
Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis

 , , , , ,
, , , , ,
Abstract
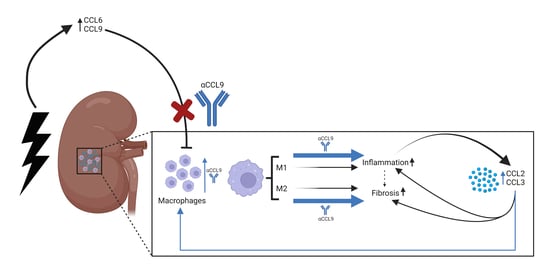
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Blood Sampling and Organ Isolation
2.3. Leukocyte Profiling and Flow Cytometry
2.4. Histological Tissue Analysis: Acid Fuchsin Orange G (AFOG) Staining
2.5. Tissue Protein Isolation, Western Blot Analysis and ELISA
2.6. Chemokine Profiling and LUNARIS Assay
2.7. Statistics
3. Results
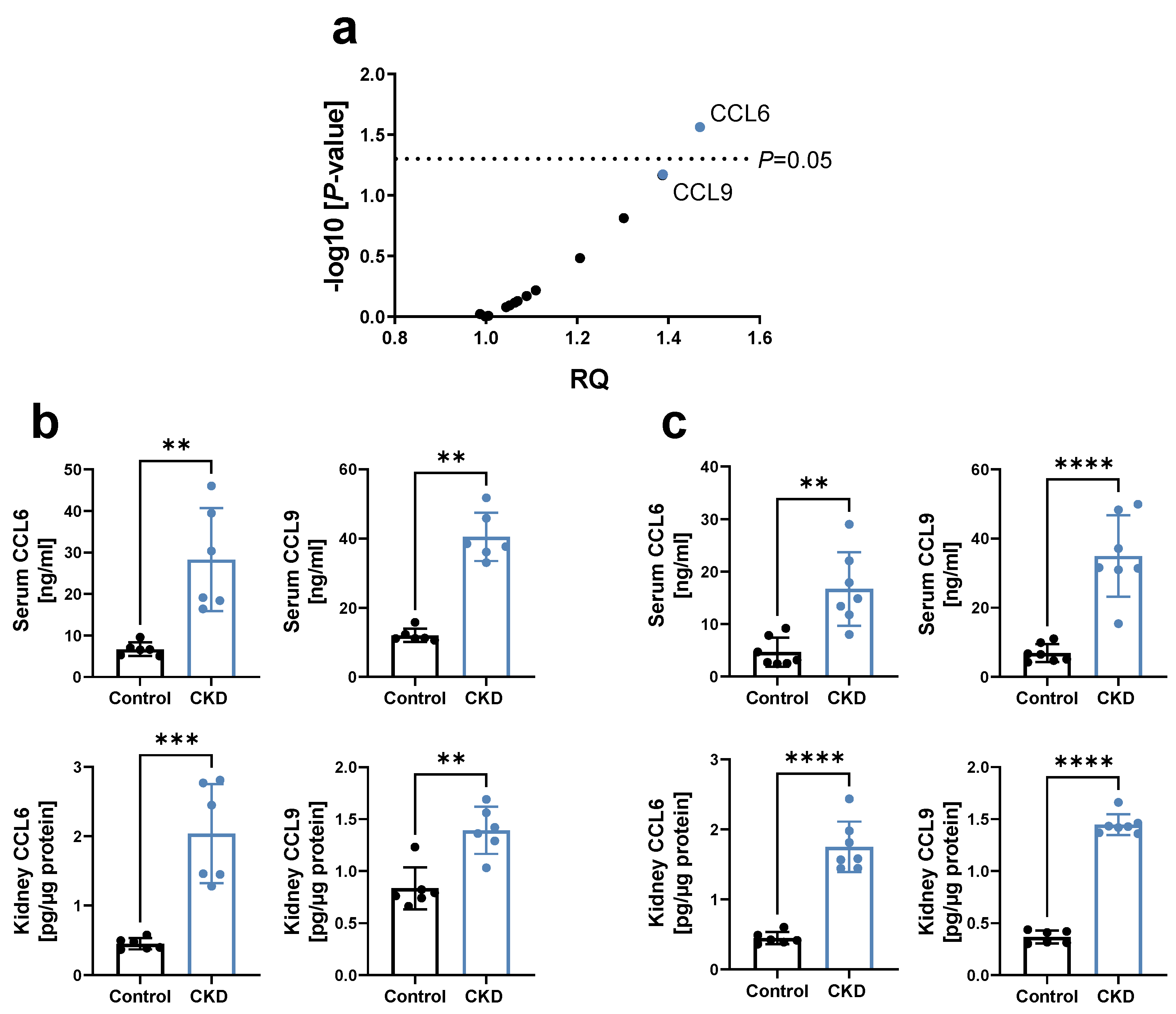
3.1. Chemokines CCL6 and CCL9 Are Increased in CKD
3.2. Blocking CCL9 Increases Kidney Fibrosis during CKD Induction
3.3. Blocking CCL9 Enhances Kidney Inflammation during CKD Induction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [Green Version]
- Lameire, N.H.; Levin, A.; Kellum, J.A.; Cheung, M.; Jadoul, M.; Winkelmayer, W.C.; Stevens, P.E. Harmonizing acute and chronic kidney disease definition and classification: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2021, 100, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Marx, N.; Noels, H.; Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M. Mechanisms of cardiovascular complications in chronic kidney disease: Research focus of the Transregional Research Consortium SFB TRR219 of the University Hospital Aachen (RWTH) and the Saarland University. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2018, 107, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Vielhauer, V.; Schlöndorff, D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int. 2003, 63, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M. Inflammatory Mediators and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 381–406. [Google Scholar] [CrossRef]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, T.D. Renal tubulointerstitial fibrosis: Common but never simple. Am. J. Physiol. Ren. Physiol. 2009, 296, F1239–F1244. [Google Scholar] [CrossRef] [Green Version]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Vielhauer, V.; Frink, M.; Linde, Y.; Cohen, C.D.; Blattner, S.M.; Kretzler, M.; Strutz, F.; Mack, M.; Grone, H.J.; et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J. Clin. Investig. 2002, 109, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, K.; Gao, J.L.; Horuk, R.; Wada, T.; Kaneko, S.; Murphy, P.M. Chemokine receptor CCR1 regulates inflammatory cell infiltration after renal ischemia-reperfusion injury. J. Immunol. 2008, 181, 8670–8676. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Correa-Costa, M.; Silva, R.C.; Cruz, M.C.; Hiyane, M.I.; da Silva, J.S.; Perez, K.R.; Cuccovia, I.M.; Camara, N.O.S. CCR2 contributes to the recruitment of monocytes and leads to kidney inflammation and fibrosis development. Inflammopharmacology 2018, 26, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Richter, R.; Forssmann, U.; Henschler, R.; Escher, S.; Frimpong-Boateng, A.; Forssmann, W.G. Increase of expression and activation of chemokine CCL15 in chronic renal failure. Biochem. Biophys. Res. Commun. 2006, 345, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Berahovich, R.D.; Miao, Z.; Wang, Y.; Premack, B.; Howard, M.C.; Schall, T.J. Proteolytic activation of alternative CCR1 ligands in inflammation. J. Immunol. 2005, 174, 7341–7351. [Google Scholar] [CrossRef]
- Berger, M.S.; Kozak, C.A.; Gabriel, A.; Prystowsky, M.B. The gene for C10, a member of the beta-chemokine family, is located on mouse chromosome 11 and contains a novel second exon not found in other chemokines. DNA Cell Biol. 1993, 12, 839–847. [Google Scholar] [CrossRef]
- Hara, T.; Bacon, K.B.; Cho, L.C.; Yoshimura, A.; Morikawa, Y.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Schall, T.J.; Miyajima, A. Molecular cloning and functional characterization of a novel member of the C-C chemokine family. J. Immunol. 1995, 155, 5352–5358. [Google Scholar]
- Noels, H.; Lehrke, M.; Vanholder, R.; Jankowski, J. Lipoproteins and fatty acids in chronic kidney disease: Molecular and metabolic alterations. Nat. Rev. Nephrol. 2021, 17, 528–542. [Google Scholar] [CrossRef]
- Soppert, J.; Lehrke, M.; Marx, N.; Jankowski, J.; Noels, H. Lipoproteins and lipids in cardiovascular disease: From mechanistic insights to therapeutic targeting. Adv. Drug Deliv. Rev. 2020, 159, 4–33. [Google Scholar] [CrossRef]
- Wenzel, U.; Schneider, A.; Valente, A.J.; Abboud, H.E.; Thaiss, F.; Helmchen, U.M.; Stahl, R.A. Monocyte chemoattractant protein-1 mediates monocyte/macrophage influx in anti-thymocyte antibody-induced glomerulonephritis. Kidney Int. 1997, 51, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Eardley, K.S.; Zehnder, D.; Quinkler, M.; Lepenies, J.; Bates, R.L.; Savage, C.O.; Howie, A.J.; Adu, D.; Cockwell, P. The relationship between albuminuria, MCP-1/CCL2, and interstitial macrophages in chronic kidney disease. Kidney Int. 2006, 69, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vielhauer, V.; Berning, E.; Eis, V.; Kretzler, M.; Segerer, S.; Strutz, F.; Horuk, R.; Grone, H.J.; Schlondorff, D.; Anders, H.J. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int. 2004, 66, 2264–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Li, F.; Zhang, C.; Li, N.; Huang, H.; Shao, Z.; Zhang, M.; Zhan, X.; He, Y.; Ju, Z.; et al. Eosinophil-derived chemokine (hCCL15/23, mCCL6) interacts with CCR1 to promote eosinophilic airway inflammation. Signal Transduct. Target. Ther. 2021, 6, 91. [Google Scholar] [CrossRef]
- Kitamura, T.; Kometani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat. Genet. 2007, 39, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef]
- Shen, B.; Liu, X.; Fan, Y.; Qiu, J. Macrophages regulate renal fibrosis through modulating TGFβ superfamily signaling. Inflammation 2014, 37, 2076–2084. [Google Scholar] [CrossRef]
- Han, Y.; Ma, F.Y.; Tesch, G.H.; Manthey, C.L.; Nikolic-Paterson, D.J. Role of macrophages in the fibrotic phase of rat crescentic glomerulonephritis. Am. J. Physiol. Ren. Physiol. 2013, 304, F1043–F1053. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.G.; Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Jo, S.K.; Cho, W. The Role of M2 Macrophages in the Progression of Chronic Kidney Disease following Acute Kidney Injury. PLoS ONE 2015, 10, e0143961. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wang, Y.; Pei, G.; Deng, X.; Jiang, H.; Wu, J.; Zhou, C.; Guo, Y.; Yao, Y.; Zeng, R.; et al. Bone marrow-derived Ly6C(-) macrophages promote ischemia-induced chronic kidney disease. Cell Death Dis. 2019, 10, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, C.M.; Minto, A.W.; Dorf, M.E.; Proudfoot, A.; Wells, T.N.; Salant, D.J.; Gutierrez-Ramos, J.C. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J. Exp. Med. 1997, 185, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Eis, V.; Luckow, B.; Vielhauer, V.; Siveke, J.T.; Linde, Y.; Segerer, S.; Perez De Lema, G.; Cohen, C.D.; Kretzler, M.; Mack, M.; et al. Chemokine receptor CCR1 but not CCR5 mediates leukocyte recruitment and subsequent renal fibrosis after unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2004, 15, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Keepers, T.R.; Gross, L.K.; Obrig, T.G. Monocyte chemoattractant protein 1, macrophage inflammatory protein 1 alpha, and RANTES recruit macrophages to the kidney in a mouse model of hemolytic-uremic syndrome. Infect. Immun. 2007, 75, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Vielhauer, V.; Anders, H.J.; Mack, M.; Cihak, J.; Strutz, F.; Stangassinger, M.; Luckow, B.; Gröne, H.J.; Schlöndorff, D. Obstructive nephropathy in the mouse: Progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J. Am. Soc. Nephrol. JASN 2001, 12, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Segerer, S.; Nelson, P.J.; Schlöndorff, D. Chemokines, chemokine receptors, and renal disease: From basic science to pathophysiologic and therapeutic studies. J. Am. Soc. Nephrol. JASN 2000, 11, 152–176. [Google Scholar] [CrossRef]
- Wada, T.; Furuichi, K.; Segawa-Takaeda, C.; Shimizu, M.; Sakai, N.; Takeda, S.I.; Takasawa, K.; Kida, H.; Kobayashi, K.I.; Mukaida, N.; et al. MIP-1alpha and MCP-1 contribute to crescents and interstitial lesions in human crescentic glomerulonephritis. Kidney Int. 1999, 56, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Rudemiller, N.P.; Patel, M.B.; Zhang, J.D.; Jeffs, A.D.; Karlovich, N.S.; Griffiths, R.; Kan, M.J.; Buckley, A.F.; Gunn, M.D.; Crowley, S.D. C-C Motif Chemokine 5 Attenuates Angiotensin II-Dependent Kidney Injury by Limiting Renal Macrophage Infiltration. Am. J. Pathol. 2016, 186, 2846–2856. [Google Scholar] [CrossRef] [Green Version]
- Crijns, H.; Vanheule, V.; Proost, P. Targeting Chemokine-Glycosaminoglycan Interactions to Inhibit Inflammation. Front. Immunol. 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Vanheule, V.; Crijns, H.; Poosti, F.; Ruytinx, P.; Berghmans, N.; Gerlza, T.; Ronsse, I.; Pörtner, N.; Matthys, P.; Kungl, A.J.; et al. Anti-inflammatory effects of the GAG-binding CXCL9(74-103) peptide in dinitrofluorobenzene-induced contact hypersensitivity in mice. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2018, 48, 1333–1344. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Campanella, G.S.; Guo, R.; Yu, S.; Xie, T.; Liu, N.; Jung, Y.; Homer, R.; Meltzer, E.B.; et al. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J. Clin. Investig. 2010, 120, 2049–2057. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmers, C.; Schulte, C.; Wollenhaupt, J.; Wong, D.W.L.; Harlacher, E.; Orth-Alampour, S.; Klinkhammer, B.M.; Schirmer, S.H.; Böhm, M.; Marx, N.; et al. Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis. Biomedicines 2022, 10, 420. https://doi.org/10.3390/biomedicines10020420
Hemmers C, Schulte C, Wollenhaupt J, Wong DWL, Harlacher E, Orth-Alampour S, Klinkhammer BM, Schirmer SH, Böhm M, Marx N, et al. Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis. Biomedicines. 2022; 10(2):420. https://doi.org/10.3390/biomedicines10020420
Chicago/Turabian StyleHemmers, Christian, Corinna Schulte, Julia Wollenhaupt, Dickson W. L. Wong, Eva Harlacher, Setareh Orth-Alampour, Barbara Mara Klinkhammer, Stephan H. Schirmer, Michael Böhm, Nikolaus Marx, and et al. 2022. "Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis" Biomedicines 10, no. 2: 420. https://doi.org/10.3390/biomedicines10020420
APA StyleHemmers, C., Schulte, C., Wollenhaupt, J., Wong, D. W. L., Harlacher, E., Orth-Alampour, S., Klinkhammer, B. M., Schirmer, S. H., Böhm, M., Marx, N., Speer, T., Boor, P., Jankowski, J., & Noels, H. (2022). Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis. Biomedicines, 10(2), 420. https://doi.org/10.3390/biomedicines10020420