
Expression Profile of CD157 Reveals Functional Heterogeneity of Capillaries in Human Dermal Skin

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture
2.2. Cell Preparation for Flow Cytometric Analysis and Cell Sorting (FACS)
2.3. Immune Cell Binding Assay In Vitro
2.4. Clonogenic Assay
2.5. Treatment of EC with Cytokines In Vitro
2.6. Immunohistochemical Staining
2.7. Quantification of CD157 Expression on Blood and Lymphatic Capillaries
2.8. Preparation of vascDESS and Non-vascDESS
2.9. Transplantation of Tissue-Engineered Skin Substitutes
2.10. Statistical Analysis
3. Results
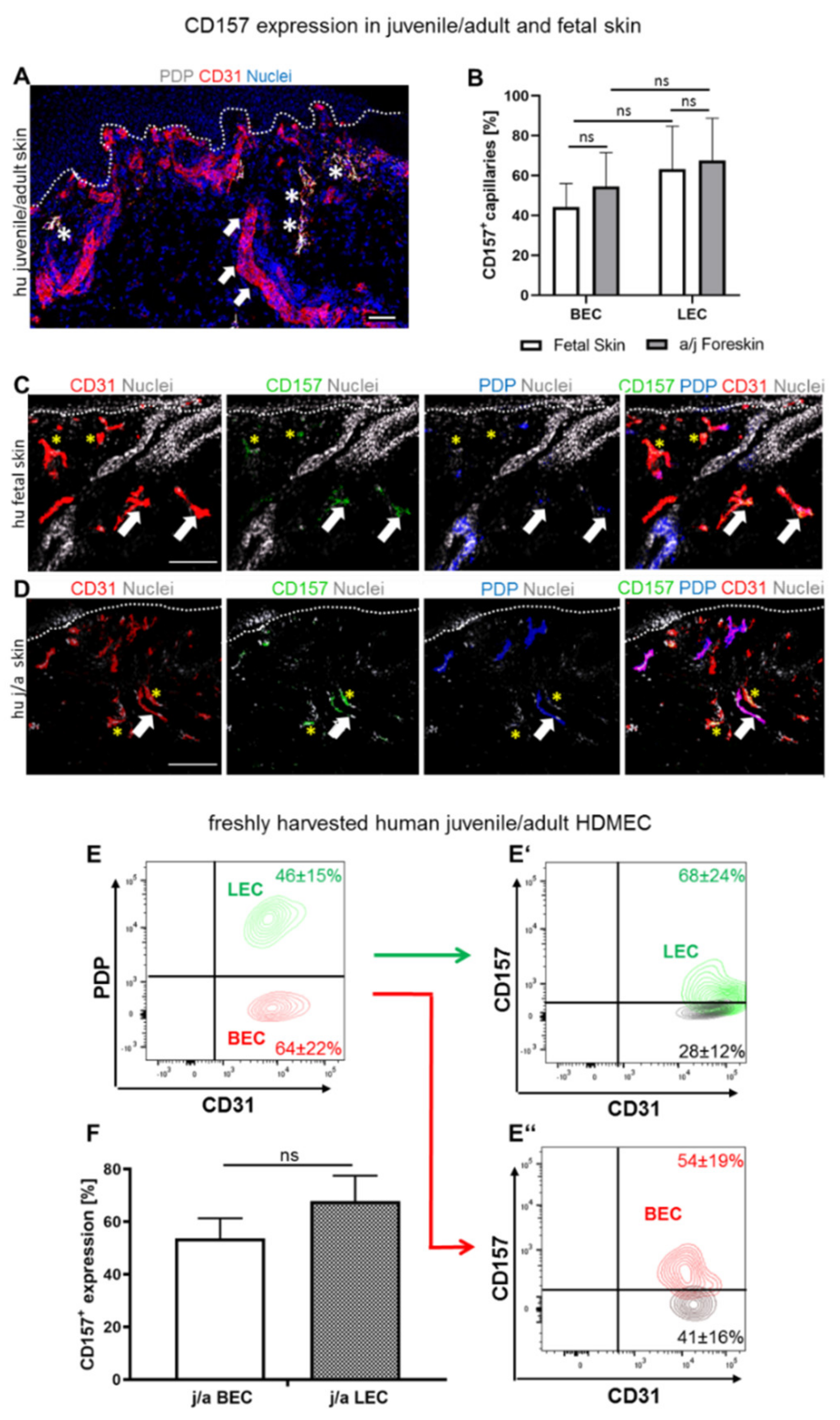
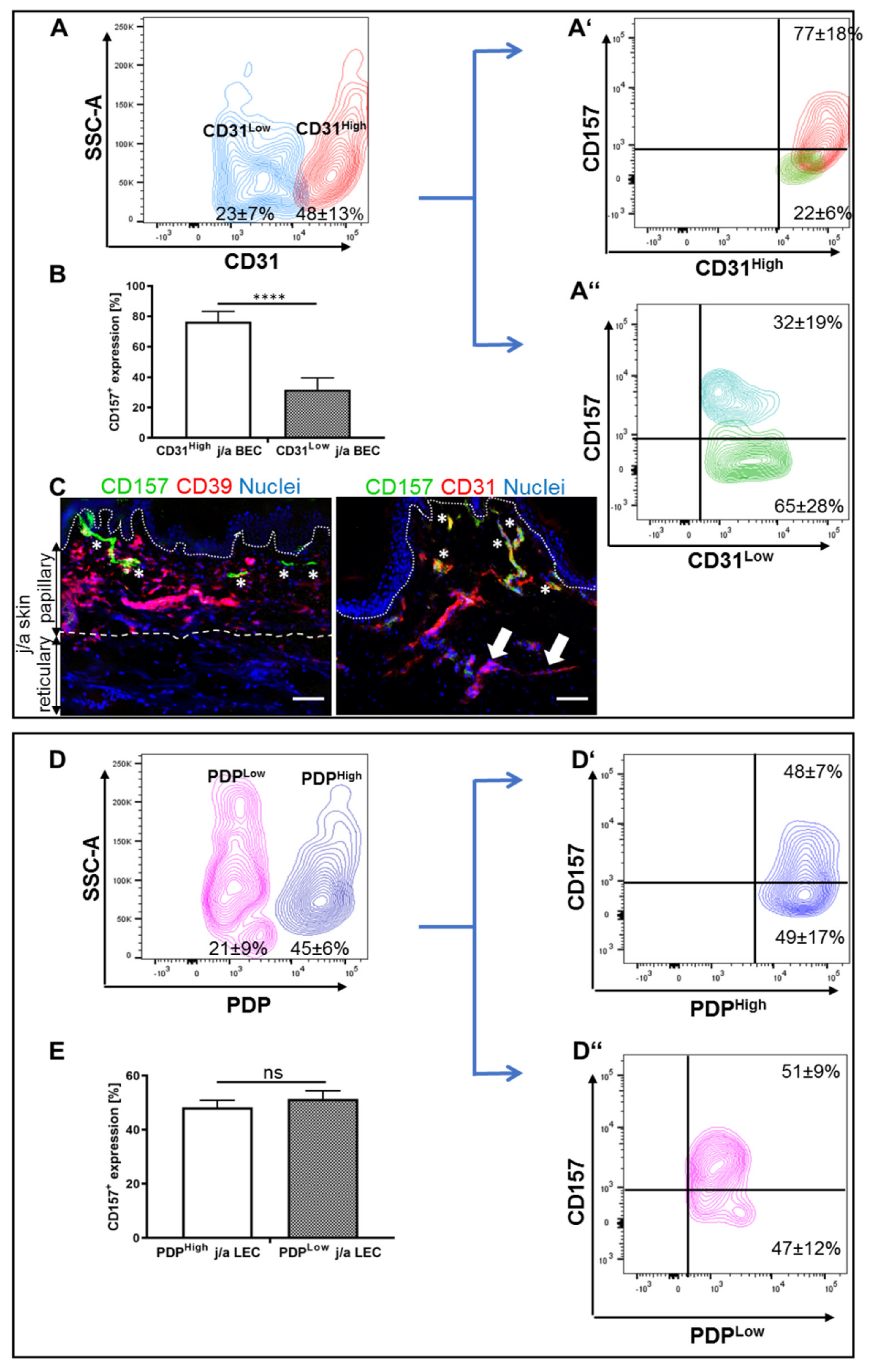
3.1. Both Blood and Lymphatic Capillaries of Juvenile/Adult and Fetal Skin Express CD157
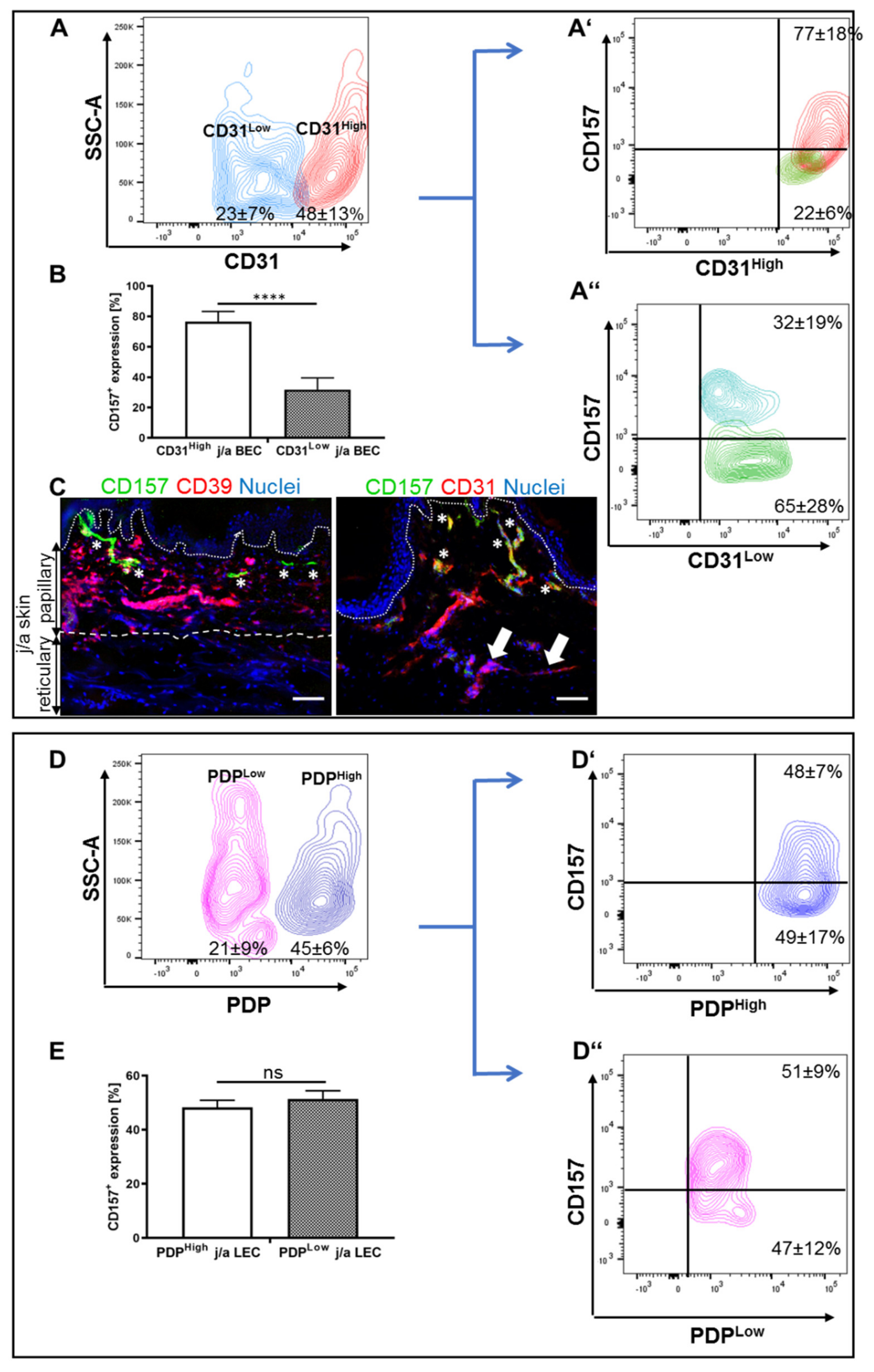
3.2. CD31Low and CD31High j/a HDMEC Differ in Their Levels of CD157 Expression
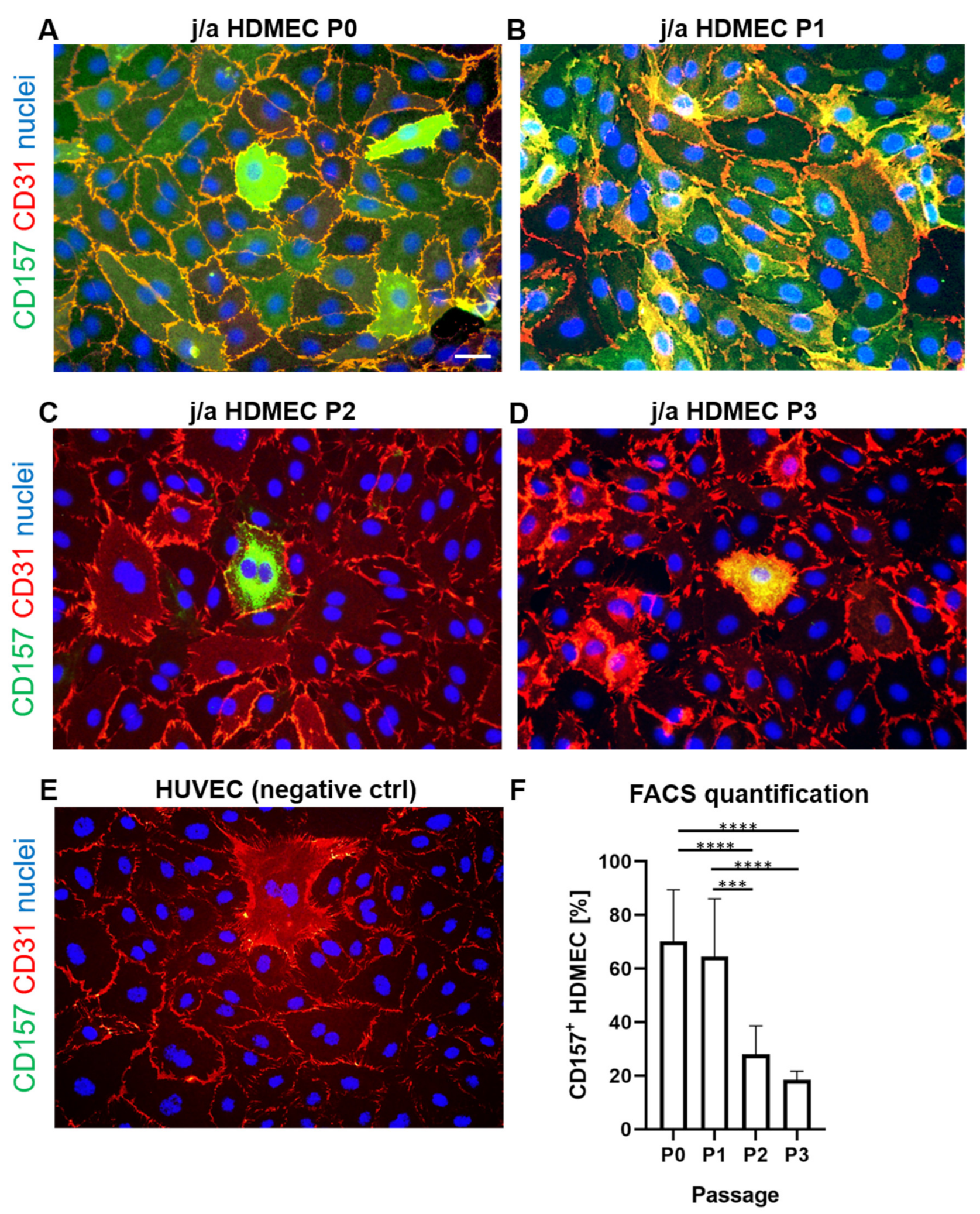
3.3. CD157 Expression Is Reduced after Prolonged Culturing In Vitro
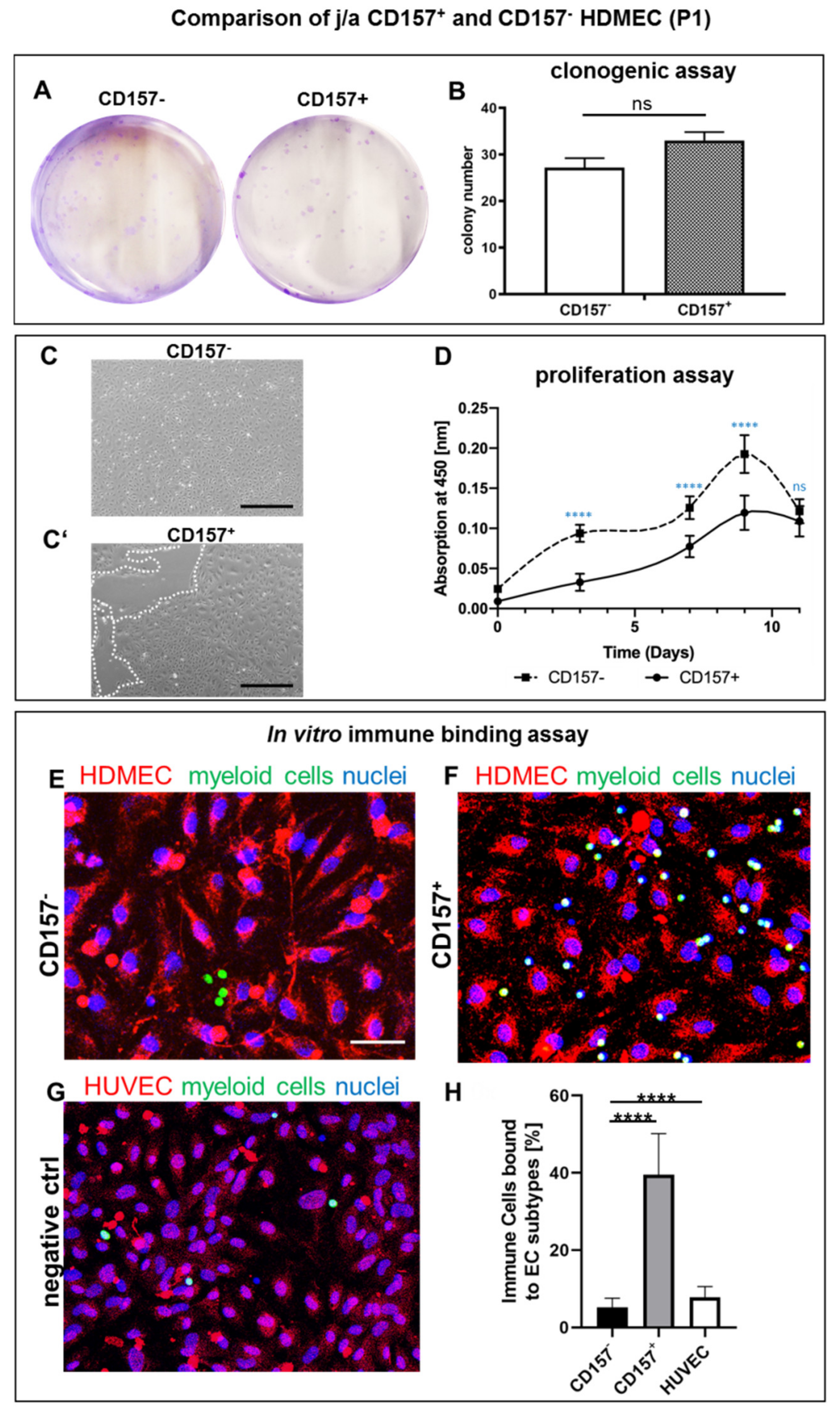
3.4. Proliferation and Clonal Expansion of CD157+ and CD157− HDMEC In Vitro
3.5. Myeloid Cells Expressing CD11b Adhere Specifically to CD157+ HDMEC In Vitro
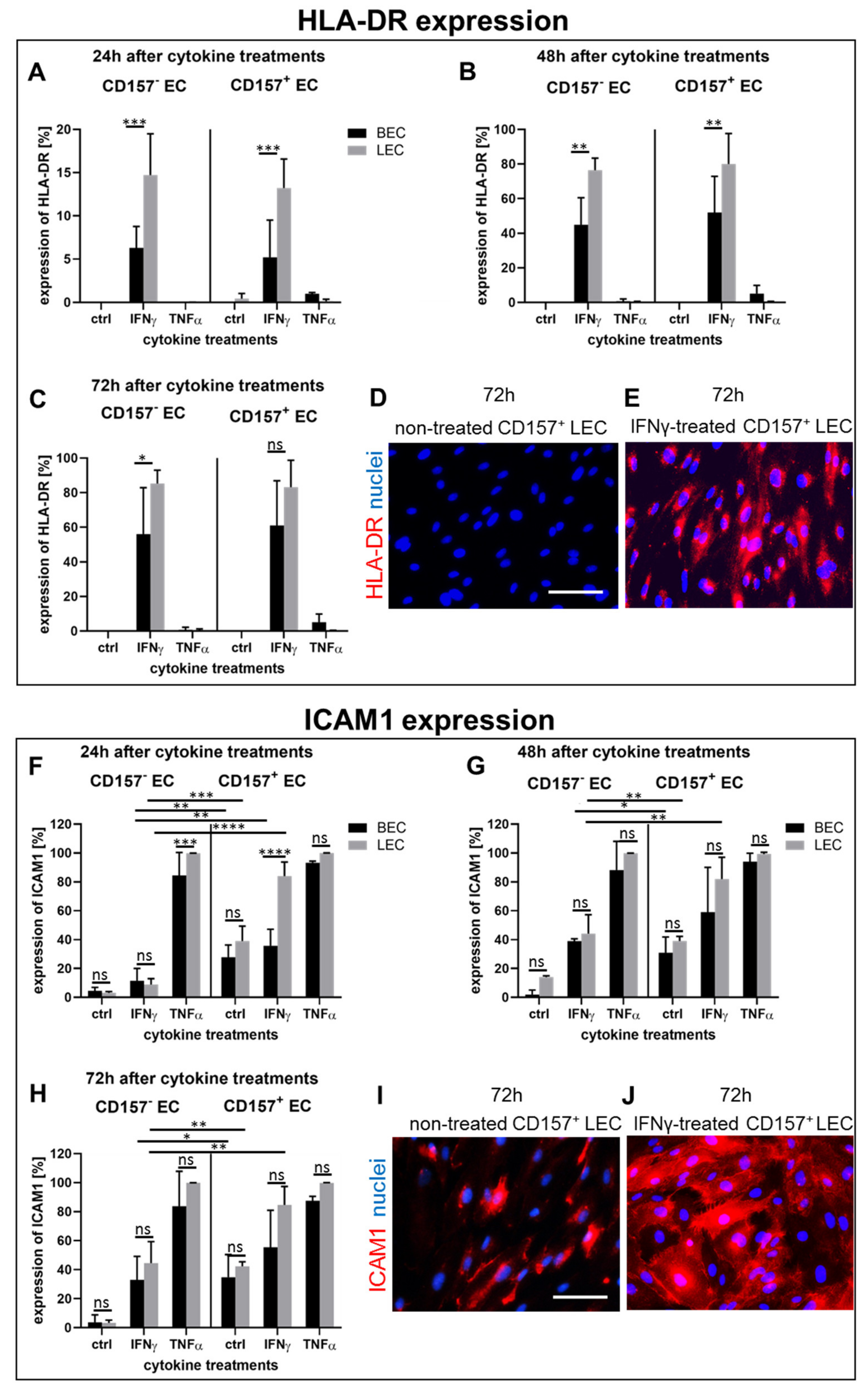
3.6. Pro-Inflammatory Cytokines Stimulate Differently HLA-DR and ICAM1 Expression in CD157− and CD157+ BEC/LEC Populations In Vitro
3.7. CD157 Interacts with Myeloid Cells in Human Skin and Skin Substitutes In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lavagno, L.; Ferrero, E.; Ortolan, E.; Malavasi, F.; Funaro, A. CD157 is part of a supramolecular complex with CD11b/CD18 on the human neutrophil cell surface. J. Biol. Regul. Homeost. Agents 2007, 21, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ortolan, E.; Augeri, S.; Fissolo, G.; Musso, I.; Funaro, A. CD157: From immunoregulatory protein to potential therapeutic target. Immunol. Lett. 2019, 205, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morone, S.; Augeri, S.; Cuccioloni, M.; Mozzicafreddo, M.; Angeletti, M.; Buono, N.L.; Giacomino, A.; Ortolan, E.; Funaro, A. Binding of CD157 Protein to Fibronectin Regulates Cell Adhesion and Spreading. J. Biol. Chem. 2014, 289, 15588–15601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortolan, E.; Tibaldi, E.V.; Ferranti, B.; Lavagno, L.; Garbarino, G.; Notaro, R.; Luzzatto, L.; Malavasi, F.; Funaro, A. CD157 plays a pivotal role in neutrophil transendothelial migration. Blood 2006, 108, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Naito, H.; Suehiro, J.; Lin, Y.; Kawaji, H.; Iba, T.; Kouno, T.; Ishikawa-Kato, S.; Furuno, M.; Takara, K.; et al. CD157 Marks Tissue-Resident Endothelial Stem Cells with Homeostatic and Regenerative Properties. Cell Stem Cell 2018, 22, 384–397. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Campo, P.M.; Almeida, J.; Sánchez, M.L.; Malvezzi, M.; Orfao, A. Normal patterns of expression of glycosylphosphatidylinositol-anchored proteins on different subsets of peripheral blood cells: A frame of reference for the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytom. Part B Clin. Cytom. 2006, 70B, 71–81. [Google Scholar] [CrossRef]
- Lo Buono, N.; Parrotta, R.; Morone, S.; Bovino, P.; Nacci, G.; Ortolan, E.; Horenstein, A.L.; Inzhutova, A.; Ferrero, E.; Funaro, A. The CD157-Integrin Partnership Controls Transendothelial Migration and Adhesion of Human Monocytes. J. Biol. Chem. 2011, 286, 18681–18691. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 12, 5379. [Google Scholar] [CrossRef]
- Coxon, A.; Rieu, P.; Barkalow, F.J.; Askari, S.; Sharpe, A.H.; Von Andrian, U.H.; Arnaout, M.A.; Mayadas, T.N. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity 1996, 5, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Farhood, A.; Bautista, A.P.; Spolarics, Z.; Spitzer, J.J.; Smith, C.W. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology 1993, 17, 915–923. [Google Scholar] [CrossRef]
- Rosetti, F.; Mayadas, T.N. The many faces of Mac-1 in autoimmune disease. Immunol. Rev. 2016, 269, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Funaro, A.; Ortolan, E.; Ferranti, B.; Gargiulo, L.; Notaro, R.; Luzzatto, L.; Malavasi, F. CD157 is an important mediator of neutrophil adhesion and migration. Blood 2004, 104, 4269–4278. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Talbott, G.A.; Juliano, R.L. Integrin-mediated signaling events in human endothelial cells. Mol. Biol. Cell 1998, 9, 1969–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Welser, J.V.; Milner, R. Absence of the alpha v beta 3 integrin dictates the time-course of angiogenesis in the hypoxic central nervous system: Accelerated endothelial proliferation correlates with compensatory increases in alpha 5 beta 1 integrin expression. J. Cereb. Blood Flow Metab. 2010, 30, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.C.; Clark, R.A.F.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Yakymiv, Y.; Augeri, S.; Bracci, C.; Marchisio, S.; Aydin, S.; D’Ardia, S.; Massaia, M.; Ferrero, E.; Ortolan, E.; Funaro, A. CD157 signaling promotes survival of acute myeloid leukemia cells and modulates sensitivity to cytarabine through regulation of anti-apoptotic Mcl-1. Sci. Rep. 2021, 11, 21230. [Google Scholar] [CrossRef]
- Chang, H.H.; Bennett, A.M.; Jin, Z.G. A Novel Role of Vascular Endothelial Cadherin in Modulating c-Src Activation and Downstream Signaling of Vascular Endothelial Growth Factor. J. Biol. Chem. 2008, 283, 7261. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Atri, D.; Eichmann, A.; Simons, M. Endothelial ERK signaling controls lymphatic fate specification. J. Clin. Invest. 2013, 123, 1202. [Google Scholar] [CrossRef]
- Naito, H.; Kidoya, H.; Sakimoto, S.; Wakabayashi, T.; Takakura, N. Identification and characterization of a resident vascular stem/progenitor cell population in preexisting blood vessels. EMBO J. 2012, 31, 1202–1215. [Google Scholar] [CrossRef] [Green Version]
- Klar, A.S.; Güven, S.; Biedermann, T.; Luginbühl, J.; Böttcher-Haberzeth, S.; Meuli-Simmen, C.; Meuli, M.; Martin, I.; Scherberich, A.; Reichmann, E. Tissue-engineered dermo-epidermal skin grafts prevascularized with adipose-derived cells. Biomaterials 2014, 35, 842–855. [Google Scholar] [CrossRef]
- Marino, D.; Luginbühl, J.; Scola, S.; Meuli, M.; Reichmann, E. Bioengineering Dermo-Epidermal Skin Grafts with Blood and Lymphatic Capillaries. Sci. Transl. Med. 2014, 6, 221ra14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, N.; Haluza, D.; Gurnhofer, E.; Raab, I.; Kasimir, M.T.; Prinz, M.; Steiner, C.W.; Reinisch, C.; Howorka, A.; Giovanoli, P.; et al. Lymphatic precollectors contain a novel, specialized subpopulation of podoplaninlow, CCL27-expressing lymphatic endothelial cells. Am. J. Pathol. 2008, 173, 1202–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.S.; Rissiek, A.; Priefler, M.; Schwedhelm, E.; Robbe, L.; Bauer, A.; Zahrte, C.; Zoellner, C.; Kluge, S.; Nierhaus, A. Human leucocyte antigen (HLA-DR) gene expression is reduced in sepsis and correlates with impaired TNFα response: A diagnostic tool for immunosuppression? PLoS ONE 2017, 12, e0182427. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Asens, M.L.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Naito, H.; Shimizu, S.; Rahmawati, F.N.; Wakabayashi, T.; Takakura, N. Isolation of tissue-resident endothelial stem cells and their use in regenerative medicine. Inflamm. Regen. 2019, 39, 9. [Google Scholar] [CrossRef]
- Boxall, S.A.; Jones, E. Markers for characterization of bone marrow multipotential stromal cells. Stem Cells Int. 2012, 2012, 975871. [Google Scholar] [CrossRef] [Green Version]
- Thitilertdecha, P.; Lohsiriwat, V.; Poungpairoj, P.; Tantithavorn, V.; Onlamoon, N. Extensive Characterization of Mesenchymal Stem Cell Marker Expression on Freshly Isolated and in Vitro Expanded Human Adipose-Derived Stem Cells from Breast Cancer Patients. Stem Cells Int. 2020, 2020, 8237197. [Google Scholar] [CrossRef]
- Newman, P.J.; Newman, D.K. Signal Transduction Pathways Mediated by PECAM-1. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 953–964. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, Z.; Li, Q.; Lin, Y.; Dang, E.; Meng, H.; Sha, N.; Bai, H.; Wang, G.; An, S.; et al. Neutrophils Enhance Cutaneous Vascular Dilation and Permeability to Aggravate Psoriasis by Releasing Matrix Metallopeptidase 9. J. Investig. Dermatol. 2021, 141, 787–799. [Google Scholar] [CrossRef]
- Avena-Woods, C. Overview of Atopic Dermatitis. Am. J. Manag. Care 2017, 23, 115–123. [Google Scholar] [CrossRef]
- Sabat, R.; Wolk, K.; Loyal, L.; Döcke, W.D.; Ghoreschi, K. T cell pathology in skin inflammation. Semin. Immunopathol. 2019, 41, 359–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proksch, E.; Brandner, J.M.; Jensen, J.-M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Millikan, L. The proposed inflammatory pathophysiology of rosacea: Implications for treatment. Skinmed 2003, 2, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Kardaun, S.H.; Kuiper, H.; Fidler, V.; Jonkman, M.F. The histopathological spectrum of acute generalized exanthematous pustulosis (AGEP) and its differentiation from generalized pustular psoriasis. J. Cutan. Pathol. 2010, 37, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Van Hatem, S.; Beerthuizen, G.I.; Kardaun, S.H. Severe flucloxacillin-induced acute generalized exanthematous pustulosis (AGEP), with toxic epidermal necrolysis (TEN)-like features: Does overlap between AGEP and TEN exist? Clinical report and review of the literature. Br. J. Dermatol. 2014, 171, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Chen, J.; Lin, Y.; Zhang, C.; Li, W.; Qiao, H.; Fu, M.; Dang, E.; Wang, G. Aryl Hydrocarbon Receptor in Cutaneous Vascular Endothelial Cells Restricts Psoriasis Development by Negatively Regulating Neutrophil Recruitment. J. Investig. Dermatol. 2020, 140, 1233–1243e9. [Google Scholar] [CrossRef]
- Johnson, L.A.; Clasper, S.; Holt, A.P.; Lalor, P.F.; Baban, D.; Jackson, D.G. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J. Exp. Med. 2006, 203, 2763. [Google Scholar] [CrossRef] [Green Version]
- Rice, T.W.; Bernard, G.R. Therapeutic intervention and targets for sepsis. Annu. Rev. Med. 2005, 56, 225–248. [Google Scholar] [CrossRef]
- Doukas, J.; Pober, J.S. IFN-gamma enhances endothelial activation induced by tumor necrosis factor but not IL-1. J. Immunol. 1990, 145, 225–248. [Google Scholar]
- Batten, P.; Yacoub, M.H.; Rose, M.L. Effect of human cytokines (IFN-gamma, TNF-alpha, IL-1 beta, IL-4) on porcine endothelial cells: Induction of MHC and adhesion molecules and functional significance of these changes. Immunology 1996, 87, 127. [Google Scholar]
- Yamamoto, T.; Matsuuchi, M.; Watanabe, K.; Katayama, I.; Nishioka, K. Correlation of soluble ICAM-1 and E-selectin in the peripheral blood of patients with generalized pustular psoriasis and their immunohistochemical localization. Eur. J. Dermatol. 2000, 7, 89–92. [Google Scholar]
- Gröger, M.; Niederleithner, H.; Kerjaschki, D.; Petzelbauer, P. A Previously Unknown Dermal Blood Vessel Phenotype in Skin Inflammation. J. Investig. Dermatol. 2007, 127, 2893–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalak-Micka, K.; Rütsche, D.; Johner, L.; Moehrlen, U.; Biedermann, T.; Klar, A.S. Expression Profile of CD157 Reveals Functional Heterogeneity of Capillaries in Human Dermal Skin. Biomedicines 2022, 10, 676. https://doi.org/10.3390/biomedicines10030676
Michalak-Micka K, Rütsche D, Johner L, Moehrlen U, Biedermann T, Klar AS. Expression Profile of CD157 Reveals Functional Heterogeneity of Capillaries in Human Dermal Skin. Biomedicines. 2022; 10(3):676. https://doi.org/10.3390/biomedicines10030676
Chicago/Turabian StyleMichalak-Micka, Katarzyna, Dominic Rütsche, Lukas Johner, Ueli Moehrlen, Thomas Biedermann, and Agnes S. Klar. 2022. "Expression Profile of CD157 Reveals Functional Heterogeneity of Capillaries in Human Dermal Skin" Biomedicines 10, no. 3: 676. https://doi.org/10.3390/biomedicines10030676
APA StyleMichalak-Micka, K., Rütsche, D., Johner, L., Moehrlen, U., Biedermann, T., & Klar, A. S. (2022). Expression Profile of CD157 Reveals Functional Heterogeneity of Capillaries in Human Dermal Skin. Biomedicines, 10(3), 676. https://doi.org/10.3390/biomedicines10030676