
An In Vivo Inflammatory Loop Potentiates KRAS Blockade

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs
2.3. Cellular Assays
2.4. Western Immunoblotting
2.5. Constructs
2.6. Mice
2.7. In Vivo Tumor Models and Drug Treatments
2.8. Microarrays, PCR, GSEA, and Kaplan-Meier Analyses
2.9. ELISA
2.10. Immunofluorescence
2.11. Bone marrow replacement
2.12. Statistics
3. Results
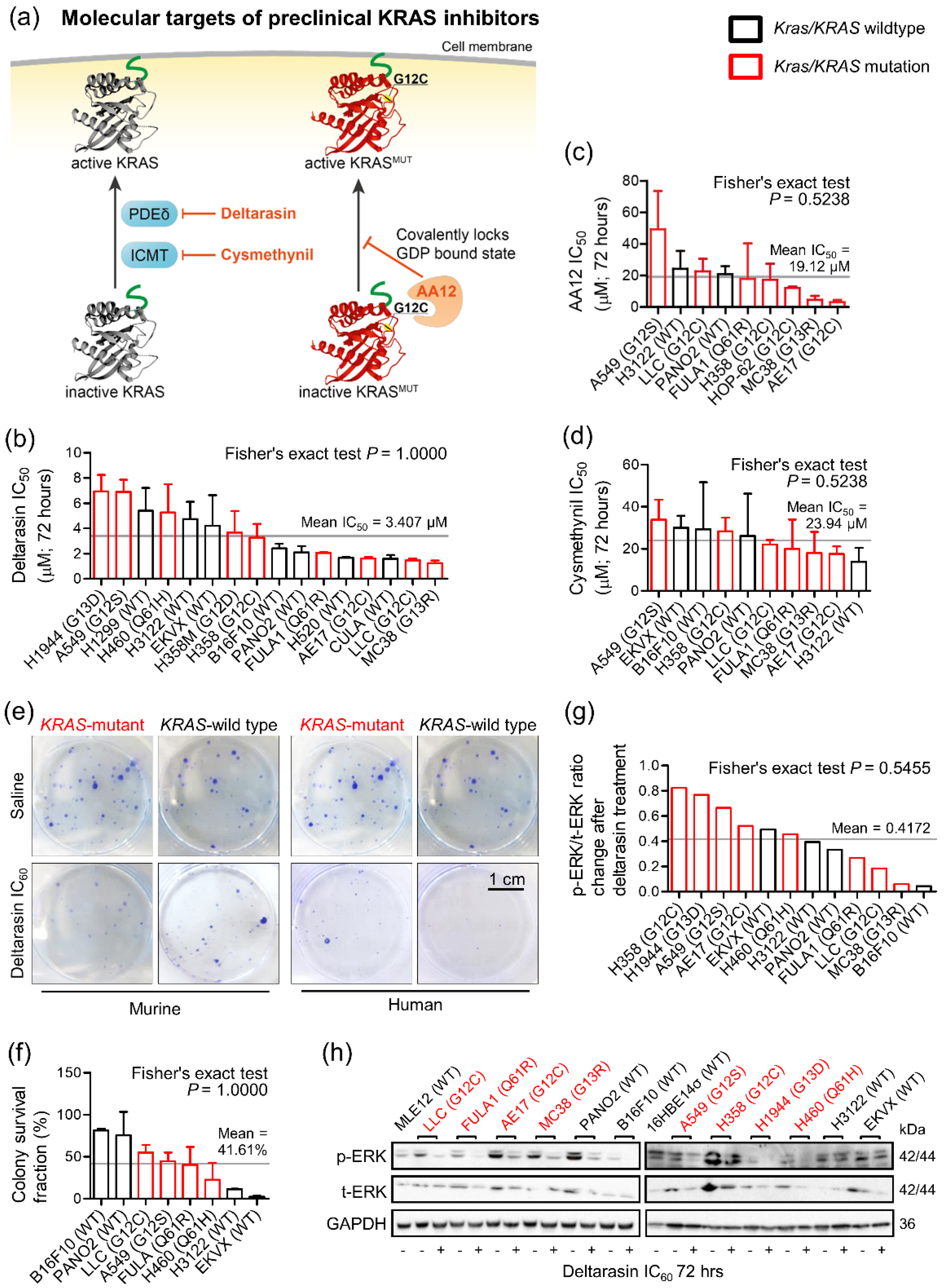
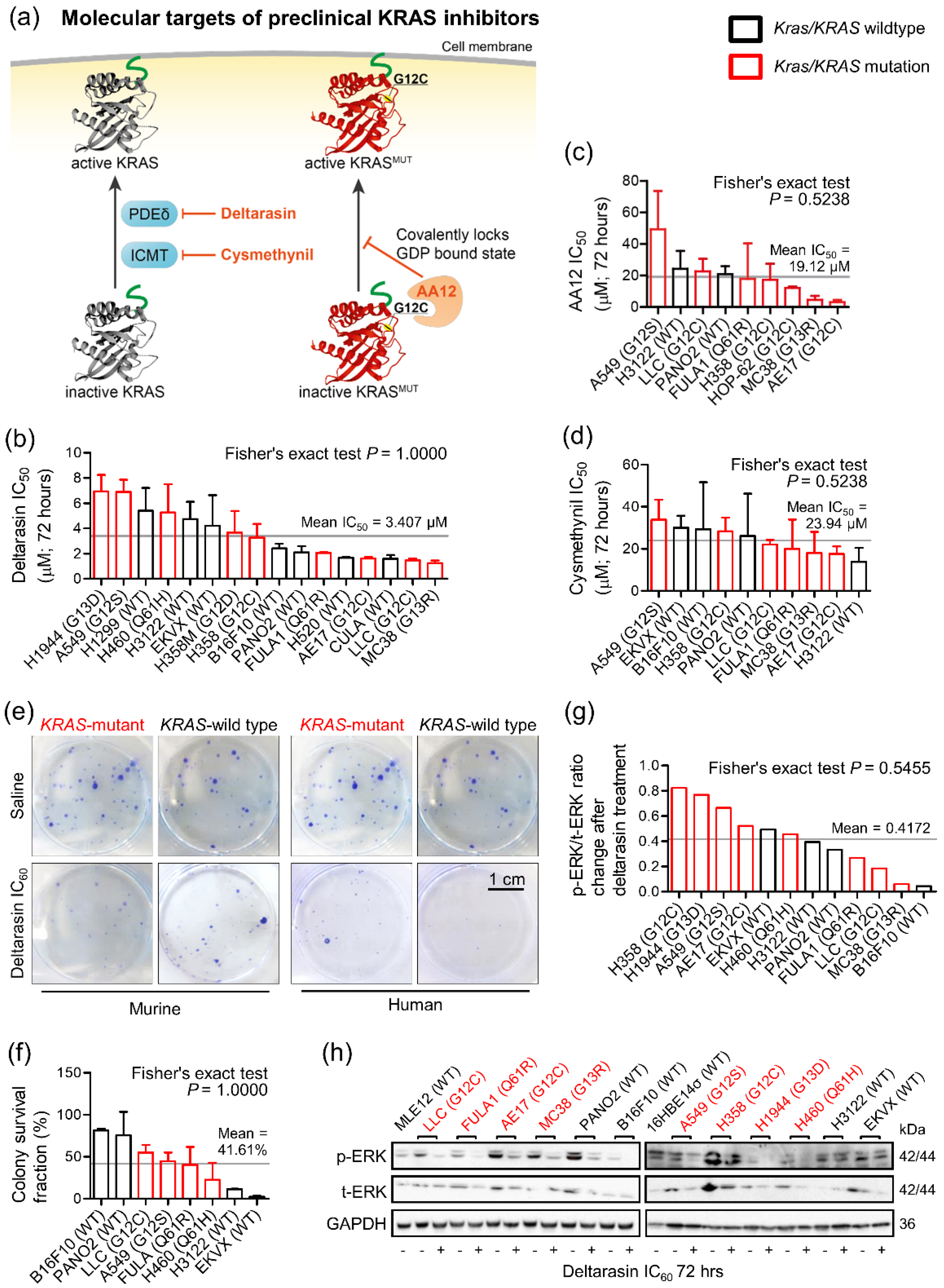
3.1. Mutation-Independent Effects of KRAS Inhibitors In Vitro
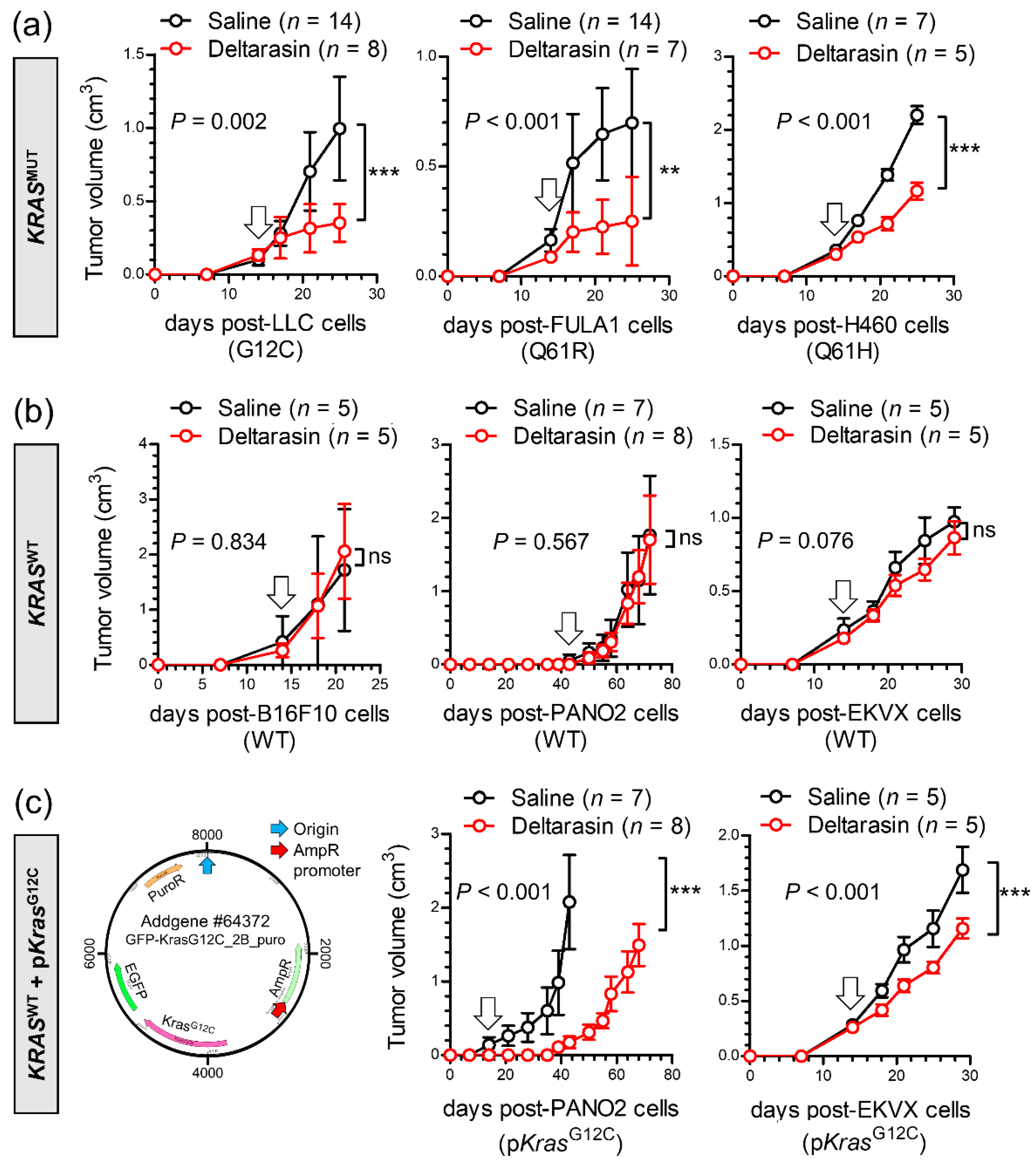
3.2. Specific In Vivo Effects of Deltarasin against KRAS-Mutant Tumors
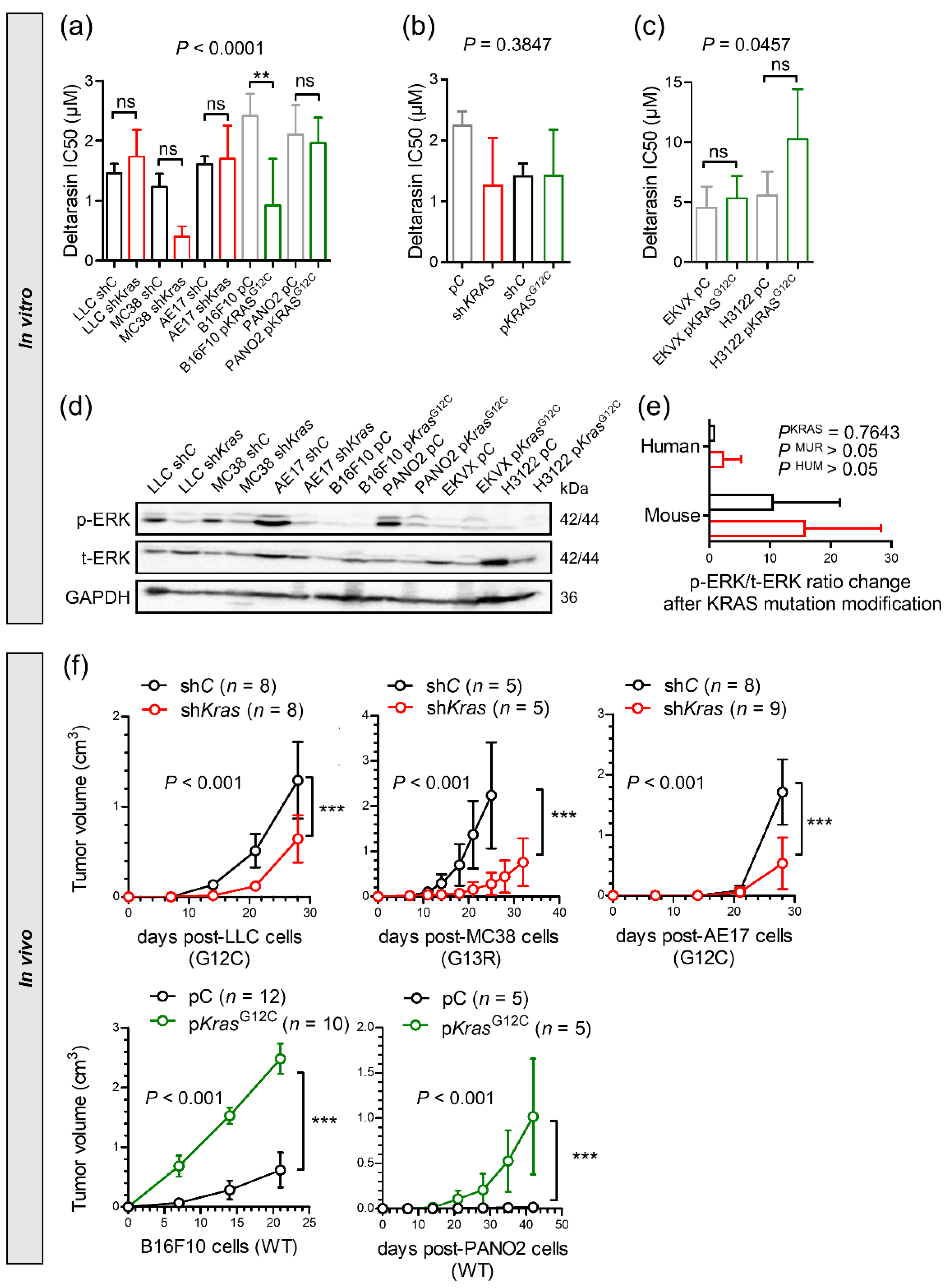
3.3. Genetic KRAS Manipulation Reveals Contrasting KRAS-Dependencies In Vitro and In Vivo
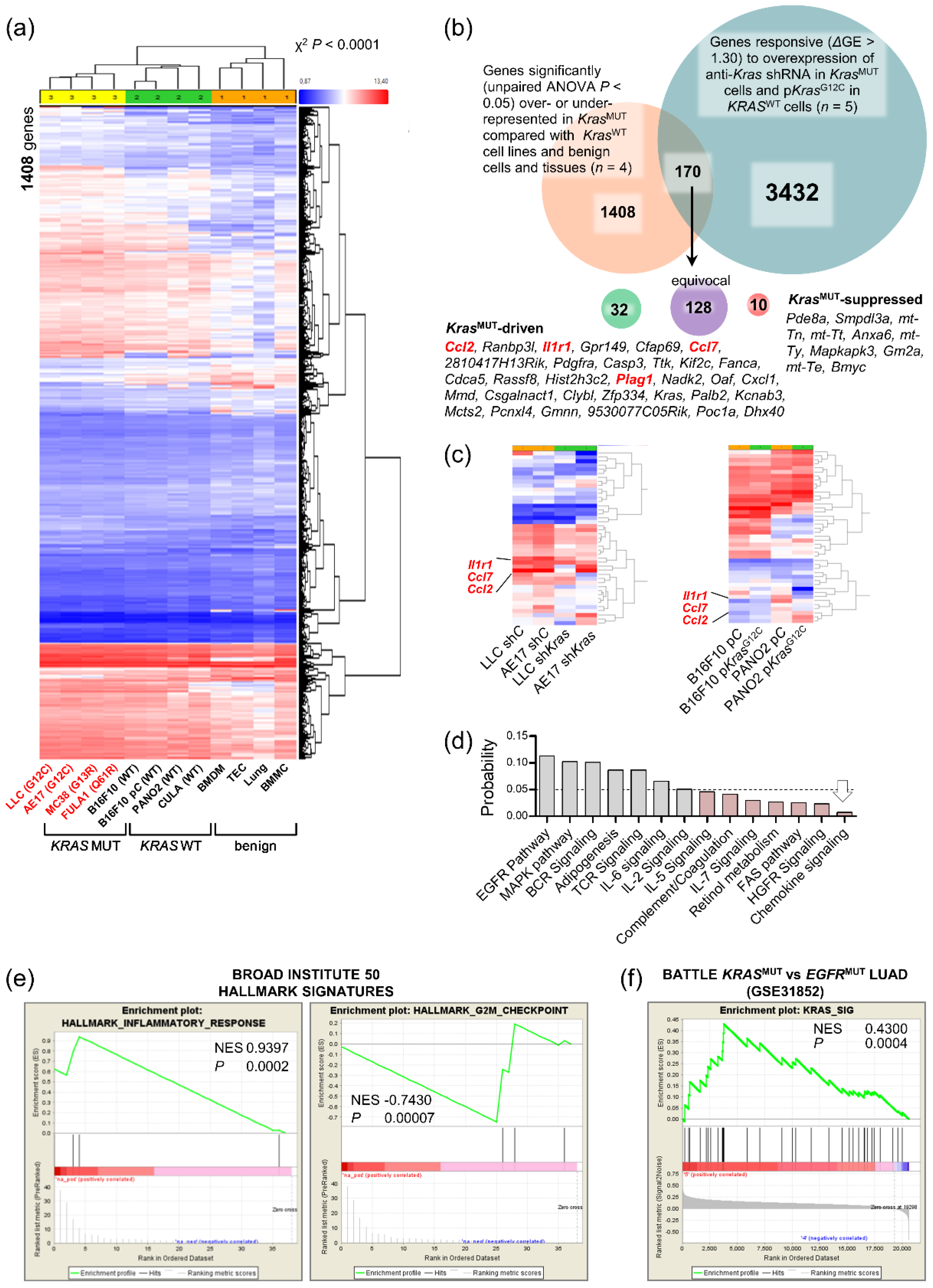
3.4. A Mutant Kras Transcriptome Signature Contains Ccl2 and Il1b
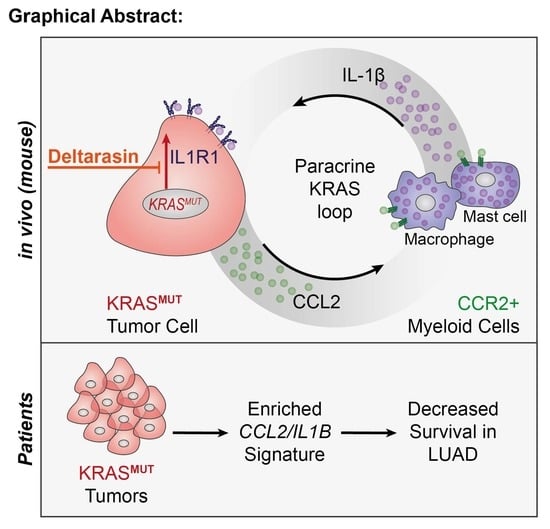
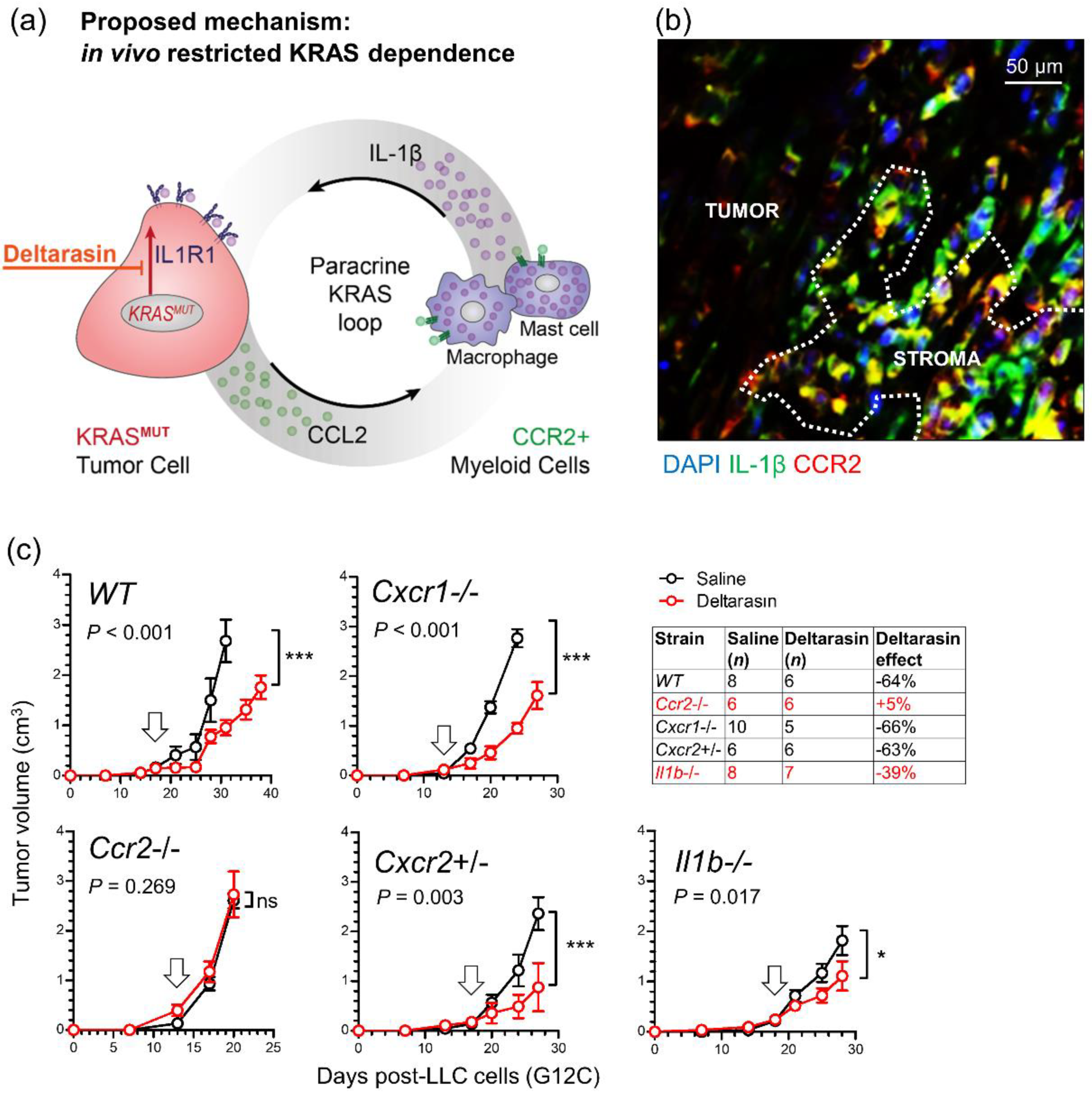
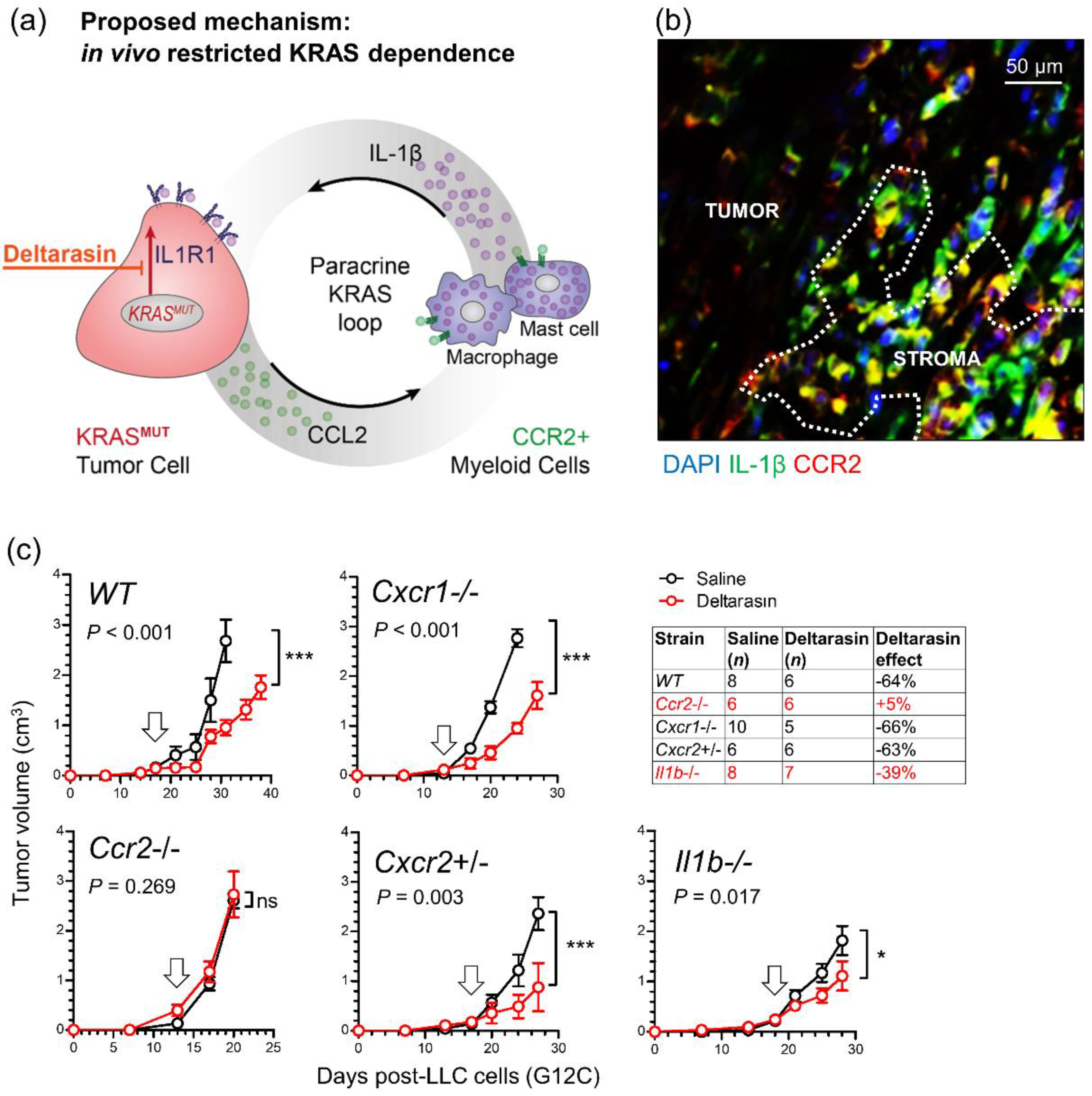
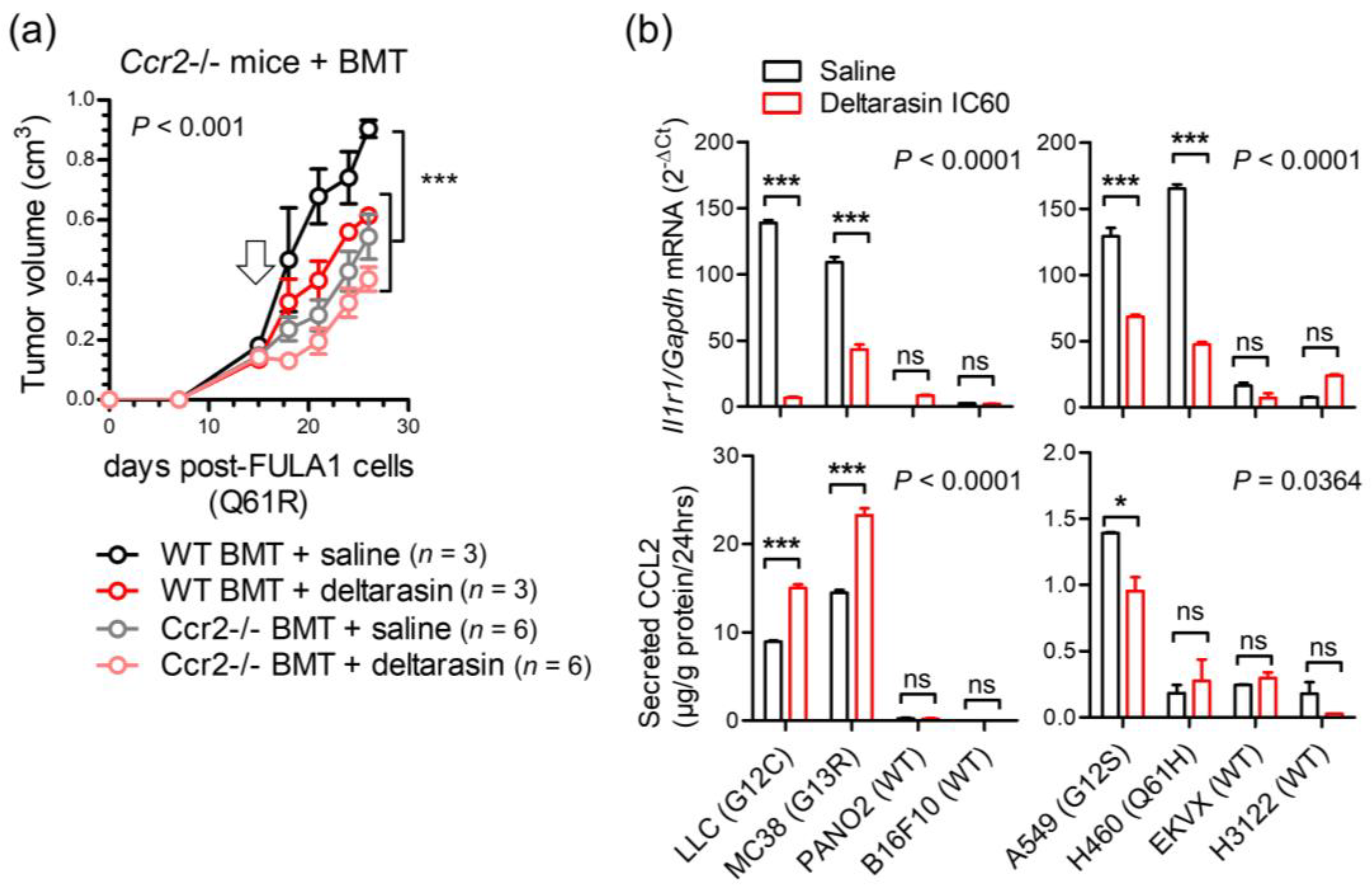
3.5. CCR2+ IL-1β-Secreting Myeloid Cells Potentiate In Vivo KRAS-Dependence
3.6. Deltarasin Limits IL-1β Sensing by KRAS-Mutant Tumor Cells
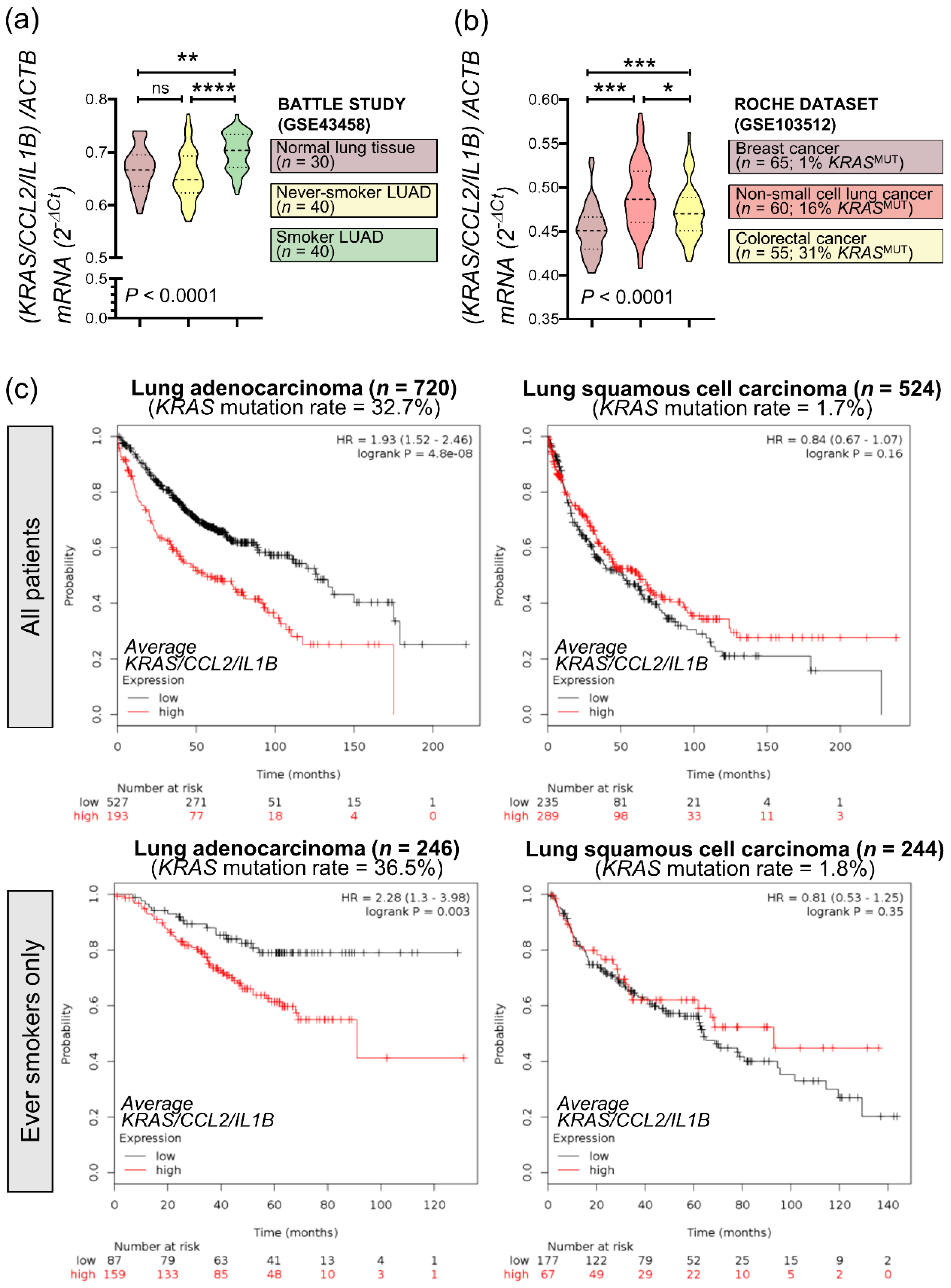
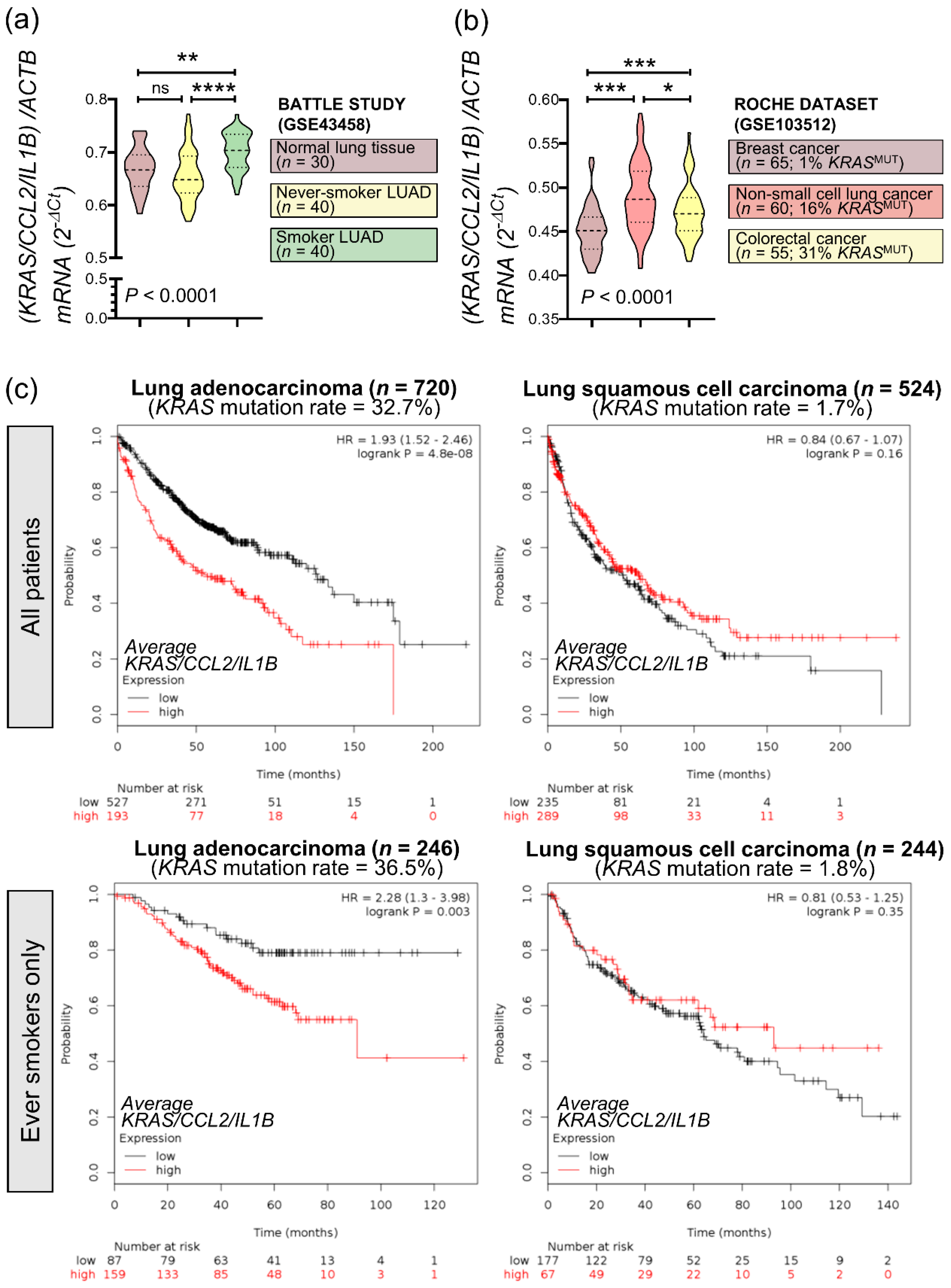
3.7. An Inflammatory CCL2/IL1B Signature in KRAS-Mutant Human Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esposito, D.; Stephen, A.G.; Turbyville, T.J.; Holderfield, M. New weapons to penetrate the armor: Novel reagents and assays developed at the NCI RAS Initiative to enable discovery of RAS therapeutics. Semin. Cancer Biol. 2019, 54, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter-Vann, A.M.; Baron, R.A.; Wong, W.; Cruz, J.D.; York, J.D.; Gooden, D.M.; Bergo, M.O.; Young, S.; Toone, E.J.; Casey, P.J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4336–4341. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.H.; et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancer with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, E.R., III; De Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef] [Green Version]
- Agalioti, T.; Giannou, A.D.; Krontira, A.; Kanellakis, N.I.; Kati, D.; Vreka, M.; Pepe, M.; Spella, M.; Lilis, I.; Zazara, D.E.; et al. Mutant KRAS promotes malignant pleural effusion formation. Nat. Commun. 2017, 8, 15205. [Google Scholar] [CrossRef]
- Marazioti, A.; Lilis, I.; Vreka, M.; Apostolopoulou, H.; Kalogeropoulou, A.; Giopanou, I.; Giotopoulou, G.A.; Krontira, A.C.; Iliopoulou, M.; Kanellakis, N.I.; et al. Myeloid-derived IL-1ß drives oncogenic KRAS-NF-kB addiction in malignant pleural effusion. Nat. Commun. 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Giannou, A.D.; Marazioti, A.; Kanellakis, N.I.; Giopanou, I.; Lilis, I.; Zazara, D.E.; Ntaliarda, G.; Kati, D.; Armenis, V.; Giotopoulou, G.A.; et al. NRAS destines tumor cells to the lungs. EMBO Mol. Med. 2017, 9, 672–686. [Google Scholar] [CrossRef]
- Giopanou, I.; Lilis, I.; Papaleonidopoulos, V.; Agalioti, T.; Kanellakis, N.I.; Spiropoulou, N.; Spella, M.; Stathopoulos, G.T. Tumor–derived osteopontin isoforms cooperate with TRP53 and CCL2 to promote lung metastasis. Oncoimmunology 2017, 6, e1256528. [Google Scholar] [CrossRef] [Green Version]
- Kanellakis, I.N.; Giannou, A.D.; Pepe, A.A.M.; Agalioti, T.; Zazara, E.D.; Giopanou, I.; Psallidas, I.; Spella, M.; Μarazioti, A.; Arendt, K.A.M.; et al. Tobacco chemical-induced mouse lung adenocarcinoma cell lines pin the prolactin orthologue proliferin as a lung tumour promoter. Carcinogenesis 2019, 40, 1352–1362. [Google Scholar] [CrossRef]
- Giannou, A.D.; Marazioti, A.; Spella, M.; Kanellakis, N.I.; Apostolopoulou, H.; Psallidas, I.; Prijovich, Z.M.; Vreka, M.; Zazara, D.E.; Lilis, I.; et al. Mast cells mediate malignant pleural effusion formation. J. Clin. Invest. 2015, 125, 2317–2334. [Google Scholar] [CrossRef]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 2012, 40, D1301–D1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Herbst, R.S.; Wistuba, I.I.; Lee, J.J.; Blumenschein, G.R., Jr.; Tsao, A.; Stewart, D.J.; Hicks, M.E.; Erasmus, J.; Gupta, S.; et al. The BATTLE trial: Personalizing therapy for lung cancer. Cancer Discov. 2011, 1, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Kabbout, M.; Garcia, M.M.; Fujimoto, J.; Liu, D.D.; Woods, D.; Chow, C.-W.; Mendoza, G.; Momin, A.A.; James, B.P.; Solis, L.; et al. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3383–3395. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Brouwer-Visser, J.; Cheng, W.-Y.; Bauer-Mehren, A.; Maisel, D.; Lechner, K.; Andersson, E.; Dudley, J.T.; Milletti, F. Regulatory T-cell Genes Drive Altered Immune Microenvironment in Adult Solid Cancers and Allow for Immune Contextual Patient Subtyping. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. Inflammation and cancer: The long reach of Ras. Nat. Med. 2005, 11, 20–21. [Google Scholar] [CrossRef]
- Vreka, M.; Lilis, I.; Papageorgopoulou, M.; Giotopoulou, G.A.; Lianou, M.; Giopanou, I.; Kanellakis, N.I.; Spella, M.; Agalioti, T.; Armenis, V.; et al. IκB Kinase α Is Required for Development and Progression of KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2018, 78, 2939–2951. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Rajewsky, K. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J. Exp. Med. 2001, 194, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; De Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lübbert, M.; et al. Oncogenic KrasG12D causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.-F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Champeris Tsaniras, S.; Villiou, M.; Giannou, A.D.; Nikou, S.; Petropoulos, M.; Pateras, I.S.; Tserou, P.; Karousi, F.; Lalioti, M.-A.; Gorgoulis, V.G.; et al. Geminin ablation in vivo enhances tumorigenesis through increased genomic instability. J. Pathol. 2018, 246, 134–140. [Google Scholar] [CrossRef]
- Marazioti, A.; Kairi, C.A.; Spella, M.; Giannou, A.D.; Magkouta, S.; Giopanou, I.; Papaleonidopolous, V.; Kalomenidis, I.; Snyder, L.A.; Kardamakis, D.; et al. Beneficial impact of CCL2 and CCL12 neutralization on experimental malignant pleural effusion. PLoS ONE 2013, 8, e71207. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Buchlis, G.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Crisanti, M.C.; Wang, L.-C.S.; Heitjan, D.; Snyder, L.A.; et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 2010, 70, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [Green Version]
- Brana, I.; Calles, A.; Lorusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; De Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin–1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double–blind, placebo–controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Song, X.; Voronov, E.; Dvorkin, T.; Fima, E.; Cagnano, E.; Benharroch, D.; Shendler, Y.; Bjorkdhal, O.; Segal, S.; Dinarello, C.A.; et al. Differential effects of IL–1 alpha and IL–1 beta on tumorigenicity patterns and invasiveness. J. Immunol. 2003, 171, 6448–6456. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL–1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [Green Version]
- Voigt, C.; May, P.; Gottschlich, A.; Markota, A.; Wenk, D.; Gerlach, I.; Voigt, S.; Stathopoulos, G.T.; Arendt, K.A.M.; Heise, C.; et al. Cancer cells induce interleukin-22 production from memory CD4+T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12994–12999. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.N.; Voronov, E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol. Rev. 2008, 222, 222–241. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [Green Version]
- Marazioti, A.; Krontira, A.C.; Behrend, S.J.; Giotopoulou, G.A.; Ntaliarda, G.; Blanquart, C.; Bayram, H.; Iliopolou, M.; Vreka, M.; Trassl, L.; et al. KRAS signaling in malignant pleural mesothelioma. EMBO Mol. Med. 2022, 14, e13631. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arendt, K.A.M.; Ntaliarda, G.; Armenis, V.; Kati, D.; Henning, C.; Giotopoulou, G.A.; Pepe, M.A.A.; Klotz, L.V.; Lamort, A.-S.; Hatz, R.A.; et al. An In Vivo Inflammatory Loop Potentiates KRAS Blockade. Biomedicines 2022, 10, 592. https://doi.org/10.3390/biomedicines10030592
Arendt KAM, Ntaliarda G, Armenis V, Kati D, Henning C, Giotopoulou GA, Pepe MAA, Klotz LV, Lamort A-S, Hatz RA, et al. An In Vivo Inflammatory Loop Potentiates KRAS Blockade. Biomedicines. 2022; 10(3):592. https://doi.org/10.3390/biomedicines10030592
Chicago/Turabian StyleArendt, Kristina A. M., Giannoula Ntaliarda, Vasileios Armenis, Danai Kati, Christin Henning, Georgia A. Giotopoulou, Mario A. A. Pepe, Laura V. Klotz, Anne-Sophie Lamort, Rudolf A. Hatz, and et al. 2022. "An In Vivo Inflammatory Loop Potentiates KRAS Blockade" Biomedicines 10, no. 3: 592. https://doi.org/10.3390/biomedicines10030592
APA StyleArendt, K. A. M., Ntaliarda, G., Armenis, V., Kati, D., Henning, C., Giotopoulou, G. A., Pepe, M. A. A., Klotz, L. V., Lamort, A.-S., Hatz, R. A., Kobold, S., Schamberger, A. C., & Stathopoulos, G. T. (2022). An In Vivo Inflammatory Loop Potentiates KRAS Blockade. Biomedicines, 10(3), 592. https://doi.org/10.3390/biomedicines10030592