyrosine Kinase-3. Int. J. Mol. Sci. 2021, 22, 12511. [Google Scholar] [CrossRef]
- Abdizadeh, R.; Hadizadeh, F.; Abdizadeh, T. QSAR analysis of coumarin-based benzamides as histone deacetylase inhibitors using CoMFA, CoMSIA and HQSAR methods. J. Mol. Struct. 2020, 1199, 126961. [Google Scholar] [CrossRef]
- Gadhe, C.G.; Kothandan, G.; Cho, S.J. Large variation in electrostatic contours upon addition of steric parameters and the effect of charge calculation schemes in CoMFA on mutagenicity of MX analogues. Mol. Simul. 2012, 38, 861–871. [Google Scholar] [CrossRef]
- Gadhe, C.G.; Madhavan, T.; Kothandan, G.; Cho, S.J. In silico quantitative structure-activity relationship studies on P-gp modulators of tetrahydroisoquinoline-ethyl-phenylamine series. BMC Struct. Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Computational Modeling of Novel Phosphoinositol-3-kinase γ Inhibitors Using Molecular Docking, Molecular Dynamics, and 3D-QSAR. Bull. Korean Chem. Soc. 2021, 42, 1093–1111. [Google Scholar] [CrossRef]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peytam, F.; Adib, M.; Mahernia, S.; Rahmanian-Jazi, M.; Jahani, M.; Masoudi, B.; Mahdavi, M.; Amanlou, M. Isoindolin-1-one derivatives as urease inhibitors: Design, synthesis, biological evaluation, molecular docking and in-silico ADME evaluation. Bioorg. Chem. 2019, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.W.; Abdulai, R.; Mogemark, M.; Petersen, J.; Thomas, M.J.; Valastro, B.; Westin Eriksson, A. Evolution of PI3Kγ and δ inhibitors for inflammatory and autoimmune diseases. J. Med. Chem. 2018, 62, 4783–4814. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Thompson, P.E.; Gabelli, S.B. Structural determinants of isoform selectivity in PI3K inhibitors. Biomolecules 2019, 9, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somoza, J.R.; Koditek, D.; Villaseñor, A.G.; Novikov, N.; Wong, M.H.; Liclican, A.; Xing, W.; Lagpacan, L.; Wang, R.; Schultz, B.E. Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase δ. J. Biol. Chem. 2015, 290, 8439–8446. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docked Compounds with PI3Kγ | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C01 | C22 | C34 | C41 | C60 | C62 | C72 | C79 | C81 | C99 | C103 | C118 | C124 | C129 | C150 | C182 | C195 | C215 | |
| Docking Score (ΔG in kcal/mol) | −10.7 | −11.0 | −11.2 | −11.4 | −10.9 | 12.7 | −13.0 | −12.2 | −12.2 | −12.4 | −13.6 | −12.4 | −14.4 | −13.3 | −14.2 | −12.3 | −11.8 | −12.9 |
| RMSD (Å) from crystal ligands Ref. V81 (6xrm) | 2.03 | 2.55 | 0.91 | 2.63 | 2.64 | 2.44 | 2.74 | 1.90 | 1.19 | 2.44 | 1.90 | 2.52 | 2.41 | 1.99 | 2.38 | 1.65 | 1.89 | 1.61 |
| Number of H-bonds | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 2 | 4 | 2 | 5 | 2 | 3 | 2 | 3 | 4 |
| H-bond-interacting residues | V882 | V882, A885 | V882, K833 | V882, A885 | V882, A885 | V882, A885 | V882, K890 | V882, K833 | V882, K833 | V882 | V882, T887, K890, K833 | V882 | V882, K833, N951, K808 | V882 | V882, T887 | K833, V882 | K833, V882 | K833, V882, K890 |
| π–π interaction | Y867 | Y867, W812 | Y867 | W812 | Y867, W812 | Y867, W812 | W812 | Y867, W812 | W812 | W812 | Y867, W812 | Y867 | Y867, F965 | Y867, F965 | Y867, F965 | Y867 | Y867 | Y867 |
| π–Sigma bonding | I963, I879 | I963 | M804, M953, I879 | I963 | I963, M953 | I881 | I963 | I879, M953, I963 | I879, M953, I963 | M953, I963 | I879, M953, I963 | I963 | I879, I963 | I879, I963 | I879, M953, I963 | I879 | I879 | I879, M953 |
| π–sulfur interaction | M953, W812 | M804, M953 | - | M804, M963 | M804 | M953 | M804, M853 | - | M804 | M804 | - | M804, W812, M953 | M804, W812, M953 | M804, W812, M953 | M804, W812 | - | - | - |
| Complexes | MM-PB/GBSA Binding Energy Terms in kcal/mol | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| VDW (±SD) | EEL (±SD) | EPB/GB (±SD) | ESURF (±SD) | ΔGgas (±SD) | ΔGsolv (±SD) | ΔTOTAL (±SD) | TΔS (±SD) | ΔGbind (±SD) | |
| PI3Kγ-C01 | −54.40 ±2.82 | −35.03 ±3.67 | 39.06 ±2.67 | −6.63 ±0.12 | −89.45 ±3.54 | 32.43 ±2.65 | −57.01 ±2.76 | 8.61 ±0.20 | −48.40 ±2.77 |
| PI3Kγ-C22 | −52.53 ±3.26 | −23.77 ±8.87 | 37.55 ±6.11 | −6.88 ±0.29 | −76.29 ±8.82 | 30.67 ±6.03 | −45.62 ±4.72 | 23.88 ±4.90 | −21.73 ±6.81 |
| PI3Kγ-C34 | −60.86 ±3.22 | −48.55 ±6.02 | 60.26 ±5.18 | −7.42 ±0.21 | −109.43 ±5.60 | 52.83 ±5.21 | −56.59 ±3.68 | 13.38 ±0.02 | −43.21 ±3.68 |
| PI3Kγ-C41 | −48.66 ±3.13 | −20.19 ±5.85 | 26.4 3±4.05 | −5.99 ±0.26 | −68.86 ±5.24 | 20.43 ±3.96 | −48.42 ±3.83 | 17.93 ±0.94 | −48.42 ±3.83 |
| PI3Kγ-C60 | −52.67 ±2.85 | −39.47 ±5.83 | 45.74 ±4.32 | −6.64 ±0.15 | −92.15 ±5.40 | 39.10 ±4.35 | −53.05 ±3.30 | 18.87 ±0.04 | −34.17 ±3.34 |
| PI3Kγ-C62 | −61.42 ±3.02 | −34.43 ±3.54 | 49.40 ±2.66 | −7.33 ±0.22 | −95.87 ±3.98 | 42.07 ±2.59 | −53.80 ±2.92 | 7.76 ±1.46 | −46.03 ±3.61 |
| PI3Kγ-C72 | −69.47 ±3.48 | −34.14 ±6.81 | 51.64 ±4.65 | −7.90 ±0.24 | −103.62 ±6.68 | 43.74 ±4.60 | −59.88 ±3.70 | 15.19 ±3.55 | −44.69 ±5.61 |
| PI3Kγ-C79 | −65.21 ±2.96 | −38.13 ±4.28 | 48.45 ±2.83 | −7.70 ±0.15 | −103.36 ±4.23 | 40.75 ±2.81 | −62.61 ±3.24 | 9.29 ±0.61 | −53.31 ±3.30 |
| PI3Kγ-C81 | −59.05 ±3.00 | −34.53 ±6.75 | 44.62 ±5.08 | −7.04 ±0.26 | −93.61 ±6.73 | 37.58 ±4.97 | −56.03 ±3.71 | 8.69 ±0.04 | −47.33 ±3.71 |
| PI3Kγ-C99 | −58.25 ±3.44 | −56.56 ±7.52 | 67.51 ±6.90 | −7.59 ±6.90 | −114.81 ±8.76 | 59.91 ±6.69 | −54.89 ±3.82 | 17.28 ±0.59 | −37.61 ±3.86 |
| PI3Kγ-C103 | −61.05 ±2.90 | −46.84 ±4.40 | 56.49 ±3.64 | −7.70 ±0.21 | −107.91 ±4.53 | 48.79 ±3.62 | −59.11 ±2.95 | 11.67 ±2.06 | −47.44 ±3.60 |
| PI3Kγ-C118 | −59.50 ±2.88 | −51.98 ±5.11 | 65.58 ±4.12 | −7.23 ±0.26 | −111.49 ±4.24 | 58.34 ±4.07 | −53.14 ±3.13 | 13.39 ±0.04 | −39.75 ±3.13 |
| PI3Kγ-C124 | −60.09 ±3.19 | −61.38 ±10.17 | 76.25 ±8.99 | −7.12 ±0.23 | −121.47 ±10.99 | 69.12 ±8.89 | −52.34 ±3.71 | 10.34 ±1.59 | −41.58 ±4.67 |
| PI3Kγ-C129 | −59.87 ±3.12 | −36.29 ±5.69 | 61.99 ±5.14 | −7.99 ±0.29 | −96.18 ±5.95 | 54.01 ±5.11 | −42.18 ±3.49 | 6.68 ±0.04 | −35.50 ±3.49 |
| PI3Kγ-C150 | −72.71 ±3.14 | −66.26 ±5.44 | 79.80 ±4.40 | −8.84 ±0.20 | −138.97 ±5.47 | 79.80 ±4.40 | −68.02 ±3.56 | 10.66 ±0.78 | −57.35 ±3.65 |
| PI3Kγ-C182 | −59.35 ±2.95 | −33.72 ±6.10 | 47.78 ±5.09 | −7.09 ±0.32 | −93.07 ±5.77 | 40.69 ±5.14 | −52.38 ±3.18 | 14.68 ±0.05 | −37.70 ±3.18 |
| PI3Kγ-C195 | −41.90 ±3.22 | −42.54 ±4.11 | 61.18 ±3.83 | −7.94 ±0.21 | −104.96 ±4.54 | 53.23 ±3.84 | −51.73 ±3.22 | 9.83 ±0.03 | −41.90 ±3.22 |
| PI3Kγ-C215 | −63.11 ±3.48 | −64.01 ±6.16 | 73.01 ±4.90 | −7.84 ±0.31 | −127.13 ±6.60 | 65.16 ±4.84 | −61.97 ±4.01 | 14.29 ±0.04 | −37.68 ±4.01 |
| Compounds in Complex with PI3Kγ | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Residues | C01 | C22 | C34 | C41 | C60 | C62 | C72 | C79 | C81 | C99 | C103 | C118 | C124 | C129 | C150 | C182 | C195 | C215 |
| M804 | - | - | −1.04 | - | - | −0.72 | −3.33 | −2.66 | −1.56 | −2.01 | −3.68 | - | - | −1.79 | −2.16 | −1.21 | −1.32 | −1.18 |
| A805 | - | - | - | - | - | - | −1.20 | - | - | - | - | - | - | - | - | - | ||
| W812 | −0.72 | −0.54 | −0.86 | −0.71 | −0.85 | −1.03 | −1.59 | - | −0.89 | −0.98 | −0.87 | - | - | −0.79 | −1.01 | −1.01 | −0.92 | −0.85 |
| I831 | −1.70 | −1.72 | −2.18 | −1.61 | −1.77 | −1.63 | −1.82 | −1.83 | −1.72 | −1.92 | - | −2.12 | −1.23 | −2.28 | −2.15 | −1.83 | - | −2.06 |
| K833 | −3.10 | −1.86 | −1.34 | −1.40 | −2.62 | −1.93 | −1.33 | −1.72 | −1.55 | −1.61 | - | −2.52 | −2.35 | −2.04 | −2.59 | −2.14 | −1.37 | −1.74 |
| Y867 | −1.75 | - | −2.03 | −1.29 | - | −1.56 | −1.37 | −1.69 | −2.10 | −1.85 | - | −1.44 | −1.41 | - | −1.60 | −2.09 | −2.21 | −2.05 |
| I879 | −2.98 | −2.53 | −2.74 | −2.75 | −2.98 | −2.85 | −2.07 | −3.05 | −3.01 | −3.03 | −2.99 | −3.09 | −3.03 | −3.19 | −3.02 | −2.74 | −3.20 | −2.79 |
| I881 | −2.29 | −0.57 | −2.53 | −2.21 | −2.15 | −2.19 | −2.07 | −3.02 | −2.44 | −2.32 | −2.17 | −2.21 | −3.48 | −1.65 | −2.44 | −2.56 | −2.60 | −2.69 |
| V882 | −3.80 | - | −3.17 | −2.90 | −3.26 | −3.22 | −3.26 | −3.45 | −3.57 | −3.27 | −3.21 | −3.90 | −3.85 | - | −3.63 | −1.63 | −1.55 | −3.88 |
| T886 | - | −3.57 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | −0.83 |
| A885 | - | - | −0.96 | −1.20 | −1.41 | - | - | - | - | - | - | - | - | - | - | - | - | |
| M953 | −1.06 | −1.81 | −1.11 | −1.04 | −1.57 | −1.44 | −1.55 | −1.24 | −1.65 | −1.15 | −1.33 | −1.31 | −1.69 | −1.01 | −2.35 | −1.39 | −1.43 | −2.04 |
| I963 | −2.42 | −2.38 | −3.08 | −2.18 | −2.01 | −2.51 | −2.93 | −2.51 | −2.27 | −2.19 | −2.19 | −2.68 | −3.73 | −3.13 | −3.40 | −2.77 | −2.90 | −2.11 |
| Complexes | MM-PB/GBSA Binding Energy Terms in kcal/mol | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| VDW (±SD) | EEL (±SD) | EGB (±SD) | ESURF (±SD) | ΔGgas (±SD) | ΔGsolv (±SD) | ΔTOTAL (±SD) | TΔS (±SD) | ΔGbind (±SD) | |
| PI3Kδ-C190 | −49.96 ±2.96 | −30.85 ±3.91 | 35.11 ±3.07 | −6.36 ±0.17 | −80.83 ±3.62 | 28.74 ±3.12 | −52.08 ±3.35 | 10.89 ±0.04 | −41.19 ±3.35 |
| PI3Kδ-Idelalisib | −51.73 ±2.26 | −18.71 ±3.18 | 36.47 ±3.21 | −5.81 ±0.17 | −70.45 ±3.84 | 30.65 ±3.20 | −39.79 ±2.60 | 7.31 ±0.03 | −32.48 ±2.60 |
| PI3Kγ-Idelalisib | −40.74 ±3.61 | −12.50 ±5.78 | 28.81 ±5.53 | −4.51 ±0.35 | −53.25 ±7.20 | 24.29 ±5.36 | −28.95 ±3.79 | 12.47 ±1.86 | −16.48 ±4.22 |
| Complexes | Residues | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M752 | W760 | I777 | L784 | Y813 | I825 | V827 | V828 | T833 | M900 | I910 | |
| PI3Kδ-C190 | −0.96 | - | −1.65 | 0.87 | - | −3.00 | −2.44 | −4.23 | - | −1.05 | - |
| PI3Kδ-Idelalisib | −2.54 | −2.98 | −2.54 | - | −1.46 | - | −3.32 | −2.42 | −1.26 | −2.21 | −1.63 |
| PI3Kγ-Idelalisib | M804 | W812 | I831 | I881 | V882 | M953 | |||||
| −2.37 | −3.38 | −2.15 | −2.82 | −1.23 | −1.50 | ||||||
| 3D-QSAR (All Compounds) | Statistical Parameters | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| q2 | ONC | SEP | r2 | SEE | F-Value | BS-r2 | BS-SD | χ2 | RMSE | MAE | k | k’ | |r02-r’02| | |||
| CoMFA | 0.612 | 6 | 0.460 | 0.800 | 0.330 | 137.507 | 0.854 | 0.025 | 0.391 | 0.324 | <0.001 | 1.000 | 0.998 | 0.050 | 0.062 | 0.621 |
| CoMSIA (SEAD) | 0.630 | 6 | 0.448 | 0.784 | 0.344 | 123.686 | 0.833 | 0.024 | 0.446 | 0.338 | <0.001 | 1.000 | 0.998 | 0.060 | 0.079 | 0.588 |
| Threshold values | >0.5 | >0.6 | <<1 | >100 | <0.5 | <0.3 | 0.85 ≤ k ≤ 1.15 | 0.85 ≤ k’ ≤ 1.15 | <0.3 | <0.1 | >0.5 | |||||
| CoMFA (Training Set Compounds) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Statistical Parameters | SET-A | SET-B | SET-C | SET-D | Threshold Values | Statistical Parameters | SET-A | SET-B | SET-C | SET-D | Threshold Values |
| q2 | 0.655 | 0.608 | 0.598 | 0.540 | >0.5 | k Test | 1.008 | 0.997 | 1.002 | 1.006 | 0.85 ≤ k ≤ 1.15 |
| ONC | 6 | 6 | 6 | 5 | k’ Test | 0.995 | 0.999 | 0.994 | 0.990 | ||
| SEP | 0.456 | 0.462 | 0.498 | 0.488 | r2 Test | 0.684 | 0.566 | 0.586 | 0.710 | ||
| r2 | 0.854 | 0.842 | 0.831 | 0.762 | >0.6 | r02 Test | 0.640 | 0.566 | 0.522 | 0.699 | ≈r2 |
| SEE | 0.296 | 0.294 | 0.323 | 0.351 | <<1 | r’02 Test | 0.664 | 0.223 | 0.540 | 0.666 | |
| F-value | 148.372 | 134.954 | 124.355 | 97.725 | >100 | |r02 − r’02| Test | 0.024 | 0.343 | 0.018 | 0.033 | <0.3 |
| BS-r2 | 0.894 | 0.889 | 0.881 | 0.818 | Test | 0.064 | NA | 0.109 | 0.015 | <0.1 | |
| BS-SD | 0.021 | 0.021 | 0.021 | 0.033 | Test | 0.030 | 0.60 | 0.078 | 0.061 | ||
| χ2 | 0.227 | 0.221 | 0.269 | 0.256 | <1.0 | Test | 0.540 | NA | 0.437 | 0.635 | >0.5 |
| RMSE | 0.289 | 0.287 | 0.315 | 0.307 | <0.5 | Test | 0.587 | 0.234 | 0.460 | 0.561 | |
| MAE | <0.001 | <0.001 | <0.001 | <0.001 | ≈ 0 | Test | 0.563 | 0.117 | 0.448 | 0.598 | >0.5 |
| RSS | 13.335 | 13.120 | 15.83 | 15.02 | Δrm2 Test | 0.024 | 0.234 | 0.012 | 0.037 | <0.2 | |
| k Train | 0.999 | 1.000 | 1.000 | 1.000 | 0.85 ≤ k ≤ 1.15 | 0.635 | 0.565 | 0.522 | 0.694 | >0.5 | |
| k’Train | 0.998 | 0.998 | 0.998 | 0.998 | 0.635 | 0.565 | 0.522 | 0.694 | |||
| r02 Train | 0.854 | 0.841 | 0.830 | 0.810 | ≈r2 | 0.628 | 0.565 | 0.520 | 0.693 | ||
| r’02 Train | 0.829 | 0.812 | 0.796 | 0.766 | 0.635 | 0.565 | 0.520 | 0.694 | |||
| |r02-r’02|Train | 0.025 | 0.029 | 0.034 | 0.044 | <0.3 | 0.820 | 0.740 | 0.730 | 0.838 | ||
| Train | 0.029 | 0.035 | 0.042 | 0.052 | <0.1 | S (%) | 44.8 | 43.7 | 43.7 | 44.9 | |
| Train | 0.718 | 0.696 | 0.676 | 0.609 | >0.5 | E (%) | 55.2 | 56.3 | 56.3 | 55.1 | |
| CoMSIA (Training Set Compounds) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Statistical Parameters | SET-A | SET-B | SET-C | SET-D | Threshold Values | ||||||||
| SED | SEAD | SEHAD | SD | SED | SEAD | SD | SED | SEAD | SD | SEAD | SHAD | ||
| q2 | 0.653 | 0.655 | 0.652 | 0.604 | 0.608 | 0.597 | 0.607 | 0.610 | 0.603 | 0.568 | 0.566 | 0.581 | >0.5 |
| ONC | 6 | 5 | 6 | 6 | 5 | 6 | 6 | 5 | 6 | 6 | 6 | 6 | |
| SEP | 0.457 | 0.454 | 0.457 | 0.465 | 0.461 | 0.469 | 0.492 | 0.489 | 0.494 | 0.475 | 0.476 | 0.468 | |
| r2 | 0.817 | 0.804 | 0.824 | 0.763 | 0.788 | 0.789 | 0.775 | 0.788 | 0.814 | 0.754 | 0.790 | 0.796 | >0.6 |
| SEE | 0.332 | 0.342 | 0.324 | 0.360 | 0.339 | 0.339 | 0.372 | 0.360 | 0.338 | 0.359 | 0.331 | 0.326 | <<1 |
| F-value | 113.148 | 125.375 | 120.235 | 81.648 | 113.465 | 94.986 | 87.143 | 113.508 | 111.033 | 77.440 | 95.264 | 98.962 | >100 |
| BS- r2 | 0.867 | 0.842 | 0.878 | 0.813 | 0.825 | 0.842 | 0.824 | 0.824 | 0.868 | 0.801 | 0.848 | 0.852 | |
| BS-SD | 0.024 | 0.027 | 0.021 | 0.028 | 0.027 | 0.025 | 0.027 | 0.027 | 0.020 | 0.031 | 0.025 | 0.024 | |
| χ2 | 0.303 | 0.325 | 0.289 | 0.359 | 0.331 | 0.326 | 0.390 | 0.370 | 0.317 | 0.363 | 0.297 | 0.292 | <1.0 |
| RMSE | 0.324 | 0.335 | 0.316 | 0.351 | 0.332 | 0.331 | 0.364 | 0.353 | 0.330 | 0.350 | 0.322 | 0.318 | <0.5 |
| MAE | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | ≈0 |
| RSS | 16.725 | 17.935 | 15.915 | 19.65 | 17.628 | 17.478 | 21.078 | 19.865 | 17.338 | 19.56 | 16.680 | 16.178 | |
| k Train | 0.999 | 1.000 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 | 1.000 | 0.999 | 0.999 | 0.999 | 0.85 ≤ k ≤ 1.15 |
| k’Train | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | 0.998 | |
| r02 Train | 0.816 | 0.803 | 0.825 | 0.781 | 0.807 | 0.791 | 0.795 | 0.821 | 0.843 | 0.764 | 0.811 | 0.805 | ≈r2 |
| r’02 Train | 0.776 | 0.756 | 0.789 | 0.677 | 0.721 | 0.708 | 0.707 | 0.740 | 0.786 | 0.645 | 0.730 | 0.735 | |
| |r02-r’02|Train | 0.04 | 0.020 | 0.036 | 0.104 | 0.085 | 0.083 | 0.087 | 0.080 | 0.057 | 0.119 | 0.081 | 0.069 | <0.3 |
| Train | 0.050 | 0.063 | 0.042 | 0.112 | 0.084 | 0.100 | 0.087 | 0.060 | 0.034 | 0.143 | 0.075 | 0.075 | <0.1 |
| Train | 0.651 | 0.627 | 0.669 | 0.539 | 0.585 | 0.565 | 0.573 | 0.616 | 0.677 | 0.505 | 0.597 | 0.600 | >0.5 |
| kTest | 1.000 | 1.001 | 1.000 | 1.003 | 1.004 | 1.000 | 0.991 | 1.004 | 1.001 | 1.003 | 0.999 | 1.000 | 0.85 ≤ k ≤ 1.15 |
| k’Test | 0.997 | 0.996 | 0.996 | 0.992 | 0.992 | 0.995 | 1.006 | 0.993 | 0.996 | 0.993 | 0.998 | 0.996 | |
| r2 Test | 0.613 | 0.631 | 0.634 | 0.539 | 0.541 | 0.559 | 0.572 | 0.582 | 0.610 | 0.716 | 0.729 | 0.695 | |
| r02 Test | 0.592 | 0.609 | 0.609 | 0.533 | 0.537 | 0.556 | 0.524 | 0.555 | 0.580 | 0.717 | 0.729 | 0.695 | ≈r2 |
| r’02 Test | 0.529 | 0.562 | 0.572 | 0.332 | 0.330 | 0.341 | 0.512 | 0.494 | 0.540 | 0.642 | 0.648 | 0.604 | |
| |r02-r’02|Test | 0.063 | 0.047 | 0.037 | 0.201 | 0.207 | 0.215 | 0.012 | 0.060 | 0.031 | 0.075 | 0.081 | 0.090 | <0.3 |
| Test | 0.034 | 0.034 | 0.039 | 0.011 | 0.007 | 0.004 | 0.083 | 0.045 | 0.049 | - | - | - | <0.1 |
| Test | 0.137 | 0.109 | 0.097 | 0.384 | 0.389 | 0.389 | 0.103 | 0.149 | 0.101 | 0.102 | 0.110 | 0.129 | |
| Test | 0.524 | 0.537 | 0.533 | 0.498 | 0.510 | 0.530 | 0.447 | 0.487 | 0.504 | - | - | - | >0.5 |
| Test | 0.435 | 0.465 | 0.476 | 0.293 | 0.292 | 0.298 | 0.432 | 0.410 | 0.458 | 0.522 | 0.522 | 0.486 | |
| Test | 0.479 | 0.501 | 0.504 | 0.395 | 0.401 | 0.414 | 0.440 | 0.448 | 0.481 | - | - | - | |
| Δrm2Test | 0.089 | 0.072 | 0.057 | 0.204 | 0.217 | 0.231 | 0.014 | 0.077 | 0.046 | - | - | - | <0.2 |
| 0.599 | 0.615 | 0.616 | 0.526 | 0.529 | 0.554 | 0.517 | 0.546 | 0.577 | 0.713 | 0.729 | 0.693 | >0.5 | |
| 0.599 | 0.615 | 0.616 | 0.526 | 0.529 | 0.554 | 0.517 | 0.546 | 0.577 | 0.713 | 0.729 | 0.693 | ||
| 0.592 | 0.608 | 0.609 | 0.526 | 0.529 | 0.554 | 0.517 | 0.546 | 0.576 | 0.713 | 0.729 | 0.693 | ||
| 0.599 | 0.615 | 0.616 | 0.526 | 0.529 | 0.554 | 0.517 | 0.546 | 0.577 | 0.713 | 0.729 | 0.693 | ||
| 0.781 | 0.793 | 0.795 | 0.722 | 0.723 | 0.733 | 0.747 | 0.761 | 0.781 | 0.840 | 0.845 | 0.693 | ||
| S (%) | 20.6 | 15.9 | 13.5 | 33.6 | 23.1 | 23.3 | 32.1 | 21.4 | 15.3 | 30.5 | 15.3 | 15.7 | |
| E (%) | 35.8 | 26.4 | 23.6 | - | 37.4 | - | - | 34.7 | 25.7 | - | 23.5 | - | |
| H (%) | - | - | 14.7 | - | - | 39.2 | - | - | - | - | 20.9 | ||
| A (%) | - | 26.7 | 21.6 | - | - | - | - | - | 24.4 | - | 25.7 | 28.4 | |
| D (%) | 43.6 | 31.0 | 26.6 | 66.4 | 39.4 | 47.2 | 67.9 | 43.9 | 34.6 | 69.5 | 35.4 | 35.1 | |
| Components | CoMFA | CoMSIA (SEHAD) | ||||
|---|---|---|---|---|---|---|
| SET-A | SET-A | |||||
| Q2 | cSDEP | dq2/dr2yy’ | Q2 | cSDEP | dq2/dr2yy’ | |
| 1 | 0.083 | 0.730 | 0.095 | 0.245 | 0.662 | 0.179 |
| 2 | 0.347 | 0.618 | 0.407 | 0.329 | 0.626 | 0.306 |
| 3 | 0.380 | 0.608 | 0.376 | 0.451 | 0.568 | 0.533 |
| 4 | 0.405 | 0.593 | 0.565 | 0.480 | 0.555 | 0.568 |
| 5 | 0.400 | 0.599 | 0.502 | 0.507 | 0.542 | 0.735 |
| 6 | 0.433 | 0.581 | 0.590 | 0.508 | 0.541 | 0.757 |
| 7 | 0.333 | 0.634 | 0.681 | 0.489 | 0.555 | 0.787 |
| 8 | 0.315 | 0.644 | 0.543 | 0.462 | 0.571 | 0.874 |
| Statistical Parameters | CoMFA | CoMSIA (SA) | Threshold Values | Statistical Parameters | CoMFA | CoMSIA | Threshold Values |
|---|---|---|---|---|---|---|---|
| SET-D | SET-D | SET-D | SET-D | ||||
| q2 | 0.547 | 0.537 | >0.5 | r2 Test | 0.627 | 0.577 | >0.5 |
| ONC | 6 | 5 | r02 Test | 0.616 | 0.566 | ||
| SEP | 0.653 | 0.658 | r’02 Test | 0.501 | 0.349 | ||
| r2 | 0.699 | 0.680 | >0.6 | |r02 − r’02|Test | 0.114 | 0.217 | <0.3 |
| SEE | 0.532 | 0.556 | <<1 | Test | 0.017 | 0.018 | <0.1 |
| F-value | 58.178 | 52.427 | Test | 0.199 | 0.394 | ||
| BS- r2 | 0.756 | 0.713 | Test | 0.561 | 0.517 | ||
| BS-SD | 0.039 | 0.042 | Test | 0.405 | 0.301 | ||
| χ2 | 0.631 | 0.683 | <1.0 | Test | 0.156 | 0.215 | |
| RMSE | 0.482 | 0.495 | <0.5 | Δrm2Test | 0.483 | 0.409 | |
| MAE | <0.001 | <0.001 | ≈0 | 0.615 | 0.562 | >0.5 | |
| RSS | 36.61 | 38.59 | 0.615 | 0.562 | |||
| k Train | 1.001 | 1.001 | 0.85 ≤ k ≤ 1.15 | 0.615 | 0.562 | ||
| k’Train | 0.994 | 0.995 | 0.615 | 0.562 | |||
| r02 Train | 0.742 | 0.728 | ≈r2 | 0.785 | 0.744 | ||
| r’02 Train | 0.633 | 0.615 | S (%) | 76.2 | 47.9 | ||
| |r02 − r’02|Train | 0.109 | 0.112 | <0.3 | E (%) | 23.8 | - | |
| Train | 0.093 | 0.094 | <0.1 | A (%) | - | 52.1 | |
| Train | 0.520 | 0.507 | >0.5 | ||||
| kTest | 0.989 | 0.985 | 0.85 ≤ k ≤ 1.15 | ||||
| k’Test | 1.005 | 1.008 |
| Components | CoMFA | CoMSIA (SA) | ||||
|---|---|---|---|---|---|---|
| SET-A | SET-A | |||||
| Q2 | cSDEP | dq2/dr2yy’ | Q2 | cSDEP | dq2/dr2yy’ | |
| 1 | 0.380 | 0.750 | 0.229 | 0.388 | 0.746 | 0.160 |
| 2 | 0.410 | 0.735 | 0.191 | 0.432 | 0.726 | 0.247 |
| 3 | 0.434 | 0.722 | 0.257 | 0.457 | 0.707 | 0.271 |
| 4 | 0.468 | 0.709 | 0.234 | 0.463 | 0.706 | 0.299 |
| 5 | 0.467 | 0.702 | 0.239 | 0.471 | 0.702 | 0.380 |
| 6 | 0.475 | 0.702 | 0.377 | 0.448 | 0.719 | 0.386 |
| 7 | 0.458 | 0.716 | 0.372 | 0.441 | 0.727 | 0.426 |
| 8 | 0.422 | 0.742 | 0.402 | 0.436 | 0.733 | 0.760 |
| Complexes | Compound’s SA Score (1–10 Scale) | MM-PB/GBSA Binding Energy Terms in kcal/mol | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VDW (±SD) | EEL (±SD) | EGB (±SD) | ESURF (±SD) | ΔGgas (±SD) | ΔGsolv (±SD) | ΔTOTAL (±SD) | TΔS (±SD) | ΔGbind (±SD) | ||
| PI3Kγ-D21 | 5.21 | −74.71 ±3.49 | −63.37 ±7.34 | 79.14 ±5.06 | −9.34 ±0.28 | −138.07 ±7.70 | 69.80 ±5.07 | −68.27 ±4.51 | 4.89 ±0.05 | −63.37 ±4.51 |
| PI3Kγ-D22 | 5.24 | −70.91 ±3.32 | −32.79 ±4.35 | 43.05 ±4.38 | −8.46 ±0.29 | −103.68 ±5.15 | 34.58 ±4.31 | −69.10 ±3.49 | 12.16 ±0.04 | −56.93 ±3.49 |
| PI3Kγ-D23 | 6.49 | −80.03 ±3.06 | −32.53 ±5.05 | 57.58 ±3.75 | −9.84 ±0.31 | −112.55 ±5.62 | 47.74 ±3.75 | −64.81 ±4.00 | 10.04 ±0.04 | −54.76 ±4.00 |
| PI3Kγ-D24 | 6.49 | −76.56 ±2.74 | −30.97 ±4.42 | 51.69 ±3.94 | −7.99 ±0.32 | −107.53 ±5.00 | 43.70 ±3.88 | −63.83 ±2.73 | 8.26 ±0.07 | −55.57 ±2.74 |
| PI3Kγ-D25 | 6.29 | −74.62 ±3.43 | −41.99 ±5.22 | 62.27 ±5.12 | −9.07 ±0.23 | −116.60 ±5.81 | 53.19 ±5.04 | −63.40 ±3.79 | 10.38 ±0.05 | −53.01 ±3.79 |
| PI3Kγ-D81 | 5.90 | −75.02 ±3.51 | −57.42 ±4.84 | 74.08 ±3.66 | −9.02 ±0.27 | −132.43 ±4.94 | 65.06 ±3.69 | −67.37 ±4.07 | 14.29 ±1.08 | −53.08 ±4.21 |
| PI3Kγ-D82 | 5.90 | −76.94 ±3.66 | −60.04 ±5.31 | 75.03 ±3.68 | −8.49 ±0.33 | −126.98 ±5.36 | 66.54 ±3.70 | −70.43 ±3.96 | 11.51 ±0.04 | −58.92 ±3.96 |
| PI3Kγ-D83 | 6.25 | −79.62 ±3.20 | −49.95 ±5.07 | 63.36 ±4.33 | −9.47 ±0.22 | −129.56 ±5.40 | 53.89 ±4.31 | −75.66 ±3.82 | 9.58 ±0.05 | −66.08 ±3.82 |
| PI3Kγ-D84 | 6.24 | −71.58 ±3.28 | −55.07 ±4.53 | 72.55 ±3.45 | −9.38 ±0.30 | −126.65 ±4.67 | 63.17 ±3.48 | −63.47 ±3.48 | 11.45 ±1.33 | −52.02 ±3.72 |
| PI3Kγ-D85 | 6.16 | −74.15 ±2.92 | −55.01 ±4.98 | 73.14 ±4.18 | −9.04 ±0.22 | −129.15 ±4.81 | 64.10 ±4.19 | −65.05 ±3.44 | 6.69 ±0.03 | −58.36 ±3.44 |
| PI3Kγ-D87 | 5.82 | −71.01 ±3.59 | −49.86 ±4.95 | 67.38 ±3.96 | −8.83 ±0.24 | −120.88 ±5.87 | 58.54 ±3.87 | −62.33 ±3.54 | 11.61 ±1.81 | −50.71 ±3.98 |
| PI3Kδ-D25 | 6.29 | −59.63 ±2.58 | −37.07 ±7.05 | 57.23 ±5.88 | −8.70 ±0.23 | −96.70 ±6.53 | 48.52 ±5.93 | −48.17 ±2.79 | 16.20 ±0.05 | −31.97 ±2.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, S.; Cho, S.J. Structural Insights from Molecular Modeling of Isoindolin-1-One Derivatives as PI3Kγ Inhibitors against Gastric Carcinoma. Biomedicines 2022, 10, 813. https://doi.org/10.3390/biomedicines10040813
Ghosh S, Cho SJ. Structural Insights from Molecular Modeling of Isoindolin-1-One Derivatives as PI3Kγ Inhibitors against Gastric Carcinoma. Biomedicines. 2022; 10(4):813. https://doi.org/10.3390/biomedicines10040813
Chicago/Turabian StyleGhosh, Suparna, and Seung Joo Cho. 2022. "Structural Insights from Molecular Modeling of Isoindolin-1-One Derivatives as PI3Kγ Inhibitors against Gastric Carcinoma" Biomedicines 10, no. 4: 813. https://doi.org/10.3390/biomedicines10040813
APA StyleGhosh, S., & Cho, S. J. (2022). Structural Insights from Molecular Modeling of Isoindolin-1-One Derivatives as PI3Kγ Inhibitors against Gastric Carcinoma. Biomedicines, 10(4), 813. https://doi.org/10.3390/biomedicines10040813