
Clinical Manifestations of Various Molecular Cytogenetic Variants of Eight Cases of “8p Inverted Duplication/Deletion Syndrome”

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Findings
3.2. Molecular Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- García-Santiago, F.A.; Martínez-Glez, V.; Santos, F.; García-Miñaur, S.; Mansilla, E.; Meneses, A.G.; Rosell, J.; Granero, Á.P.; Vallespín, E.; Fernández, L.; et al. Analysis of invdupdel(8p) rearrangement: Clinical, cytogenetic and molecular characterization. Am. J. Med. Genet. Part A 2015, 167, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.orpha.net (accessed on 10 January 2022).
- Weleber, R.G.; Verma, R.S.; Kimberling, W.J.; Fieger, H.G., Jr. Duplication deficiency of the short arm of chromosome 8 following artificial insemination. Ann. Genet. 1976, 19, 241–247. [Google Scholar] [PubMed]
- Okur, V.; Hamm, L.; Kavus, H.; Mebane, C.; Robinson, S.; Levy, B.; Chung, W.K. Clinical and genomic characterization of 8p cytogenomic disorders. Genet. Med. 2021, 23, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Bianco, M.L.; Vecchio, D.; Timpanaro, T.A.; Arena, A.; Macchiaiolo, M.; Bartuli, A.; Sciuto, L.; Presti, S.; Sciuto, S.; Sapuppo, A.; et al. Deciphering the Invdupdel(8p) Genotype–Phenotype Correlation: Our Opinion. Brain Sci. 2020, 10, 451. [Google Scholar] [CrossRef]
- Vibert, R.; Mignot, C.; Keren, B.; Chantot-Bastaraud, S.; Portnoï, M.; Nouguès, M.; Moutard, M.; Faudet, A.; Whalen, S.; Haye, D.; et al. Neurodevelopmental phenotype in 36 new patients with 8p inverted duplication–deletion: Genotype–phenotype correlation for anomalies of the corpus callosum. Clin. Genet. 2021, 101, 307–316. [Google Scholar] [CrossRef]
- Weckselblatt, B.; Rudd, M.K. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015, 31, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Zuffardi, O.; Bonaglia, M.C.; Ciccone, R.; Giorda, R. Inverted duplications deletions: Underdiagnosed rearrangements?? Clin. Genet. 2009, 75, 505–513. [Google Scholar] [CrossRef]
- Shimokawa, O.; Kurosawa, K.; Ida, T.; Harada, N.; Kondoh, T.; Miyake, N.; Yoshiura, K.; Kishino, T.; Ohta, T.; Niikawa, N.; et al. Molecular characterization of inv dup del(8p): Analysis of five cases. Am. J. Med. Genet. 2004, 128A, 133–137. [Google Scholar] [CrossRef]
- Kato, T.; Inagaki, H.; Miyai, S.; Suzuki, F.; Naru, Y.; Shinkai, Y.; Kato, A.; Kanyama, K.; Mizuno, S.; Muramatsu, Y.; et al. The involvement of U-type dicentric chromosomes in the formation of terminal deletions with or without adjacent inverted duplications. Hum. Genet. 2020, 139, 1417–1427. [Google Scholar] [CrossRef]
- Rowe, L.R.; Lee, J.-Y.; Rector, L.; Kaminsky, E.B.; Brothman, A.R.; Martin, C.L.; South, S.T. U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J. Med. Genet. 2009, 46, 694–702. [Google Scholar] [CrossRef]
- Yu, S.; Graf, W. Telomere Capture as a Frequent Mechanism for Stabilization of the Terminal Chromosomal Deletion Associated with Inverted Duplication. Cytogenet. Genome Res. 2010, 129, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) [published correction appears in Genet Med. 2021 Nov;23(11):2230]. Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, H.; Harada, N.; Ida, T.; Ishida, T.; Ledbetter, D.H.; Yoshiura, K.-I.; Ohta, T.; Kishino, T.; Niikawa, N.; Matsumoto, N. Complex low-copy repeats associated with a common polymorphic inversion at human chromosome 8p23. Genomics 2003, 82, 238–244. [Google Scholar] [CrossRef]
- Giglio, S.; Broman, K.W.; Matsumoto, N.; Calvari, V.; Gimelli, G.; Neumann, T.; Ohashi, H.; Voullaire, L.; Larizza, D.; Giorda, R.; et al. Olfactory Receptor–Gene Clusters, Genomic-Inversion Polymorphisms, and Common Chromosome Rearrangements. Am. J. Hum. Genet. 2001, 68, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Tabarés-Seisdedos, R.; Rubenstein, J.L.R. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: Implications for schizophrenia, autism and cancer. Mol. Psychiatry 2009, 14, 563–589. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://dosage.clinicalgenome.org/ (accessed on 10 January 2022).
- Hollox, E. Copy number variation of beta-defensins and relevance to disease. Cytogenet. Genome Res. 2008, 123, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Nucaro, A.; Pisano, T.; Chillotti, I.; Montaldo, C.; Pruna, D. Chromosome 8p23.2-pter: A critical region for mental retardation, autism and epilepsy? Clin. Genet. 2011, 79, 394–395. [Google Scholar] [CrossRef]
- Ozgen, H.M.; Van Daalen, E.; Bolton, P.F.; Maloney, V.K.; Huang, S.; Cresswell, L.; Van Den Boogaard, M.J.; Eleveld, M.J.; Van‘t Slot, R.; Hochstenbach, R.; et al. Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin. Genet. 2009, 76, 348–356. [Google Scholar] [CrossRef]
- Chen, C.-P.; Ko, T.-M.; Huang, W.-C.; Chern, S.-R.; Wu, P.-S.; Chen, Y.-N.; Chen, S.-W.; Lee, C.-C.; Pan, C.-W.; Yang, C.-W.; et al. Molecular cytogenetic characterization of inv dup del(8p) in a fetus associated with ventriculomegaly, hypoplastic left heart, polyhydramnios and intestinal obstruction. Taiwan J. Obstet. Gynecol. 2016, 55, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Glancy, M.; Barnicoat, A.; Vijeratnam, R.; De Souza, S.; Gilmore, J.; Huang, S.; Maloney, V.K.; Thomas, N.S.; Bunyan, D.J.; Jackson, A.; et al. Transmitted duplication of 8p23.1–8p23.2 associated with speech delay, autism and learning difficulties. Eur. J. Hum. Genet. 2008, 17, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Davis, R.; Youngblom, J.; Gregg, J. Genotype–Phenotype Association Studies of Chromosome 8p Inverted Duplication Deletion Syndrome. Behav. Genet. 2011, 41, 373–380. [Google Scholar] [CrossRef] [PubMed]
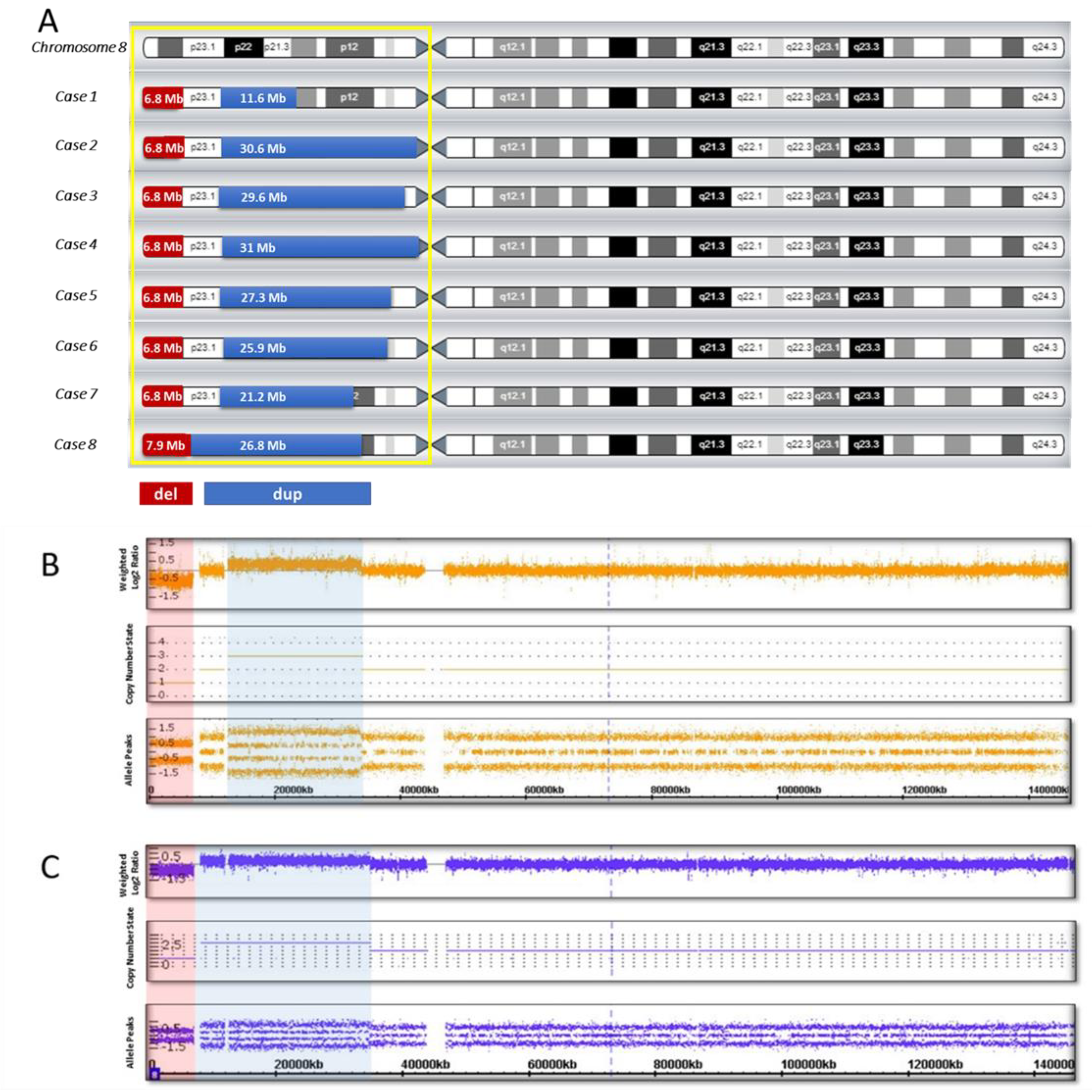
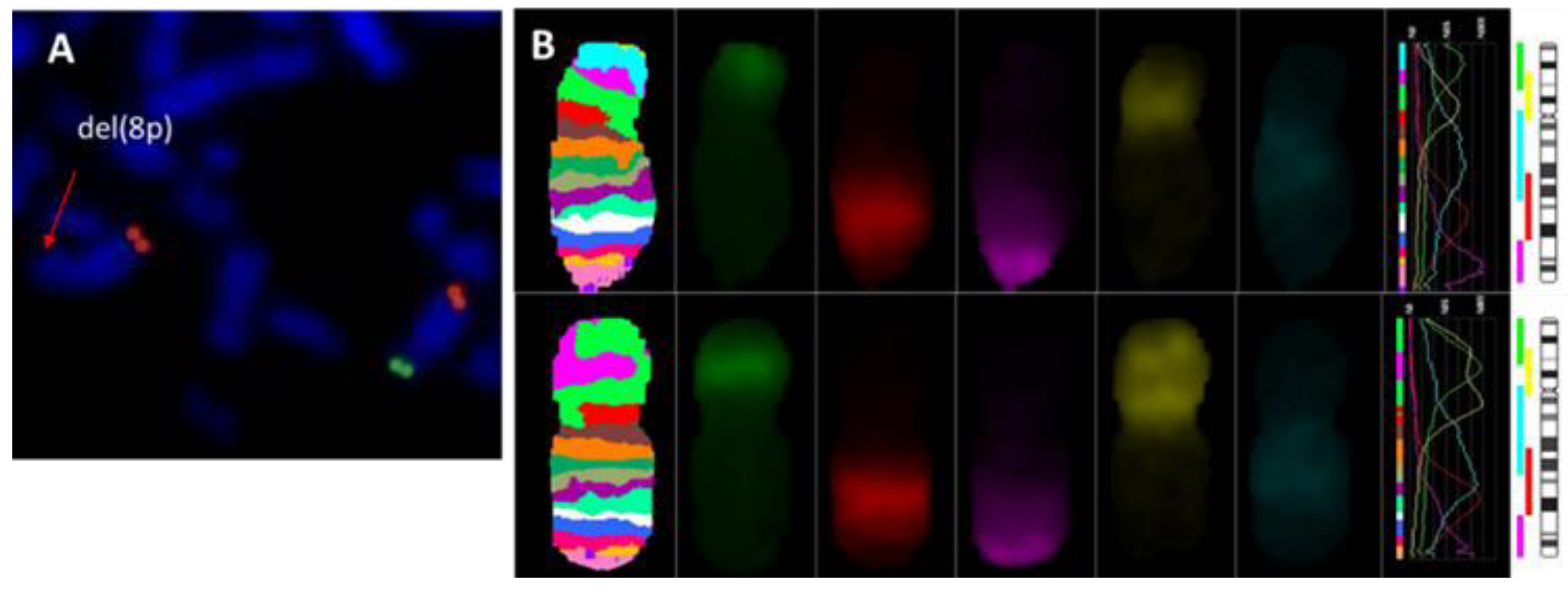

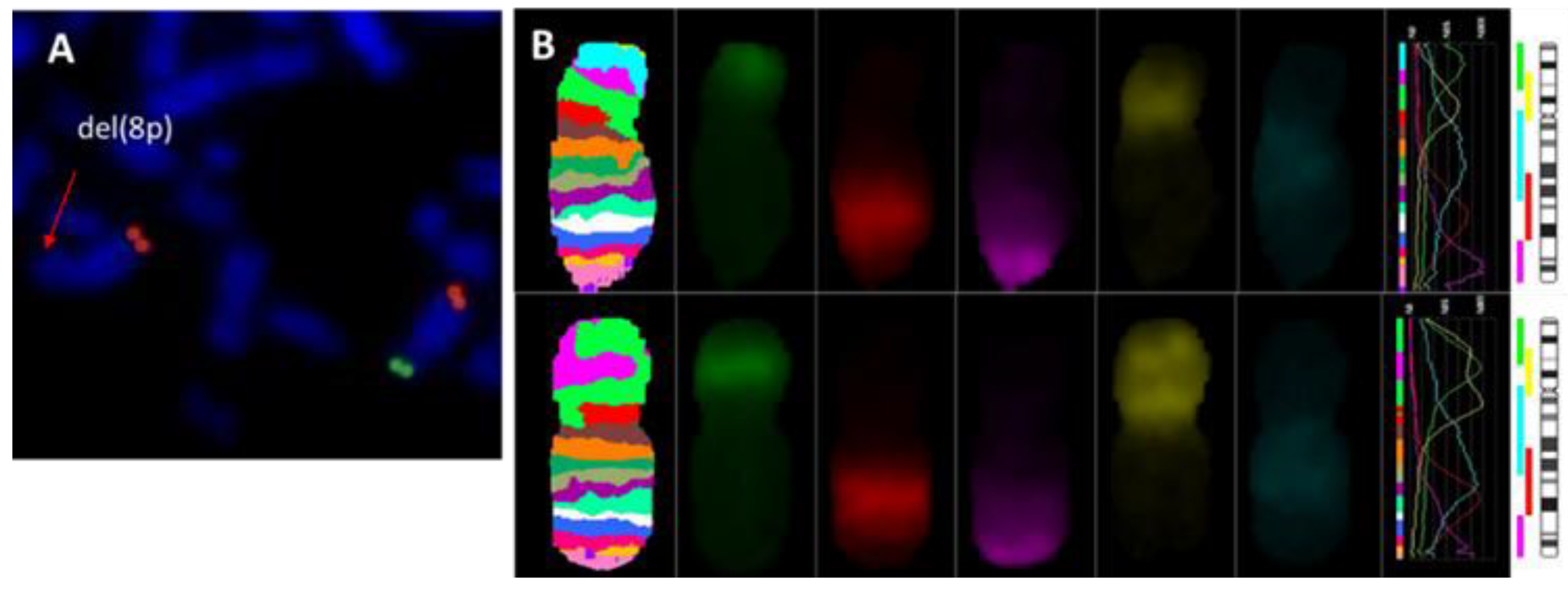
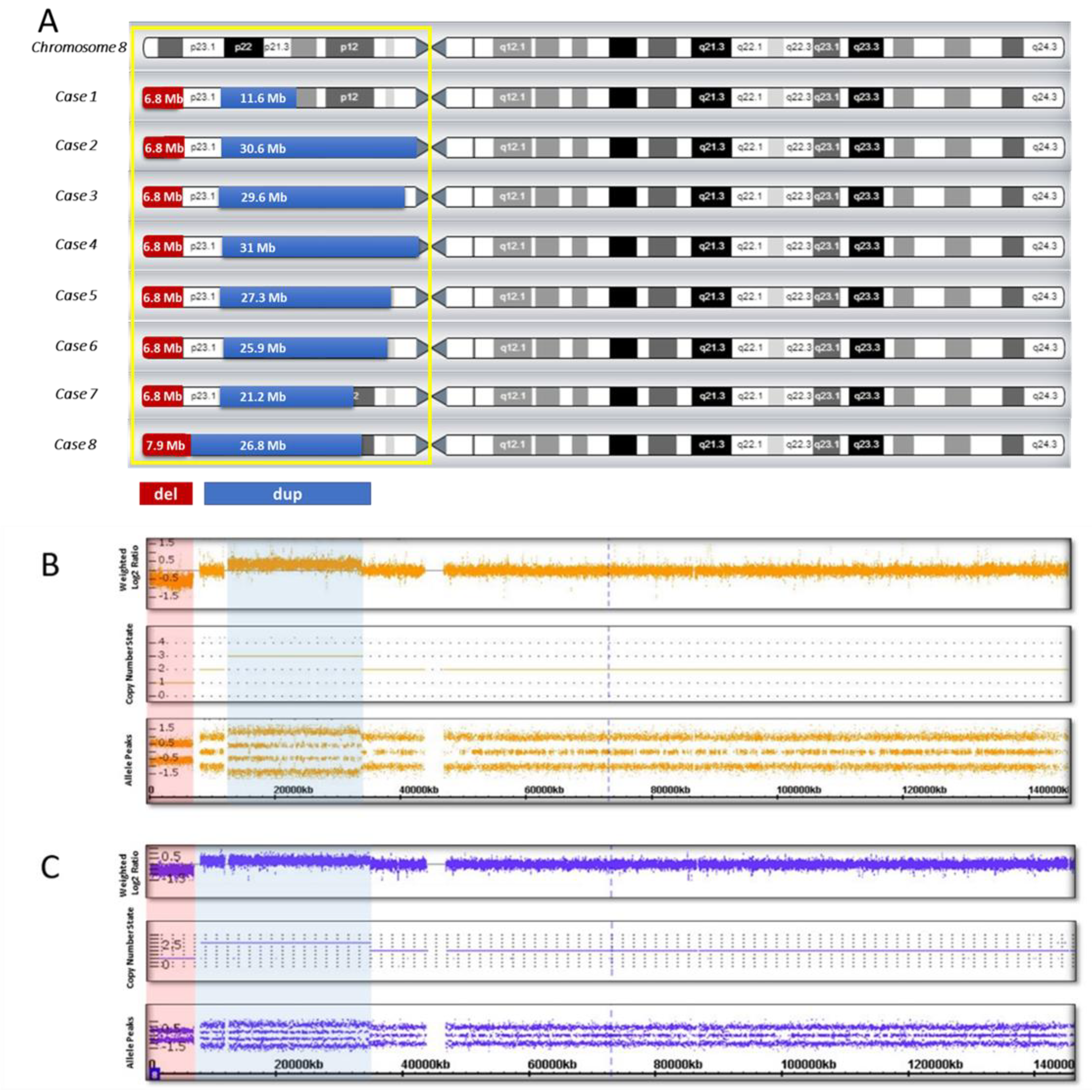{kind=link}
{kind=link}
| Cases | Sex | Age (Years) | Mental and Language Development | Motor Development * | Craniofacial Dysmorphism | CNS Malformations on MRI | Coordinates ** of the Chromosomal Alterations and Size of the Rearrangement (CMA) |
|---|---|---|---|---|---|---|---|
| Case 1 | M | 7 | Severe delay | Mild delay (unsupported walking since 18 months) | Sloping forehead, hypertelorism, upslanted palpebral fissures, wide nasal base, thin upper lip, micrognathia, malocclusion, large and deformed ears | No | arr[hg19] 8p23.3p23.1 (158048_6982980) x 1,8p23.1p21.2 (12490998_24107465) x 3 del: 6.8 Mb dup: 11.6 Mb spacer: 5.5 Mb |
| Case 2 | M | 2 | Mild delay | Severe delay (not walking) | Hydrocephalic skull, large and prominent forehead, mild facial asymmetry, epicanthus, exophthalmos, strabismus, ptosis, wide nasal base, thin upper lip, mild micrognathia (more pronounced in infancy), wide interdental spaces, large and deformed low-set ears | Retrocerebellar cyst | arr[hg19] 8p23.3p23.1 (158048_6982980) x 1,8p23.1p11.1 (12527948_43169003) x 3 del: 6.8 Mb dup: 30.6 Mb spacer: 5.5 Mb |
| Case 3 | F | 9 | Severe delay | Severe delay (unsupported walking since 7 years) | Epicanthus, wide nasal bridge, nares anteverted, abnormal growth of teeth, large dysplastic ears | Corpus callosum agenesis | arr[hg19] 8p23.3p23.1 (158048_6982257) x 1,8p23.1p11.21 (11936000_41509224) x 3 del: 6.8 Mb dup: 29.6 Mb spacer: 4.95 Mb |
| Case 4 | M | 5 | Severe delay | Moderate delay (unsupported walking since 3 years 6 months) | Brachycephaly, large and prominent forehead, enophthalmos, mildly arched eyebrows, wide nasal base, nares anteverted, short philtrum, protruding lower lip, deformed low-set ears | Corpus callosum hypoplasia | arr[hg19] 8p23.3p23.1 (158048_6999114) x 1,8p23.1p11.1 (12592122_43673602) x 3 del: 6.8 Mb dup: 31 Mb spacer: 5.59 Mb |
| Case 5 | F | 3 | Moderate delay | Moderate delay (walking with support since 2 years) | Moderate acrocephaly, flat occiput, wide interdental spaces, micrognathia, protruding low-set ears | No | arr[hg19] 8p23.3p23.1 (158048_6940661) x 1,8p23.1p11.22 (11935023_39246760) x 3 del: 6.8 Mb dup: 27.3 Mb spacer: 4.99 Mb |
| Case 6 | M | 2 | Mild delay | Severe delay (not walking) | Prominent forehead, temporal balding, downslanted palpebral fissures, mildly arched eye-brows, wide nasal base, depressed nasal bridge, full cheeks, wide mouth, thin upper lip, micrognathia, low-set ears | Corpus callosum hypoplasia, pineal cyst | arr[hg19] 8p23.3p23.1 (158048_6944774) x 1,8p23.1p11.22 (12528482_38476919) x 3 del: 6.8 Mb dup: 25.95 Mb spacer: 5.58 Mb |
| Case 7 | F | 6 | Severe delay | Moderate delay (unsupported walking since 4 years) | Narrow forehead, low anterior and posterior hairlines, upslanted palpebral fissures, mildly arched eye-brows, wide nasal base, nares anteverted, short philtrum, full cheeks, short chin, dysplastic ears | Corpus callosum hypoplasia | arr[hg19] 8p23.3p23.1 (158048_6982257) x 1,8p23.1p12 (12528482_33760127) x 3 del: 6.8 Mb dup: 21 Mb spacer: 5.5 Mb |
| Case 8 | M | 5 | Severe delay | Moderate delay (unsupported walking since 2 years 3 months) | Prominent forehead, hypertelorism, upslanted palpebral fissures, thin upper lip, wide interdental spaces, large low-set ears | Corpus callosum agenesis | arr[hg19] 8p23.3p23.1 (158048_8093169) x 1,8p23.1p12 (8093169_34866530) x 3 del: 7.9 Mb dup: 26.8 Mb spacer is missing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yurchenko, D.A.; Minzhenkova, M.E.; Dadali, E.L.; Markova, Z.G.; Rudenskaya, G.E.; Matyushchenko, G.N.; Kanivets, I.V.; Shilova, N.V. Clinical Manifestations of Various Molecular Cytogenetic Variants of Eight Cases of “8p Inverted Duplication/Deletion Syndrome”. Biomedicines 2022, 10, 567. https://doi.org/10.3390/biomedicines10030567
Yurchenko DA, Minzhenkova ME, Dadali EL, Markova ZG, Rudenskaya GE, Matyushchenko GN, Kanivets IV, Shilova NV. Clinical Manifestations of Various Molecular Cytogenetic Variants of Eight Cases of “8p Inverted Duplication/Deletion Syndrome”. Biomedicines. 2022; 10(3):567. https://doi.org/10.3390/biomedicines10030567
Chicago/Turabian StyleYurchenko, Darya A., Marina E. Minzhenkova, Elena L. Dadali, Zhanna G. Markova, Galina E. Rudenskaya, Galina N. Matyushchenko, Ilya V. Kanivets, and Nadezda V. Shilova. 2022. "Clinical Manifestations of Various Molecular Cytogenetic Variants of Eight Cases of “8p Inverted Duplication/Deletion Syndrome”" Biomedicines 10, no. 3: 567. https://doi.org/10.3390/biomedicines10030567
APA StyleYurchenko, D. A., Minzhenkova, M. E., Dadali, E. L., Markova, Z. G., Rudenskaya, G. E., Matyushchenko, G. N., Kanivets, I. V., & Shilova, N. V. (2022). Clinical Manifestations of Various Molecular Cytogenetic Variants of Eight Cases of “8p Inverted Duplication/Deletion Syndrome”. Biomedicines, 10(3), 567. https://doi.org/10.3390/biomedicines10030567