
Drug Design Targeting the Muscarinic Receptors and the Implications in Central Nervous System Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Drug Design Targeting the mAChRs: Alzheimer’s Disease
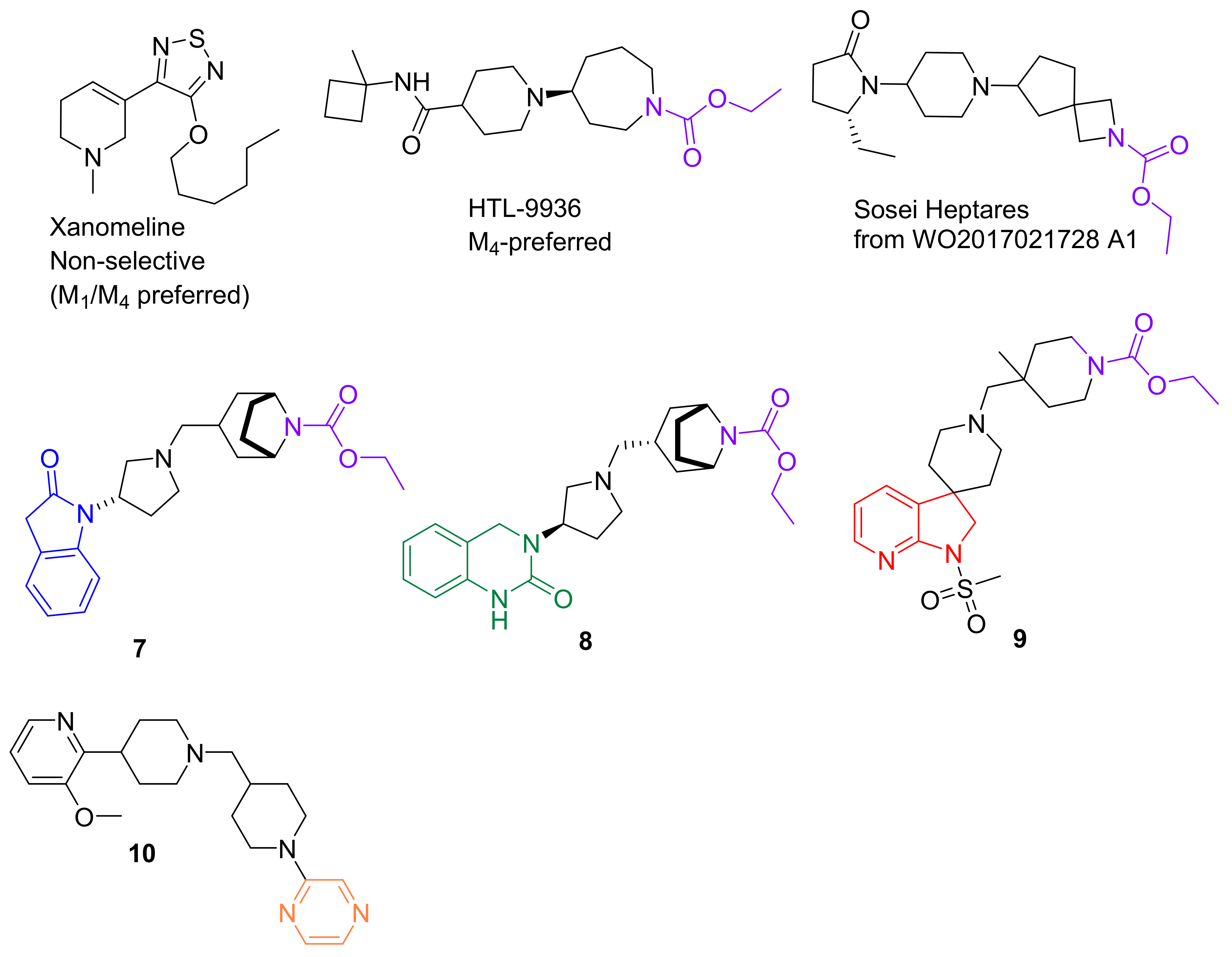
2.1. Orthosteric Agonists
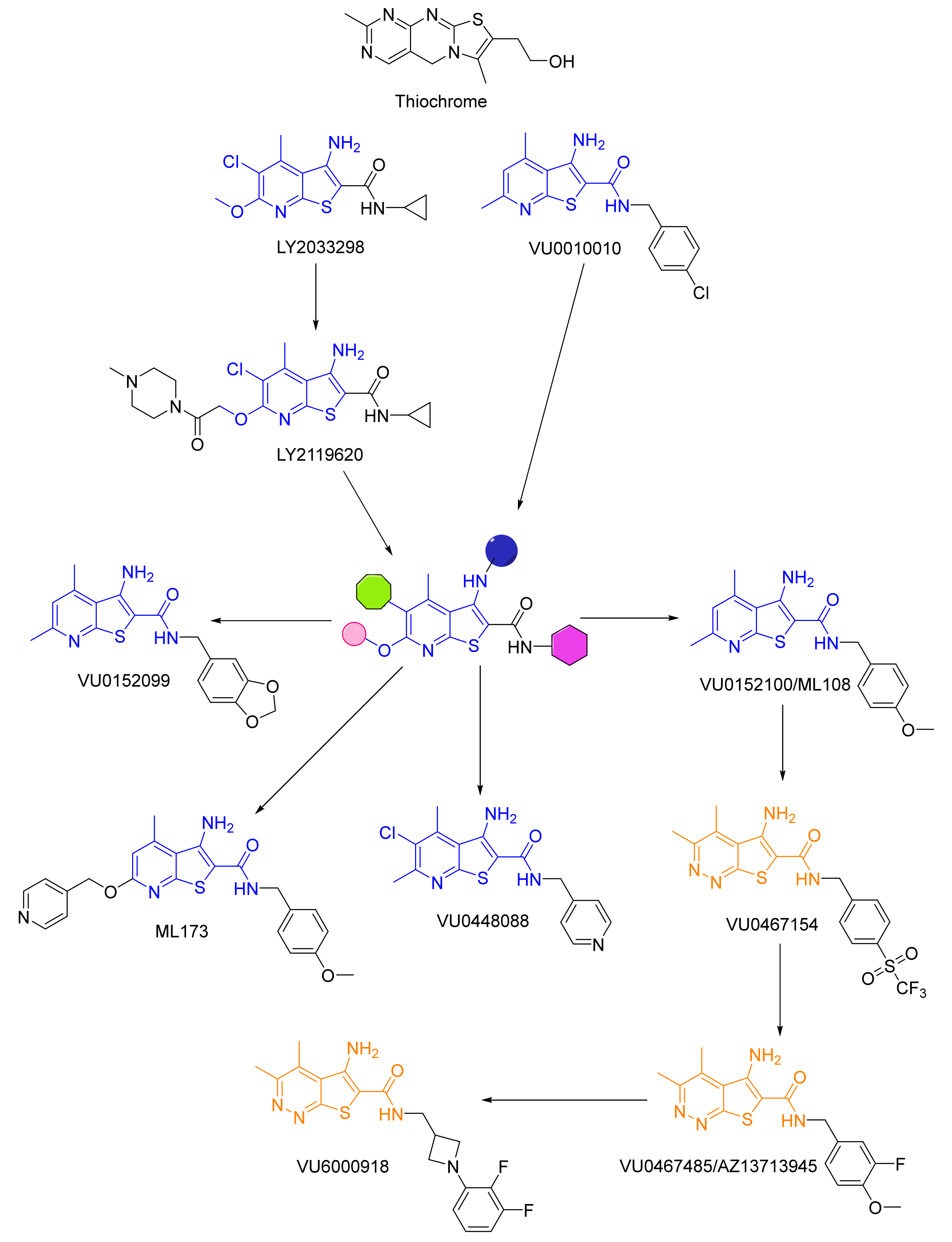
2.2. Allosteric Modulators
3. Drug Design Targeting the mAChRs: Schizophrenia
3.1. Orthosteric Agonists
3.2. M4-Positive Allosteric Modulators
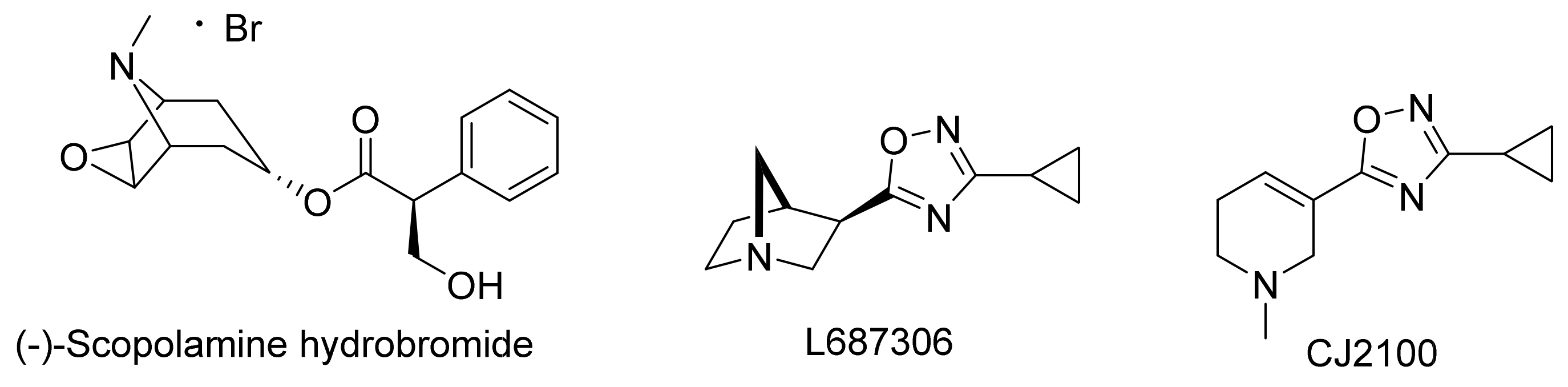
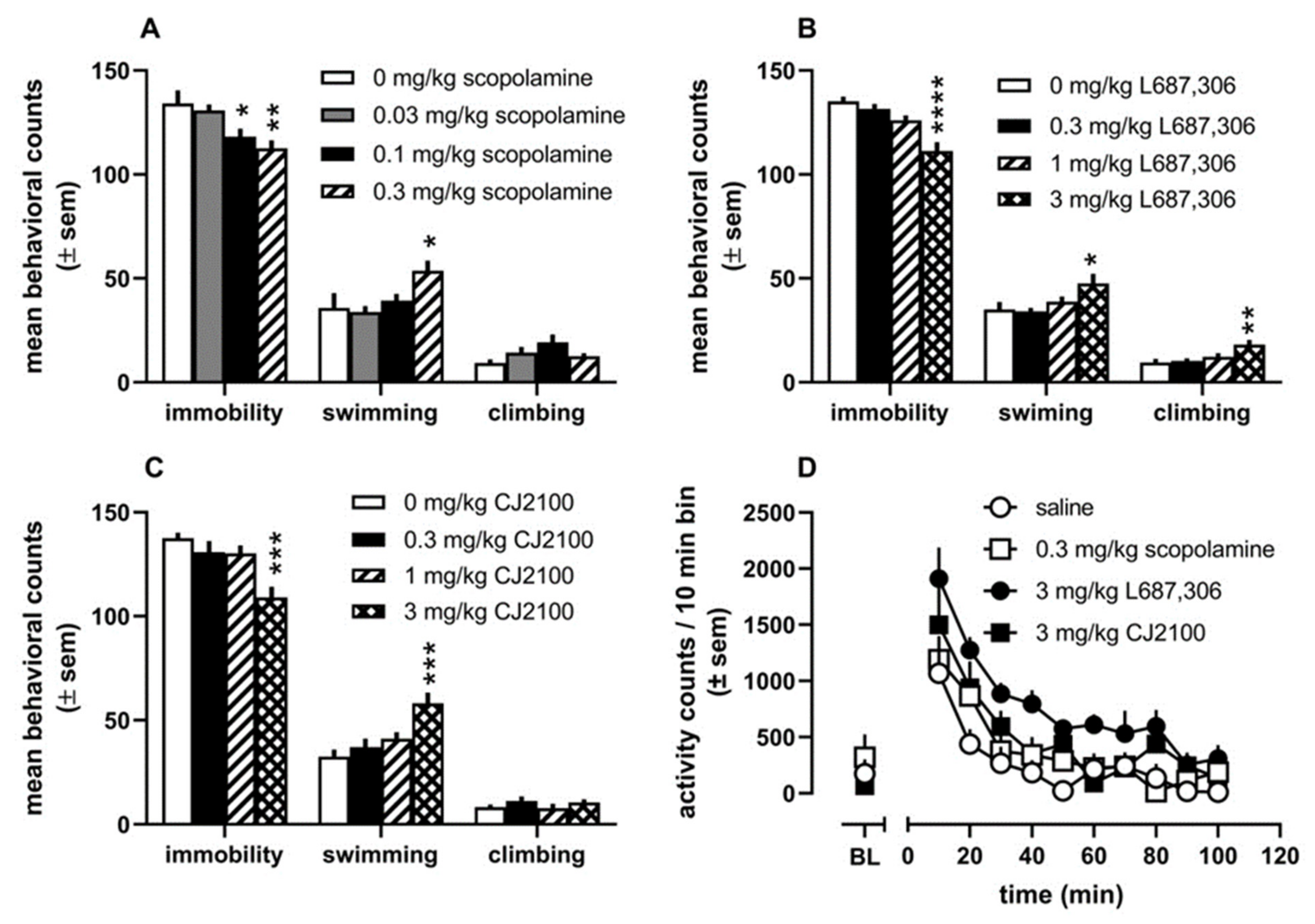
4. Drug Design Targeting the mAChRs: Major Depressive Disorder
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupling receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed]
- Lagerström, M.C.; Schiöth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef]
- Conn, P.M.; Ulloa-Aguirre, A.; Ito, J.; Janovick, J.A. G Protein-Coupled Receptor Trafficking in Health and Disease: Lessons Learned to Prepare for Therapeutic Mutant Rescue in Vivo. Pharmacol. Rev. 2007, 59, 225–250. [Google Scholar] [CrossRef]
- Fang, Y.; Kenakin, T.; Liu, C. Editorial: Orphan GPCRs As Emerging Drug Targets. Front. Pharmacol. 2015, 6, 295. [Google Scholar] [CrossRef]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic acetylcholine receptors: Mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar] [CrossRef]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef]
- Wess, J. Novel Muscarinic Receptor Mutant Mouse Models. Handb. Exp. Pharmacol. 2011, 95–117. [Google Scholar] [CrossRef]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia n senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurones in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Nilsson, L.; Nordberg, A.; Hardy, J.; Wester, P.; Winblad, B. Physostigmine restores3H-acetylcholine efflux from Alzheimer brain slices to normal level. J. Neural Transm. 1986, 67, 275–285. [Google Scholar] [CrossRef]
- Rylett, R.; Ball, M.; Colhoun, E. Evidence for high affinity choline transport in synaptosomes prepared from hippocampus and neocortex of patients with Alzheimer’s disease. Brain Res. 1983, 289, 169–175. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean III, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Fillit, H.; Hill, J.W.; Futterman, R. Health care utilization and costs of Alzheimer’s disease: The role of co-morbid conditions, disease stage and pharmacotherapy. Fam. Med. 2002, 34, 528–535. [Google Scholar]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Mouradian, M.M.; Mohr, E.; Williams, J.A.; Chase, T.N. No response to high-dose muscarinic agonist therapy in Alzheimer’s disease. Neurology 1988, 38, 606. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Thinakaran, G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 2006, 66 (Suppl. 1), S69–S73. [Google Scholar] [CrossRef] [PubMed]
- Caine, E.D. Cholinomimetic Treatment Fails to Improve Memory Disorders. New Engl. J. Med. 1980, 303, 585–586. [Google Scholar] [CrossRef]
- Barten, D.M.; Albright, C.F. Therapeutic strategies for Alzheimer’s disease. Mol. Neurobiol. 2008, 37, 171–186. [Google Scholar] [CrossRef]
- Soukup, O.; Winder, M.; Killi, U.K.; Wsól, V.; Jun, D.; Kuca, K.; Tobin, G. Acetylcholinesterase Inhibitors and Drugs Acting on Muscarinic Receptors-Potential Crosstalk of Cholinergic Mechanisms During Pharmacological Treatment. Curr. Neuropharmacol. 2017, 15, 637–653. [Google Scholar] [CrossRef]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 41, 615–631. [Google Scholar] [CrossRef]
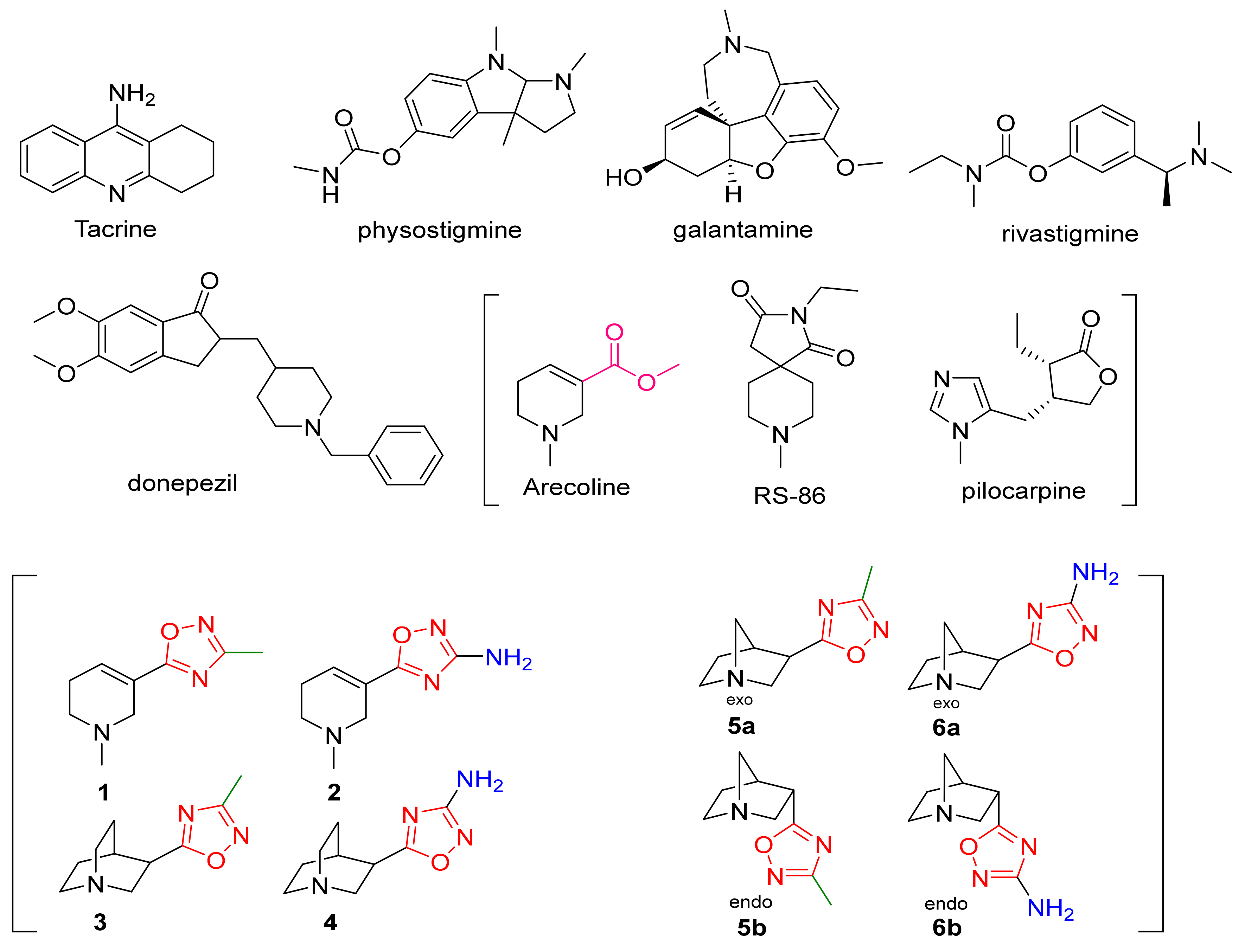
- Street, L.J.; Baker, R.; Book, T.; Kneen, C.O.; MacLeod, A.M.; Merchant, K.J.; Showell, G.A.; Saunders, J.; Herbert, R.H.; Freedman, S.B.; et al. Synthesis and biological activity of 1,2,4-oxadiazole derivatives: Highly potent and efficacious agonists for cortical muscarinic receptors. J. Med. Chem. 1990, 33, 2690–2697. [Google Scholar] [CrossRef]
- Perry, E.K. The cholinergic hypothesis--ten years on. Br. Med. Bull. 1986, 42, 63–69. [Google Scholar] [CrossRef]
- Perry, E. Acetylcholine and Alzheimer’s Disease. Br. J. Psychiatry 1988, 152, 737–740. [Google Scholar] [CrossRef]
- Sims, N.R.; Bowen, D.M.; Allen, S.J.; Smith, C.C.; Neary, D.; Thomas, D.J.; Davidson, A.N. Presynaptic cholinergic dysfunction in patients with dementia. J. Neurochem. 1983, 40, 503–509. [Google Scholar] [CrossRef]
- Wei, J.; Walton, E.A.; Milici, A.; Buccafusco, J.J. m1-m5 Muscarinic Receptor Distribution in Rat CNS by RT-PCR and HPLC. J. Neurochem. 2002, 63, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Davoren, J.E.; Garnsey, M.; Pettersen, B.; Brodney, M.A.; Edgerton, J.R.; Fortin, J.P.; Grimwood, S.; Harris, A.R.; Jenkinson, S.; Kenakin, T.; et al. Design and Synthesis of γ- and δ-Lactam M1 Positive Allosteric Modulators (PAMs): Convulsion and Cholinergic Toxicity of an M1-Selective PAM with Weak Agonist Activity. J. Med. Chem. 2017, 60, 6649–6663. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, M.; Neugebauer, N.M.; Sato, T.; Ardehali, H.; Meltzer, H.Y. Muscarinic receptor signaling contributes to atypical anti-psychotic drug reversal of the phencyclidine-induced deficit in novel object recognition in rats. J. Psychopharmacol. 2017, 31, 1588–1604. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.W.; Dencker, D.; Grannan, M.; Bubser, M.; Zhan, X.; Wess, J.; Xiang, Z.; Locuson, C.; Lindsley, C.W.; Conn, P.J.; et al. Role for the M1 Muscarinic Acetylcholine Receptor in Top-Down Cognitive Processing Using a Touchscreen Visual Discrimination Task in Mice. ACS Chem. Neurosci. 2015, 6, 1683–1695. [Google Scholar] [CrossRef]
- Anagnostaras, S.G.; Murphy, G.G.; Hamilton, S.E.; Mitchell, S.L.; Rahnama, N.P.; Nathanson, N.M.; Silva, A.J. Selective cognitive dys-function in acetylcholine M1 muscarinic receptor mutant mice. Nat. Neurosci. 2003, 6, 51–58. [Google Scholar] [CrossRef]
- Lebois, E.P.; Thorn, C.; Edgerton, J.R.; Popiolek, M.; Xi, S. Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer’s disease. Neuropharmacology 2018, 136, 362–373. [Google Scholar] [CrossRef]
- Foster, D.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef]
- Bubser, M.; Byun, N.; Wood, M.R.; Jones, C.K. Muscarinic Receptor Pharmacology and Circuitry for the Modulation of Cognition. Handb. Exp. Pharmacol. 2011, 121–166. [Google Scholar] [CrossRef]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martínez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 Receptors Play a Central Role in Modulating AD-like Pathology in Transgenic Mice. Neuron 2006, 49, 671–682. [Google Scholar] [CrossRef]
- Jones, C.K.; Brady, A.E.; Davis, A.A.; Xiang, Z.; Bubser, M.; Tantawy, M.N.; Kane, A.S.; Bridges, T.M.; Kennedy, J.P.; Bradley, S.R.; et al. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 2008, 28, 10422–10433. [Google Scholar] [CrossRef]
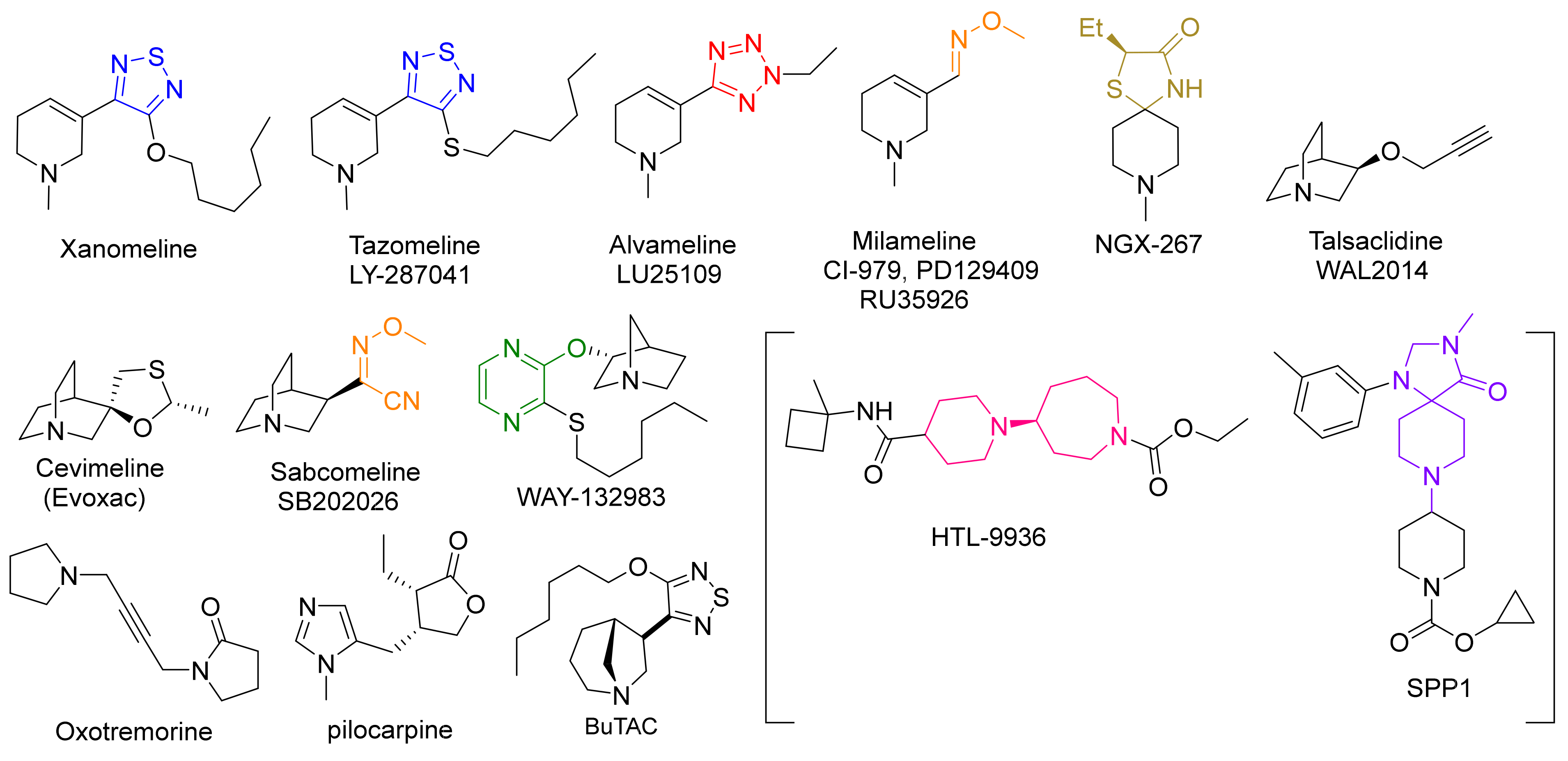
- Sauerberg, P.; Olesen, P.H.; Nielson, S.; Treppendahl, S.; Sheardown, M.J.; Honore, T.; Mitch, C.H.; Ward, J.S.; Pike, A.J. Novel functional M1 selective muscarinic agonists. Synthesis and structure-activity relationships of 3-(1,2,5-thiadiazolyl)-1,2,5,6-tetrahydro-1-methylpyridines. J. Med. Chem. 1992, 35, 2274–2283. [Google Scholar] [CrossRef]
- Ward, J.S.; Merritt, L.; Klimkowski, V.J.; Lamb, M.L.; Mitch, C.H.; Bymaster, F.P.; Sawyer, B.; Shannon, H.E.; Olesen, P.H. Novel functional M1 selective muscarinic agonists. Synthesis and structure-activity relationships of 3-pyrazinyl-1,2,5,6-tetrahydro-1-methylpyridines. Construction of a molecular model for the M1 pharmacophore. J. Med. Chem. 1992, 35, 4011–4019. [Google Scholar] [CrossRef]
- Bodick, N.C.; Offen, W.W.; Levey, A.I.; Cutler, N.R.; Gauthier, S.G.; Satlin, A.; Shannon, H.E.; Tollefson, G.D.; Rasmussen, K.; Bymaster, F.P.; et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer’s disease. Arch. Neurol. 1997, 54, 465–473. [Google Scholar] [CrossRef]
- Shekhar, A.; Potter, W.Z.; Lightfoot, J.; Lienemann, J.; Dube, S.; Mallinckrodt, C.; Bymaster, F.P.; McKinzie, D.L.; Felder, C.C. Selective Muscarinic Receptor Agonist Xanomeline as a Novel Treatment Approach for Schizophrenia. Am. J. Psychiatry 2008, 165, 1033–1039. [Google Scholar] [CrossRef]
- Melancon, B.J.; Tarr, J.C.; Panarese, J.D.; Wood, M.R.; Lindsley, C.W. Allosteric modulation of the M1 muscarinic acetylcholine re-ceptor: Improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discov. Today 2013, 18, 1185–1199. [Google Scholar] [CrossRef]
- Noetzel, M.J.; Grant, M.K.O.; El-Fakahany, E.E. Immediate and Delayed Consequences of Xanomeline Wash-Resistant Binding at the M3 Muscarinic Receptor. Neurochem. Res. 2008, 34, 1138–1149. [Google Scholar] [CrossRef]
- Messer, W.S. The utility of muscarinic agonists in the treatment of alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 187–193. [Google Scholar] [CrossRef]
- Mirza, N.R.; Peters, D.; Sparks, R.G. Xanomeline and the Antipsychotic Potential of Muscarinic Receptor Subtype Selective Agonists. CNS Drug Rev. 2006, 9, 159–186. [Google Scholar] [CrossRef]
- Ward, J.S.; Merritt, L.; Calligaro, D.O.; Bymaster, F.P.; Shannon, H.E.; Sawyer, B.D.; Mitch, C.H.; Deeter, J.B.; Peters, S.C. Functionally selective M1 muscarinic agonists. Side chain and azacycles contributing to functional muscarinic selectivity among pyrazinylazacycles. J. Med. Chem. 1995, 38, 3469–3481. [Google Scholar] [CrossRef]
- Moltzen, E.K.; Pedersen, H.; Boegesoe, K.P.; Meier, E.; Frederiksen, K.; Sanchez, C.; Lemboel, H.L. Bioisosteres of Arecoline: 1,2,3,6-Tetrahydro-5-pyridyl-Substituted and 3-Piperidyl-Substituted Derivatives of Tetrazoles and 1,2,3-Triazoles. Synthesis and Muscarinic Activity. J. Med. Chem. 1994, 37, 4085–4099. [Google Scholar] [CrossRef]
- Bromidge, S.M.; Brown, F.; Cassidy, F.; Clark, M.S.; Dabbs, S.; Hadley, M.S.; Loudon, J.M.; Orlek, B.S.; Riley, G.J. Design and synthesis of azabicyclic muscarinic agonists incorporating an oxime ether functionality. Bioorganic Med. Chem. Lett. 1992, 2, 787–790. [Google Scholar] [CrossRef]
- Bromidge, S.M.; Brown, F.; Cassidy, F.; Clark, M.S.G.; Dabbs, S.; Hadley, M.S.; Hawkins, J.; Loudon, J.M.; Naylor, C.B.; Orlek, B.S.; et al. Design of [R-(Z)]-(+)-α-(Methoxyimino)-1-azabicyclo[2.2.2]octane-3-acetonitrile (SB 202026), a Functionally Selective Azabicyclic Muscarinic M1 Agonist Incorporating the N-Methoxy Imidoyl Nitrile Group as a Novel Ester Bioisostere. J. Med. Chem. 1997, 40, 4265–4280. [Google Scholar] [CrossRef] [PubMed]
- Korczyn, A.D. Muscarinic M1 agonists in the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2000, 9, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Sramek, J.J.; Forrest, M.; Mengel, H.; Jhee, S.S.; Hourani, J.; Cutler, N.R. A Bridging Study of LU 25-109 in Patients with Probably Alzheimer’s Disease. Life Sci. 1997, 62, 195–202. [Google Scholar] [CrossRef]
- Ono, M.; Takamura, E.; Shinozaki, K.; Tsumura, T.; Hamano, T.; Yagi, Y.; Tsubota, K. Therapeutic effect of cevimeline on dry eye in patients with Sjogren’s syndrome: A randomized, double-blind clinical study. Am. J. Ophthalmol. 2004, 138, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Sedman, A.J.; Bockbrader, H.; Schwarz, R.D. Preclinical and phase 1 clinical characterization of CI-979/RU35926, a novel muscarinic agonist for the treatment of Alzheimer’s diseases. Life Sci. 1995, 56, 877–882. [Google Scholar] [CrossRef]
- TorreyPines Therapeutics Muscarinic Agonist NGX267 Meets Primary Endpoint in a Phase II Clinical Trial in Patients with Xerostom. Available online: https://www.fiercebiotech.com/biotech/torreypines-therapeutics-muscarinic-agonist-ngx267-meets-primary-endpoint-a-phase-ii (accessed on 18 November 2021).
- Adamus, W.S.; Leonard, J.; Troger, W. Phase I clinical trials with WAL 2014, a new muscarinic agonist for the treatment of Alz-heimer’s disease. Life Sci. 1995, 56, 883–890. [Google Scholar] [CrossRef]
- Loudon, J.M.; Bromidge, S.M.; Brown, F.; Clark, M.S.G.; Hatcher, J.P.; Hawkins, J.; Riley, G.J.; Noy, G.; Orlek, B.S. SB 202026: A Novel Muscarinic Partial Agonist with Functional Selectivity for M1 Receptors. J. Pharmacol. Exp. Ther. 1997, 283, 1059–1068. [Google Scholar]
- Heinrich, J.N.; Butera, J.A.; Carrick, T.; Kramer, A.; Kowal, D.; Lock, T.; Marquis, K.L.; Pausch, M.H.; Popiolek, M.; Sun, S.C.; et al. Pharmacological comparison of muscarinic ligands: Historical versus more recent muscarinic M1-preferring receptor agonists. Eur. J. Pharmacol. 2009, 605, 53–56. [Google Scholar] [CrossRef]
- Collingwood, S.P.; Ratcliffe, A.J.; Pryde, D.; Porter, R. Recent disclosures of clinical candidates: Highlights from the Society of Medicines research symposium. Drugs Future 2015, 40, 81–91. [Google Scholar] [CrossRef]
- Brown, A.J.H.; Bradley, S.J.; Marshall, F.H.; Brown, G.A.; Bennett, K.A.; Brown, J.; Cansfield, J.E.; Cross, D.M.; De Graaf, C.; Hudson, B.D.; et al. From structure to clinic: Design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer’s disease. Cell 2021, 184, 5886–5901.e22. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Goldsmith, P.; Jackson, K.; Sanger, H.E.; Evans, D.A.; Mogg, A.J.; Broad, L.M. Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology 2018, 136, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.D.; Patel, H.M.; Surana, S.J.; Belgamwar, V.S.; Pardeshi, C.V. Brain-blood ratio: Implications in brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Schrage, R.; Seemann, W.; Klöckner, J.; Dallanoce, C.; Racké, K.; Kostenis, E.; De Amici, M.; Holzgrabe, U.; Mohr, K. Agonists with supraphysiological efficacy at the muscarinic M2 ACh receptor. J. Cereb. Blood Flow Metab. 2013, 169, 357–370. [Google Scholar] [CrossRef]
- Rajagopal, S.; Ahn, S.; Rominger, D.H.; Gowen-MacDonald, W.; Lam, C.M.; DeWire, S.M.; Violin, J.D.; Lefkowitz, R.J. Quantifying Ligand Bias at Seven-Transmembrane Receptors. Mol. Pharmacol. 2011, 80, 367–377. [Google Scholar] [CrossRef]
- Buchwald, P. A Receptor Model With Binding Affinity, Activation Efficacy, and Signal Amplification Parameters for Complex Fractional Response Versus Occupancy Data. Front. Pharmacol. 2019, 10, 605. [Google Scholar] [CrossRef]
- Sosei Provides Update on HTL0018318. Available online: https://soseiheptares.com/news/109/129/Sosei-Provides-Update-on-HTL0018318.html) (accessed on 1 December 2021).
- Bakker, C.; Tasker, T.; Liptrot, J.; Hart, E.P.; Klaassen, E.S.; Prins, S.; van der Doef, T.F.; Brown, G.A.; Brown, A.; Congreve, M.; et al. First-in-man study to investigate safety, pharmacokinetics and exploratory pharmacodynamics of HTL0018318, a novel M1 -receptor partial agonist for the treatment of dementias. Br. J. Clin. Pharmacol. 2021, 87, 2945–2955. [Google Scholar] [CrossRef]
- Bakker, C.; Tasker, T.; Liptrot, J.; Hart, E.P.; Klaassen, E.S.; Doll, R.J.; Brown, G.A.; Brown, A.; Congreve, M.; Weir, M.; et al. Safety, pharmacokinetics and exploratory pro-cognitive effects of HTL0018318, a selective M1 receptor agonist, in healthy younger adult and elderly subjects: A multiple ascending dose study. Alzheimer’s Res. Ther. 2021, 13, 87. [Google Scholar] [CrossRef]
- Congreve, M.; Brown, G.; Cansfield, J.; Tehan, B. Muscarinic M1 Receptor Agonists. U.S. Patent WO/2013/072705, 23 May 2013. [Google Scholar]
- Broad, L.M.; Sanger, H.E.; Mogg, A.J.; Colvin, E.M.; Zwart, R.; Evans, D.A.; Pasqui, F.; Sher, E.; Wishart, G.N.; Barth, V.N.; et al. Identification and pharmacological profile of SPP1, a potent, functionally selective and brain penetrant agonist at mus-carinic M1 receptors. Br. J. Pharmacol. 2019, 176, 110–126. [Google Scholar] [CrossRef]
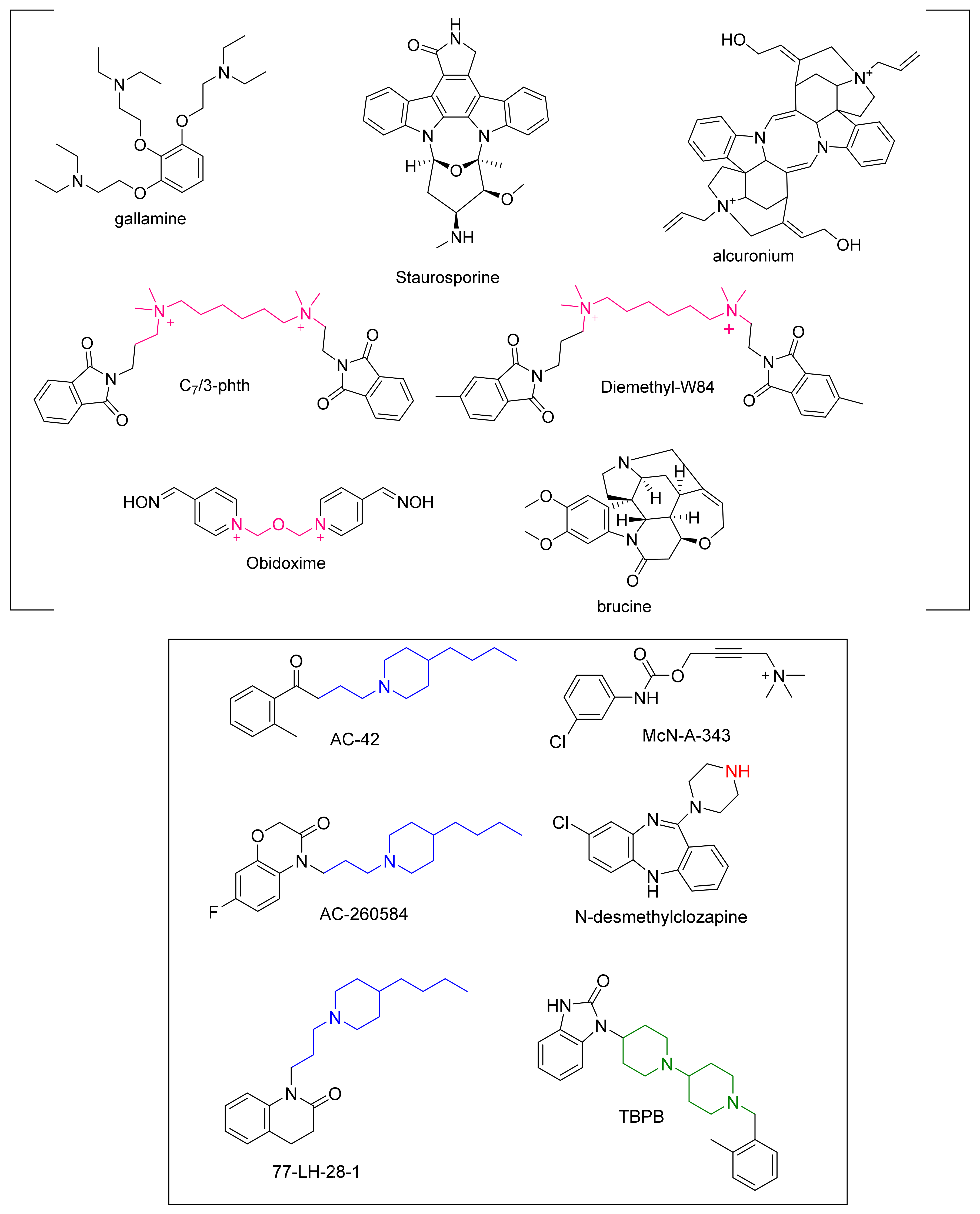
- Clark, A.; Mitchelson, F. The inhibitory effect of gallamine on muscarinic receptors. J. Cereb. Blood Flow Metab. 1976, 58, 323–331. [Google Scholar] [CrossRef]
- Fischer, A. Cholingeric treatments with emphasis on M1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 433–442. [Google Scholar] [CrossRef]
- Fisher, A.; Brandeis, R.; Bar-Ner, R.H.; Kliger-Spatz, M.; Natan, N.; Sonego, H.; Marcovitch, I.; Pittel, Z.A. 150(S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 145–153. [Google Scholar] [CrossRef]
- Lazareno, S.; Dolezal, V.; Popham, A.; Birdsall, N.J.M. Thiochrome Enhances Acetylcholine Affinity at Muscarinic M4 Receptors: Receptor Subtype Selectivity via Cooperativity Rather than Affinity. Mol. Pharmacol. 2004, 65, 257–266. [Google Scholar] [CrossRef]
- Bock, A.; Schrage, R.; Mohr, K. Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 2018, 136, 427–437. [Google Scholar] [CrossRef]
- Kenakin, T.P.; Beek, D. The effects on Schild regressions of antagonist removal from the receptor compartment by a saturable process. Naunyn-Schmiedebergs Arch. fur Exp. Pathol. Pharmakol. 1987, 335, 103–108. [Google Scholar] [CrossRef]
- Proska, J.; Tucek, S. Mechanisms of steric and cooperative actions of alcuronium on cardiac muscarinic acetylcholine receptors. Mol. Pharmacol. 1994, 45, 709–717. [Google Scholar]
- Tucek, S.; Musilkova, J.; Nedoma, J.; Proska, J.; Shelkovnikov, S.; Vorlícek, J. Positive cooperativity in the binding of alcuronium and N-methylscopolamine to muscarinic acetylcholine receptors. Mol. Pharmacol. 1990, 38, 674–680. [Google Scholar]
- Lanzafame, A.; Christopoulos, A.; Mitchelson, F. Three allosteric modulators act a common site, distinct from that of competitive antagonists, at muscarinic acetylcholine M2 receptors. J. Pharmacol. Exp. Ther. 1997, 282, 278–285. [Google Scholar]
- Lazareno, S.; Popham, A.; Birdsall, N.J.M. Allosteric Interactions of Staurosporine and Other Indolocarbazoles withN-[methyl-3H]Scopolamine and Acetylcholine at Muscarinic Receptor Subtypes: Identification of a Second Allosteric Site. Mol. Pharmacol. 2000, 58, 194–207. [Google Scholar] [CrossRef]
- Birdsall, N.J.; Burgen, A.S.; Hulme, E.C.; Stockton, J.M.; Zigmond, M.J. The effect of McN-A-343 on muscarinic receptors in the cerebral cortex and heart. Br. J. Pharmacol. 1983, 78, 257–259. [Google Scholar] [CrossRef]
- Waelbroeck, M. Identification of drugs competing with d-tubocurarine for an allosteric site on cardiac muscarinic receptors. Mol. Pharmacol. 1994, 46, 685–692. [Google Scholar]
- Mitchelson, F.J. The pharmacology of McN-A-343. Pharmacol. Ther. 2012, 135, 216–245. [Google Scholar] [CrossRef]
- Langmead, C.J.; Fry, V.A.; Forbes, I.T.; Branch, C.L.; Christopoulos, A.; Wood, M.D.; Herdon, H.J. Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the muscarinic M1 receptor: Di-rect pharmacological evidence that AC-42 is an allosteric agonist. Mol. Pharmacol. 2006, 69, 236–246. [Google Scholar] [CrossRef]
- Spalding, T.A.; Ma, J.N.; Ott, T.R.; Friberg, M.; Bajpai, A.; Bradley, S.R.; Davis, R.E.; Brann, M.R.; Burstein, E.S. Structural requirements of transmembrane domain 3 for activation by the M1 muscarinic receptor agonists AC-42, AC-260584, Clozapine, and N-Desmethylclozapine: Evidence for three distinct modes of receptor activation. Mol. Pharmacol. 2006, 70, 1974–1983. [Google Scholar] [CrossRef]
- Bradley, S.R.; Lameh, J.; Ohrmund, L.; Son, T.; Bajpai, A.; Nguyen, D.; Friberg, M.; Burstein, E.S.; Spalding, T.A.; Ott, T.R.; et al. AC-260584, an orally bioavailable M1 muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology 2010, 58, 365–373. [Google Scholar] [CrossRef]
- Vanover, K.E.; Veinbergs, I.; Davis, R.E. Antipsychotic-like behavioral effects and cognitive enhancement by a potent and selective muscarinic M-sub-1 receptor agonist, AC-260584. Behav. Neurosci. 2008, 122, 570–575. [Google Scholar] [CrossRef]
- Langmead, C.J.; Austin, N.E.; Branch, C.L.; Brown, J.T.; Buchanan, K.A.; Davies, C.H.; Forbes, I.T.; Fry, V.A.H.; Hagan, J.J.; Herdon, H.J.; et al. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Brit. J. Pharmacol. 2008, 154, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.K.; Brady, A.E.; Bubser, M.; Deutch, A.Y.; Williams, L.C.; Hammond, A.; Willliams, R.; Conn, P.J. TBPB is a highly selective M1 allosteric muscarinic receptor agonist in vitro and produces robust antipsychotic-like effects in vivo. Neuropsychopharmacology 2006, 31, S116. [Google Scholar]
- Conn, P.J.; Jones, C.K.; Lindsley, C.W. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 2009, 30, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Seager, M.A.; Wittmann, M.; Jacobson, M.; Bickel, D.; Burno, M.; Jones, K.; Graufelds, V.K.; Xu, G.; Pearson, M.; et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 15950–15955. [Google Scholar] [CrossRef] [PubMed]
- Shirey, J.K.; Brady, A.E.; Jones, P.J.; Davis, A.A.; Bridges, T.M.; Kennedy, J.P.; Jadhav, S.B.; Menon, U.N.; Xiang, Z.; Watson, M.L.; et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci. 2009, 29, 14271–14286. [Google Scholar] [CrossRef]
- Kuduk, S.D.; Chang, R.K.; Di Marco, C.N.; Ray, W.J.; Ma, L.; Wittmann, M.; Seager, M.A.; Koeplinger, K.A.; Thompson, C.D.; Hartman, G.D.; et al. Quinolizidinone carboxylic acids as CNS penetrant, selective m1 allosteric mus-carinic receptor modulators. ACS Med. Chem. Lett. 2010, 1, 263–267. [Google Scholar] [CrossRef]
- Kuduk, S.D.; Di Marco, C.N.; Chang, R.K.; Ray, W.J.; Ma, L.; Wittmann, M.; Seager, M.A.; Koeplinger, K.A.; Thompson, C.D.; Hartman, G.D.; et al. Heterocyclic fused pyridone carboxylic acid M(1) positive allosteric modulators. Bioorg. Med. Chem. Lett. 2010, 20, 2533–2537. [Google Scholar] [CrossRef]
- Kuduk, S.D.; Di Marco, C.N.; Cofre, V.; Pitts, D.R.; Ray, W.J.; Ma, L.; Wittmann, M.; Seager, M.A.; Koeplinger, K.; Thompson, C.D.; et al. Pyridine containing M(1) positive allosteric modulators with reduced plasma protein binding. Bioorg. Med. Chem. Lett. 2010, 20, 657–661. [Google Scholar] [CrossRef]
- Kuduk, S.D.; Chang, R.K.; Greshock, T.J.; Ray, W.J.; Ma, L.; Wittmann, M.; Seager, M.A.; Koeplinger, K.A.; Thompson, C.D.; Hartman, G.D.; et al. Identification of Amides as Carboxylic Acid Surrogates for Quinolizidinone-Based M1 Positive Allosteric Modulators. ACS Med. Chem. Lett. 2012, 3, 1070–1074. [Google Scholar] [CrossRef][Green Version]
- Yang, Z.Q.; Shu, Y.; Ma, L.; Wittmann, M.; Ray, W.J.; Seager, M.A.; Koeplinger, K.A.; Thompson, C.D.; Hartman, G.D.; Bilodeau, M.T.; et al. Discovery of naphthyl-fused 5-membered lactams as a new class of m1 positive allosteric mod-ulators. ACS Med. Chem. Lett. 2014, 5, 604–608. [Google Scholar] [CrossRef]
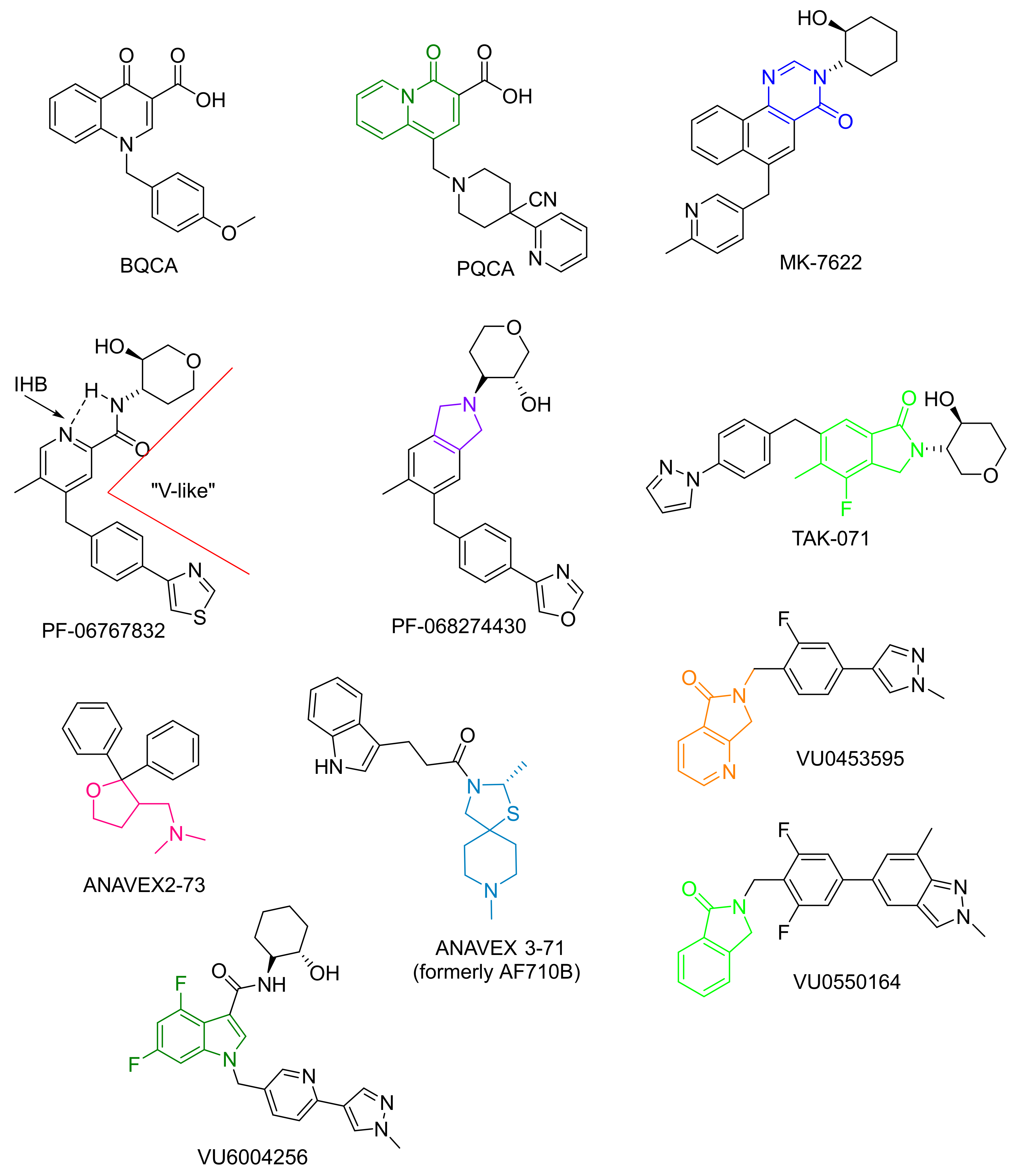
- Beshore, D.C.; Di Marco, N.C.; Chang, R.K.; Greshock, T.J.; Ma, L.; Wittmann, M.; Seager, M.A.; Koeplinger, K.A.; Thompson, C.D.; Fuerst, J.; et al. MK-7622: A First-in-Class M1 Positive Allosteric Modulator Development Candidate. ACS Med. Chem. Lett. 2018, 9, 652–656. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front Oncol. 2014, 4, 41. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Brown, P.W.; Ozer, J.S.; Lai, Y. Liquid chromatography/tandem mass spectrometry based targeted proteomics quantification of P-glycoprotein in various biological samples. Rapid Commun. Mass Spectrom. 2011, 25, 1715–1724. [Google Scholar] [CrossRef]
- Watanabe, R.; Esaki, T.; Ohashi, R.; Kuroda, M.; Kawashima, H.; Komura, H.; Natsume-Kitatani, Y.; Mizuguchi, K. Development of an In Silico Prediction Model for P-glycoprotein Efflux Potential in Brain Capillary Endothelial Cells toward the Prediction of Brain Penetration. J. Med. Chem. 2021, 64, 2725–2738. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.; Li, J.; Cummings, J.; Farlow, M.; Assaid, C.; Froman, S.; Leibensperger, H.; Snow-Adami, L.; McMahon, K.B.; Egan, M.; et al. Randomized, controlled, proof-of-concept trial of MK-7622 in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Rook, J.M.; Abe, M.; Cho, H.P.; Nance, K.D.; Luscombe, V.B.; Adams, J.J.; Dickerson, J.W.; Remke, D.H.; Garcia-Barrantes, P.M.; Engers, D.W.; et al. Diverse Effects on M1 Signaling and Adverse Effect Liability within a Series of M1 Ago-PAMs. ACS Chem. Neurosci. 2017, 8, 866–883. [Google Scholar] [CrossRef] [PubMed]
- Alt, A.; Pendri, A.; Bertekap, R.L., Jr.; Li, G.; Benitex, Y.; Nophsker, M.; Rockwell, K.L.; Burford, N.T.; Sum, C.S.; Chen, J.; et al. Evidence for Classical Cholinergic Toxicity Associated with Selective Activation of M1 Muscarinic Receptors. J. Pharmacol. Exp. Ther. 2016, 356, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Kuduk, S.D.; Beshore, D.C. Novel M1allosteric ligands: A patent review. Expert Opin. Ther. Patents 2012, 22, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2018, 62, 88–127. [Google Scholar] [CrossRef] [PubMed]
- Moran, S.P.; Dickerson, J.W.; Cho, H.P.; Xiang, Z.; Maksymetz, J.; Remke, D.H.; Lv, X.; Doyle, C.A.; Rajan, D.H.; Niswender, C.M.; et al. M1-positive allosteric modulators lacking agonist activity provide the op-timal profile for enhancing cognition. Neuropsychopharmacology 2018, 43, 1763–1771. [Google Scholar] [CrossRef]
- Panarese, J.D.; Cho, H.P.; Adams, J.J.; Nance, K.D.; Garcia-Barrantes, P.M.; Chang, S.; Morrison, R.D.; Blobaum, A.L.; Niswender, C.M.; Stauffer, S.R.; et al. Further optimization of the M1 PAM VU0453595: Discovery of novel hetero-bicyclic core motifs with improved CNS penetration. Bioorg Med. Chem. Lett. 2016, 26, 3822–3825. [Google Scholar] [CrossRef][Green Version]
- Conley, A.C.; Key, A.P.; Blackford, J.U.; Rook, J.M.; Conn, J.; Lindsley, C.W.; Jones, C.K.; Newhouse, P.A. Cognitive performance effects following a single dose of the M 1 muscarinic positive allosteric modulator VU319. Alzheimer’s Dement. 2020, 16, e045339. [Google Scholar] [CrossRef]
- Newhouse, P.A.; Conley, A.C.; Key, A.P.; Blackford, J.U.; Rook, J.M.; Conn, J.; Lindsley, C.W.; Jones, C.K. Safety and pharmacokinetics of the muscarinic positive allosteric modulator VU319: A phase 1 single dose study. Alchaimer’s Dement. 2020, 16, e045359. [Google Scholar] [CrossRef]
- Vamvakidès, A. Effets anticonvulsivant et anti-immobilité (nage forcée) de la tétrahydro-N, N-diméthyl-2, 2-diphényl-3-furaneméthanamine (AE37) [Anticonvulsant and forced swim anti-immobility effects of tetrahy-dro-N, N-dimethyl-2,2-diphenyl-3-furanemethanamine (AE37): Common action mechanism]. Ann. Pharm. Fr. 2002, 60, 88–92. [Google Scholar]
- Lahmy, V.; Meunier, J.; Malmström, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of Tau hyperphosphorylation and A1−42; generation by the ami-notetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ1; receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723. [Google Scholar] [CrossRef]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s dis-ease model. Front. Cell Neurosci. 2015, 8, 463. [Google Scholar] [CrossRef]
- ANAVEX®2-73 (Blarcamesine) Currently in Phase 2b/3 Early Alzheimer’s Disease (AD): Analysis of Cognitive Outcome Measures Relevant to AD of Double-Blind, Multicenter, Placebo-Controlled Phase 2 Clinical Trial in 132 Patients with Parkinson’s Disease Dementia. Available online: https://www.globenewswire.com/en/news-release/2020/11/06/2122112/29248/en/Proof-of-Concept-Controlled-Phase-2-Clinical-Trial-Data-Evaluating-ANAVEX-2-73-blarcamesine-in-Parkinson-s-Disease-Dementia-Presented-at-CTAD-2020-Conference.html (accessed on 2 December 2021).
- Fisher, A.; Bezprozvanny, I.; Wu, L.; Ryskamp, D.A.; Bar-Ner, N.; Natan, N.; Brandeis, R.; Elkon, H.; Nahum, V.; Gershonov, E.; et al. AF710B, a Novel M1/σ1 Agonist with Therapeutic Efficacy in Animal Models of Alzheimer’s Disease. Neurodegener Dis. 2016, 16, 95–110. [Google Scholar] [CrossRef]
- Sako, Y.; Kurimoto, E.; Mandai, T.; Suzuki, A.; Tanaka, M.; Suzuki, M.; Shimizu, Y.; Yamada, M.; Kimura, H. TAK-071, a novel M1 positive allosteric modulator with low cooperativity, im-proves cognitive function in rodents with few cholinergic side effects. Neuropsychopharmacology 2019, 44, 950–960. [Google Scholar] [CrossRef]
- Harvard Medical School, Psychiatry Neuroimaging Laboratory. Available online: http://pnl.bwh.harvard.edu/education/what-is/schizophrenia/ (accessed on 3 December 2021).
- Larson, M.K.; Walker, E.F.; Compton, M.T. Early signs, diagnosis, and therapeutics of the prodromal phase of schizo-phrenia and related psychotic disorders. Expert Rev. Neurother. 2010, 10, 1347–1359. [Google Scholar] [CrossRef]
- Andreasen, N.C.; Carpenter, W.T., Jr. Diagnosis and classification of schizophrenia. Schizophr. Bull. 1993, 19, 199–214. [Google Scholar] [CrossRef]
- Rook, J.M.; Choi, D.L.; Foster, D.; Conn, P.J. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatr. Dis. Treat. 2014, 10, 183–191. [Google Scholar] [CrossRef]
- Borroni, B.; Costanzi, C.; Padovani, A. Genetic susceptibility to behavioural and psychological symptoms in Alzheimer disease. Curr. Alzheimer Res. 2010, 7, 158–164. [Google Scholar] [CrossRef]
- Carlsson, A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1988, 1, 179–186. [Google Scholar] [CrossRef]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.; Kapur, S. The Dopamine Hypothesis of Schizophrenia: Version III—The Final Common Pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, H.; Pavey, G.; Dean, B. Altered levels of dopamine transporter in the frontal pole and dorsal striatum in schizophrenia. npj Schizophr. 2019, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, S.F.; Gomes, F.V.; Grace, A.A. Dysregulation of Midbrain Dopamine System and the Pathophysiology of Schizophrenia. Front. Psychiatry 2020, 11, 613. [Google Scholar] [CrossRef]
- Kesby, J.P.; Eyles, D.; McGrath, J.J.; Scott, J. Dopamine, psychosis and schizophrenia: The widening gap between basic and clinical neuroscience. Transl. Psychiatry 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Lieberman, J.A.; Stroup, T.S.; McEvoy, J.P.; Swartz, M.S.; Rosenheck, R.A.; Perkins, D.O.; Keefe, R.S.; Davis, S.M.; Davis, C.E.; Le-bowitz, B.D.; et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Ef-fectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005, 353, 1209–1223. [Google Scholar] [CrossRef]
- Lally, J.; MacCabe, J.H. Antipsychotic medication in schizophrenia: A review. Br. Med. Bull. 2015, 114, 169–179. [Google Scholar] [CrossRef]
- Raedler, T.J.; Bymaster, F.P.; Tandon, R.; Copolov, D.; Dea, B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 2007, 12, 232–246. [Google Scholar] [CrossRef]
- Foster, D.J.; Jones, C.K.; Conn, P.J. Emerging approaches for treatment of schizophrenia: Modulation of cholinergic sig-naling. Discov. Med. 2012, 14, 413–420. [Google Scholar]
- Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Ac-tions. Int. J. Mol. Sci. 2017, 18, 135. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F.; de Bartolomeis, A. The Glutamatergic Aspects of Schizophrenia Molecular Pathophysiology: Role of the Postsynaptic Density, and Implications for Treatment. Curr. Neuropharmacol. 2014, 12, 219–238. [Google Scholar] [CrossRef]
- De Bartolomeis, A.; Avagliano, C.; Vellucci, L.; D’Ambrosio, L.; Manchia, M.; D’Urso, G.; Buonaguro, E.F.; Iasevoli, F. Translating preclinical findings in clinically relevant new anti-psychotic targets: Focus on the glutamatergic postsynaptic density. Implications for treatment resistant schizophrenia. Neurosci. Biobehav. Rev. 2019, 107, 795–827. [Google Scholar] [CrossRef]
- Coyle, J.T.; Basu, A.; Benneyworth, M.; Balu, D.; Konopaske, G. Glutamatergic Synaptic Dysregulation in Schizophrenia: Therapeutic Implications. Nov. Antischizophrenia Treat. 2012, 267–295. [Google Scholar] [CrossRef]
- Noetzel, M.J.; Jones, C.K.; Conn, P.J. Emerging approaches for treatment of schizophrenia: Modulation of glutamatergic signaling. Discov. Med. 2012, 14, 335–343. [Google Scholar]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Coyle, J.T. Schizophrenia: Basic and Clinical. Neurodegener. Dis. 2017, 15, 255–280. [Google Scholar] [CrossRef]
- Perry, E.K.; Perry, R.H. Acetylcholine and hallucinations: Disease-related compared to drug-induced alterations in human con-sciousness. Brain Cogn. 1995, 28, 240–258. [Google Scholar] [CrossRef]
- Minzenberg, M.J.; Poole, J.H.; Benton, C.; Vinogradov, S. Association of Anticholinergic Load With Impairment of Complex Attention and Memory in Schizophrenia. Am. J. Psychiatry 2004, 161, 116–124. [Google Scholar] [CrossRef]
- Klinkenberg, I.; Blokland, A. The validity of scopolamine as a pharmacological model for cognitive impairment: A review of animal behavioral studies. Neurosci. Biobehav. Rev. 2010, 34, 1307–1350. [Google Scholar] [CrossRef]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Pae, C.-U. Role of the cholinesterase inhibitors in the treatment of schizophrenia. Expert Opin. Investig. Drugs 2013, 22, 293–298. [Google Scholar] [CrossRef]
- Ribeiz, S.R.I.; Bassitt, D.P.; Arrais, J.A.; Avila, R.; Steffens, D.C.; Bottino, C.M.C. Cholinesterase Inhibitors as Adjunctive Therapy in Patients with Schizophrenia and Schizoaffective Disorder. CNS Drugs 2010, 24, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Thakurathi, N.; Vincenzi, B.; Henderson, D.C. Assessing the prospect of donepezil in improving cognitive impairment in patients with schizophrenia. Expert Opin. Investig. Drugs 2012, 22, 259–265. [Google Scholar] [CrossRef] [PubMed]
- McDermott, C.L.; Gray, S.L. Cholinesterase Inhibitor Adjunctive Therapy for Cognitive Impairment and Depressive Symptoms in Older Adults with Depression. Ann. Pharmacother. 2012, 46, 599–605. [Google Scholar] [CrossRef]
- Gomeza, J.; Zhang, L.; Kostenis, E.; Felder, C.; Bymaster, F.; Brodkin, J.; Shannon, H.; Xia, B.; Deng, C.-X.; Wess, J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M4 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 10483–10488. [Google Scholar] [CrossRef]
- Miyakawa, T.; Yamada, M.; Duttaroy, A.; Wess, J. Hyperactivity and Intact Hippocampus-Dependent Learning in Mice Lacking the M1Muscarinic Acetylcholine Receptor. J. Neurosci. 2001, 21, 5239–5250. [Google Scholar] [CrossRef]
- Koshimizu, H.; Leiter, L.M.; Miyakawa, T. M4 muscarinic receptor knockout mice display abnormal social behavior and decreased prepulse inhibition. Mol. Brain 2012, 5, 10. [Google Scholar] [CrossRef]
- Gerber, D.J.; Sotnikova, T.D.; Gainetdinov, R.; Huang, S.Y.; Caron, M.G.; Tonegawa, S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 15312–15317. [Google Scholar] [CrossRef]
- Dean, B.; Hopper, S.; Conn, P.J.; Scarr, E. Changes in BQCA Allosteric Modulation of [3H]NMS Binding to Human Cortex within Schizophrenia and by Divalent Cations. Neuropsychopharmacology 2015, 41, 1620–1628. [Google Scholar] [CrossRef]
- Tzavara, E.T.; Bymaster, F.P.; Davis, R.J.; Wade, M.R.; Perry, K.W.; Wess, J.; McKinzie, D.L.; Felder, C.; Nomikos, G.G. M4 muscarinic re-ceptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: Relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J. 2004, 18, 1410–1412. [Google Scholar] [CrossRef]
- Jeon, J.; Dencker, D.; Wörtwein, G.; Woldbye, D.P.D.; Cui, Y.; Davis, A.A.; Levey, A.I.; Schütz, G.; Sager, T.N.; Mørk, A.; et al. A Subpopulation of Neuronal M4 Muscarinic Acetylcholine Receptors Plays a Critical Role in Modulating Dopamine-Dependent Behaviors. J. Neurosci. 2010, 30, 2396–2405. [Google Scholar] [CrossRef]
- Nair, A.; Castro, L.R.V.; El Khoury, M.; Gorgievski, V.; Giros, B.; Tzavara, E.T.; Hellgren-Kotaleski, J.; Vincent, P. The high efficacy of muscarinic M4 receptor in D1 medium spiny neurons reverses striatal hyperdopaminergia. Neuropharmacology 2018, 146, 74–83. [Google Scholar] [CrossRef]
- Hopper, S.; Udawela, M.; Scarr, E.; Dean, B. Allosteric modulation of cholinergic system: Potential approach to treating cognitive deficits of schizophrenia. World J. Pharmacol. 2016, 5, 1. [Google Scholar] [CrossRef]
- Crook, J.M.; Tomaskovic-Crook, E.; Copolov, D.L.; Dean, B. Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: A study of Brodmann’s areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am. J. Psychiatry 2001, 158, 918–925. [Google Scholar] [CrossRef]
- Zavitsanou, K.; Katsifis, A.; Mattner, F.; Huang, X.F. Investigation of M1/M4 muscarinic receptors in the anterior cingulate cortex in schizophrenia, bipolar, disorder, and major depression disorder. Neuropsychopharmacology 2004, 29, 619–625. [Google Scholar] [CrossRef]
- Crook, J.M.; Tomaskovic-Crook, E.; Copolov, D.L.; Dean, B. Decreased muscarinic receptor binding in subjects with schizophrenia: A study of the human hippocampal formation. Biol. Psychiatry 2000, 48, 381–388. [Google Scholar] [CrossRef]
- Dean, B.; Crook, J.M.; Opeskin, K.; Hill, C.; Keks, N.; Copolov, D.L. The density of muscarinic M1 receptors is decreased in the cau-date-putamen of subjects with schizophrenia. Mol Psychiatry 1996, 1, 54–58. [Google Scholar]
- Raedler, T.J.; Knable, M.B.; Jones, D.W.; Urbina, R.A.; Gorey, J.G.; Lee, K.S.; Egan, M.F.; Coppola, R.; Weinberger, D.R. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am. J. Psychiatry 2003, 160, 118–127. [Google Scholar] [CrossRef]
- Dean, B.; Scarr, E. Muscarinic M1 and M4 receptors: Hypothesis driven drug development for schizophrenia. Psychiatry Res. 2020, 288, 112989. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; McLeod, M.; Keriakous, D.; McKenzie, J.; Scarr, E. Decreased muscarinic1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2002, 7, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Sundram, S.; Keriakous, D.; Dean, B. Altered Hippocampal Muscarinic M4, but Not M1, Receptor Expression from Subjects with Schizophrenia. Biol. Psychiatry 2007, 61, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Craig, J.M.; Cairns, M.J.; Seo, M.S.; Galati, J.C.; Beveridge, N.J.; Gibbons, A.; Juzva, S.; Weinrich, B.; Parkinson-Bates, M.; et al. Decreased cortical muscarinic M1 receptors in schizophrenia are associated with changes in gene promoter methylation, mRNA and gene targeting microRNA. Transl. Psychiatry 2013, 3, e230. [Google Scholar] [CrossRef]
- Scarr, E.; Um, J.Y.; Cowie, T.F.; Dean, B. Cholinergic muscarinic M4 receptor gene polymorphisms: A potential risk factor and pharmacogenomic marker for schizophrenia. Schizophr. Res. 2013, 146, 279–284. [Google Scholar] [CrossRef]
- Salah-Uddin, H.; Scarr, E.; Pavey, G.; Harris, K.; Hagan, J.J.; Dean, B.; Challiss, R.A.; Watson, J.M. Altered M(1) muscarinic acetylcholine receptor (CHRM1)-Galpha(q/11) coupling in a schizophrenia endophenotype. Neuropsychopharmacology 2009, 34, 2156–2166. [Google Scholar] [CrossRef]
- Scarr, E.; Cowie, T.F.; Kanellakis, S.; Sundram, S.; Pantelis, C.; Dean, B. Decreased cortical muscarinic receptors define a subgroup of subjects with schizophrenia. Mol. Psychiatry 2008, 14, 1017–1023. [Google Scholar] [CrossRef]
- Scarr, E.; Udawela, M.; Thomas, A.E.; Dean, B. Changed gene expression in subjects with schizophrenia and low cortical muscarinic M1 receptors predicts disrupted upstream pathways interacting with that receptor. Mol. Psychiatry 2016, 23, 295–303. [Google Scholar] [CrossRef]
- Hopper, S.; Pavey, G.M.; Gogos, A.; Dean, B.; Udawela, M.; Conn, P.J. Widespread Changes in Positive Allosteric Modulation of the Muscarinic M1 Receptor in Some Participants with Schizophrenia. Int. J. Neuropsychopharmacol. 2019, 22, 640–650. [Google Scholar] [CrossRef]
- Bymaster, F.P.; Whitesiit, C.A.; Shannon, H.E.; DeLapp, N.; Ward, J.S.; Calligaro, D.O.; Shipley, L.A.; Buelke-Sam, J.L.; Bodick, N.C.; Farde, L.; et al. Xanomeline: A selective muscarinic agonist for the treatment of Alzheimer’s Disease. Drug Dev. Res. 1997, 40, 158–170. [Google Scholar] [CrossRef]
- Targum, S.D.; Murphy, C.; Breier, A.; Brannan, S.K. Site-independent confirmation of primary site-based PANSS ratings in a schizophrenia trial. J. Psychiatr. Res. 2021, 144, 241–246. [Google Scholar] [CrossRef]
- Brannan, S.K.; Sawchak, S.; Miller, A.C.; Lieberman, J.A.; Paul, S.M.; Breier, A. Muscarinic Cholinergic Receptor Agonist and Peripheral Antagonist for Schizophrenia. N. Engl. J. Med. 2021, 384, 717–726. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Enomoto, T.; Takai, K.; Takahashi, Y.; Konishi, Y.; Uruno, Y.; Tojo, K.; Suwa, A.; Matsuda, H.; Nakako, T.; et al. Discovery of novel N-substituted oxindoles as selective M1 and M4 muscarinic acetylcholine re-ceptors partial agonists. ACS Med. Chem. Lett. 2013, 4, 244–248. [Google Scholar] [CrossRef]
- Uruno, Y.; Konishi, Y.; Suwa, A.; Takai, K.; Tojo, K.; Nakako, T.; Sakai, M.; Enomoto, T.; Matsuda, H.; Kitamura, A.; et al. Discovery of dihydroquinazolinone derivatives as potent, selective, and CNS-penetrant M1 and M4 muscarinic acetylcholine receptors agonists. Bioorg. Med. Chem. Lett. 2015, 25, 5357–5361. [Google Scholar] [CrossRef]
- Suwa, A.; Konishi, Y.; Uruno, Y.; Takai, K.; Nakako, T.; Sakai, M.; Enomoto, T.; Ochi, Y.; Matsuda, H.; Kitamura, A.; et al. Discovery of N-sulfonyl-7-azaindoline derivatives as potent, orally available and selective M4 muscarinic ac-etylcholine receptor agonists. Bioorg. Med. Chem. Lett. 2014, 24, 2909–2912. [Google Scholar] [CrossRef]
- Yang, Q.; Lachapelle, E.A.; Kablaoui, N.M.; Webb, D.; Popiolek, M.; Grimwood, S.; Kozak, R.; O’Connor, R.E.; Lazzaro, J.T.; Butler, C.R.; et al. Discovery of Selective M4 Muscarinic Acetylcholine Receptor Agonists with Novel Carbamate Isosteres. ACS Med. Chem. Lett. 2019, 10, 941–948. [Google Scholar] [CrossRef]
- Chan, W.Y.; McKinzie, D.L.; Bose, S.; Mitchell, S.N.; Witkin, J.M.; Thompson, R.C.; Christopoulos, A.; Lazareno, S.; Birdsall, N.J.M.; Bymaster, F.P.; et al. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc. Natl. Acad. Sci. USA 2008, 105, 10978–10983. [Google Scholar] [CrossRef]
- Leach, K.; Loiacono, E.R.; Felder, C.C.; McKinzie, D.L.; Mogg, A.; Shaw, D.B.; Sexton, P.M.; Christopoulos, A. Molecular Mechanisms of Action and In Vivo Validation of an M4 Muscarinic Acetylcholine Receptor Allosteric Modulator with Potential Antipsychotic Properties. Neuropsychopharmacology 2009, 35, 855–869. [Google Scholar] [CrossRef]
- Suratman, S.; Leach, K.; Sexton, P.; Felder, C.; Loiacono, R.; Christopoulos, A. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. J. Cereb. Blood Flow Metab. 2010, 162, 1659–1670. [Google Scholar] [CrossRef]
- Croy, C.H.; Schober, D.A.; Xiao, H.; Quets, A.; Christopoulos, A.; Felder, C.C. Characterization of the Novel Positive Allosteric Modulator, LY2119620, at the Muscarinic M2and M4Receptors. Mol. Pharmacol. 2014, 86, 106–115. [Google Scholar] [CrossRef]
- Shirey, J.K.; Xiang, Z.; Orton, D.; Brady, E.A.; Johnson, K.A.; Williams, R.; Ayala, E.J.; Rodriguez, A.L.; Wess, J.; Weaver, D.; et al. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat. Chem. Biol. 2007, 4, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.P.; Bridges, T.M.; Gentry, P.R.; Brogan, J.T.; Kane, A.S.; Jones, C.K.; Brady, A.E.; Shirey, J.K.; Conn, P.J.; Lindsley, C.W. Synthesis and Structure-Activity Relationships of Allosteric Potentiators of the M4Muscarinic Acetylcholine Receptor. ChemMedChem 2009, 4, 1600–1607. [Google Scholar] [CrossRef]
- Brady, A.E.; Jones, C.K.; Bridges, T.M.; Kennedy, J.P.; Thompson, A.D.; Heiman, J.U.; Breininger, M.L.; Gentry, P.R.; Yin, H.; Jadhav, S.B.; et al. Centrally Active Allosteric Potentiators of the M4 Muscarinic Acetylcholine Receptor Reverse Amphetamine-Induced Hyperlocomotor Activity in Rats. J. Pharmacol. Exp. Ther. 2008, 327, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Byun, N.E.; Grannan, M.; Bubser, M.; Barry, R.L.; Thompson, A.; Rosanelli, J.; Gowrishankar, R.; Kelm, N.D.; Damon, S.; Bridges, T.M.; et al. Antipsychotic Drug-Like Effects of the Selective M4 Muscarinic Acetylcholine Receptor Positive Allosteric Modulator VU0152100. Neuropsychopharmacology 2014, 39, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Bridges, T.M.; Niswender, C.M.; Jones, C.K.; Lewis, L.M.; Weaver, C.D.; Wood, M.R.; Daniels, J.S.; Conn, J.; Lindsley, C.W. Discovery of a Highly Selective in vitro and in vivo M4 Positive Allosteric Modulator (PAM) Series with Greatly Improved Human Receptor Activity. 2010 Aug 31 [Updated 2013 Mar 14]. In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. Available online: https://www.ncbi.nlm.nih.gov/books/NBK143196/ (accessed on 4 December 2021).
- Le, U.; Melancon, B.J.; Bridges, T.M.; Vinson, P.N.; Utley, T.J.; Lamsal, A.; Rodriguez, A.L.; Venable, D.; Sheffler, D.J.; Jones, C.K.; et al. Discovery of a selective M4 positive allosteric modulator based on the 3-amino-thieno[2,3-b]pyridine-2-carboxamide scaffold: Development of ML253, a potent and brain penetrant compound that is active in a preclinical model of schizophrenia. Bioorganic Med. Chem. Lett. 2012, 23, 346–350. [Google Scholar] [CrossRef]
- Wood, M.R.; Noetzel, M.J.; Poslusney, M.S.; Melancon, B.J.; Tarr, J.C.; Lamsal, A.; Chang, S.; Luscombe, V.B.; Weiner, R.L.; Cho, H.P.; et al. Challenges in the development of an M 4 PAM in vivo tool compound: The discovery of VU0467154 and unexpected DMPK profiles of close analogs. Bioorganic Med. Chem. Lett. 2016, 27, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.R.; Noetzel, M.J.; Melancon, B.J.; Poslusney, M.S.; Nance, K.D.; Hurtado, M.A.; Luscombe, V.B.; Weiner, R.L.; Rodriguez, A.L.; Lamsal, A.; et al. Discovery of VU0467485/AZ13713945: An M4 PAM Evaluated as a Preclinical Candidate for the Treatment of Schizophrenia. ACS Med. Chem. Lett. 2016, 8, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Salovich, J.M.; Vinson, P.N.; Sheffler, D.J.; Lamsal, A.; Utley, T.J.; Blobaum, A.L.; Bridges, T.M.; Le, U.; Jones, C.K.; Wood, M.R.; et al. Discovery of N-(4-methoxy-7-methylbenzo[d]thiazol-2-yl)isonicatinamide, ML293, as a novel, selective and brain penetrant positive allosteric modulator of the muscarinic 4 (M4) receptor. Bioorganic Med. Chem. Lett. 2012, 22, 5084–5088. [Google Scholar] [CrossRef]
- Wood, M.R.; Noetzel, M.J.; Engers, J.L.; Bollinger, K.A.; Melancon, B.J.; Tarr, J.C.; Han, C.; West, M.; Gregro, A.R.; Lamsal, A.; et al. Discovery and optimization of a novel series of highly CNS penetrant M4 PAMs based on a 5,6-dimethyl-4-(piperidin-1-yl)thieno[2,3-d]pyrimidine core. Bioorganic Med. Chem. Lett. 2016, 26, 3029–3033. [Google Scholar] [CrossRef]
- Engers, J.L.; Bender, A.M.; Kalbfleisch, J.J.; Cho, H.P.; Lingenfelter, K.S.; Luscombe, V.B.; Han, C.; Melancon, B.J.; Blobaum, A.L.; Dickerson, J.W.; et al. Discovery of Tricyclic Triazolo- and Imidazopyridine Lactams as M1 Positive Allosteric Modulators. ACS Chem. Neurosci. 2018, 10, 1035–1042. [Google Scholar] [CrossRef]
- Schubert, J.W.; Harrison, S.T.; Mulhearn, J.; Gomez, R.; Tynebor, R.; Jones, K.; Bunda, J.; Hanney, B.; Wai, J.M.; Cox, C.; et al. Discovery, Optimization, and Biological Characterization of 2,3,6-Trisubstituted Pyridine-Containing M4 Positive Allosteric Modulators. ChemMedChem 2019, 14, 943–951. [Google Scholar] [CrossRef]
- Tong, L.; Li, W.; Lo, M.M.; Gao, X.; Wai, J.M.; Rudd, M.; Tellers, D.; Joshi, A.; Zeng, Z.; Miller, P.; et al. Discovery of [11C]MK-6884: A Positron Emission Tomography (PET) Imaging Agent for the Study of M4 Muscarinic Receptor Positive Allosteric Modulators (PAMs) in Neurodegenerative Dis-eases. J. Med. Chem. 2020, 63, 2411–2425. [Google Scholar] [CrossRef]
- Mazola, R. Moving Chemistry From Bench to Market. In Proceedings of the Fall 2020 ACS National Meeting (in Part), Virtual meeting, online, 17–20 August 2020. [Google Scholar]
- Marino, M.J.; Rouse, S.T.; Levey, A.I.; Potter, L.T.; Conn, P.J. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11465–11470. [Google Scholar] [CrossRef]
- Marinò, M.; Conn, P. Direct and Indirect Modulation of the N-Methyl D-Aspartate Receptor: Potential for the Development of Novel Antipsychotic Therapies. Curr. Drug Target. -CNS Neurol. Disord. 2002, 1, 1–16. [Google Scholar] [CrossRef]
- Digby, G.J.; Noetzel, M.J.; Bubser, M.; Utley, T.J.; Walker, A.G.; Byun, N.E.; Lebois, E.P.; Xiang, Z.; Sheffler, D.J.; Cho, H.P.; et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain re-gion-specific responses that correspond with behavioral effects in animal models. J. Neurosci. 2012, 32, 8532–8544. [Google Scholar] [CrossRef]
- Ghoshal, A.; Rook, J.M.; Dickerson, J.W.; Roop, G.N.; Morrison, R.D.; Jalansakrikar, N.; Lamsal, A.; Noetzel, M.J.; Poslusney, M.S.; Wood, M.R.; et al. Potentiation of M1 Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology 2015, 41, 598–610. [Google Scholar] [CrossRef]
- Grannan, M.D.; Mielnik, C.A.; Moran, S.P.; Gould, R.W.; Ball, J.; Lu, Z.; Bubser, M.; Ramsey, A.J.; Abe, M.; Cho, H.P.; et al. Prefrontal Cortex-Mediated Impairments in a Genetic Model of NMDA Receptor Hypofunction Are Reversed by the Novel M1 PAM VU6004256. ACS Chem. Neurosci. 2016, 7, 1706–1716. [Google Scholar] [CrossRef]
- Sur, C.; Mallorga, P.J.; Wittmann, M.; Jacobson, M.A.; Pascarella, D.; Williams, J.B.; Brandish, P.E.; Pettibone, D.J.; Scolnick, E.M.; Conn, P.J. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc. Natl. Acad. Sci. USA 2003, 100, 13674–13679. [Google Scholar] [CrossRef]
- Uslaner, J.M.; Eddins, D.; Puri, V.; Cannon, C.E.; Sutcliffe, J.; Chew, C.S.; Pearson, M.; Vivian, J.A.; Chang, R.K.; Ray, W.J.; et al. The muscarinic M1 receptor positive allosteric modulator PQCA improves cognitive measures in rat, cynomolgus ma-caque, and rhesus macaque. Psychopharmacology 2013, 225, 21–30. [Google Scholar] [CrossRef]
- Lange, H.S.; Cannon, C.E.; Drott, J.T.; Kuduk, S.D.; Uslaner, J.M. The M1 Muscarinic Positive Allosteric Modulator PQCA Improves Performance on Translatable Tests of Memory and Attention in Rhesus Monkeys. J. Pharmacol. Exp. Ther. 2015, 355, 442–450. [Google Scholar] [CrossRef]
- Davoren, J.E.; O’Neil, S.V.; Anderson, D.P.; Brodney, M.A.; Chenard, L.; Dlugolenski, K.; Edgerton, J.R.; Green, M.; Garnsey, M.; Grimwood, S.; et al. Design and optimization of selective azaindole amide M1 positive allosteric modulators. Bioorg. Med. Chem. Lett. 2016, 26, 650–655. [Google Scholar] [CrossRef]
- Davoren, J.E.; Lee, C.W.; Garnsey, M.; Brodney, M.A.; Cordes, J.; Dlugolenski, K.; Edgerton, J.R.; Harris, A.R.; Helal, C.J.; Jenkinson, S.; et al. Discovery of the Potent and Selective M1 PAM-Agonist N-[(3R,4S)-3-Hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): Evaluation of Efficacy and Cholinergic Side Effects. J. Med. Chem. 2016, 59, 6313–6328. [Google Scholar] [CrossRef]
- Hosp, J.A.; Luft, A.R. Dopaminergic Meso-Cortical Projections to M1: Role in Motor Learning and Motor Cortex Plasticity. Front. Neurol. 2013, 4, 145. [Google Scholar] [CrossRef]
- Janowsky, D.; Davis, J.; El-Yousef, M.; Sekerke, H. A cholinergic-adrenergic hypothesis of mania and depression. Lancet 1972, 300, 632–635. [Google Scholar] [CrossRef]
- Dilsaver, S.C. Cholinergic mechanisms in depression. Brain Res. Rev. 1986, 11, 285–316. [Google Scholar] [CrossRef]
- Dagytė, G.; Boer, J.A.D.; Trentani, A. The cholinergic system and depression. Behav. Brain Res. 2011, 221, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Janowsky, D.S.; El-Yousef, M.K.; Davis, J.M. Acetylcholine and depression. Psychosom. Med. 1974, 36, 248–257. [Google Scholar] [CrossRef]
- Risch, S.C.; Kalin, N.H.; Janowsky, D.S. Cholinergic challenges in affective illness: Behavioral and neuroendocrine correlates. J. Clin. Psychopharmacol. 1981, 1, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Nurnberger, J.I., Jr.; Jimerson, D.C.; Simmons-Alling, S.; Tamminga, C.; Nadi, N.S.; Lawrence, D.; Sitaram, N.; Gillin, J.C.; Gershon, E.S. Behavioral, physiological, and neuroendocrine responses to arecoline in normal twins and “well state” bipolar patients. Psychiatry Res. 1983, 9, 191–200. [Google Scholar] [CrossRef]
- Sunderland, T.; Tariot, P.N.; Newhouse, P.A. Differential responsivity of mood, behavior, and cognition to cholinergic agents in elderly neuropsychiatric populations. Brain Res. 1988, 472, 371–389. [Google Scholar] [CrossRef]
- Davis, K.L.; Hollander, E.; Davidson, M.; Davis, B.M.; Mohs, R.C.; Horvath, T.B. Induction of depression with oxotremorine in patients with Alzheimer’s disease. Am. J. Psychiatry 1987, 144, 468–471. [Google Scholar] [CrossRef]
- Janowsky, D.S.; Risch, S.C.; Kennedy, B.; Ziegler, M.; Huey, L. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology 1986, 89, 150–154. [Google Scholar] [CrossRef]
- Riemann, D.; Hohagan, F.; Bahro, M.; Lis, S.; Stadmuller, G.; Gann, H.; Berger, M. Cholinergic neurotransmission, REM sleep and depression. J. Psychosom. Res. 1994, 38 (Suppl. 1), 15–25. [Google Scholar] [CrossRef]
- Poland, R.E.; Tondo, L.; Rubin, R.T.; Trelease, R.B.; Lesser, I.M. Differential effects of scopolamine on nocturnal cortisol secretion, sleep architecture, and REM latency in normal volunteers: Relation to sleep and cortisol abnormalities in depression. Biol. Psychiatry 1989, 25, 403–412. [Google Scholar] [CrossRef]
- Sagales, T.; Erill, S.; Domino, E.F. Differential effects of scopolamine and chlorpromazine on REM and NREM sleep in normal male subjects. Clin. Pharmacol. Ther. 1969, 10, 522–529. [Google Scholar] [CrossRef]
- Cannon, D.M.; Klaver, J.K.; Gandhi, S.K.; Solorio, G.; Peck, S.A.; Erikson, K.; Savitz, J.; Akula, N.; Eckelman, W.C.; Furey, M.L.; et al. Genetic variation in cholinergic muscarinic-2 receptor gene modulates M2 receptor binding in vivo and accounts for reduced binding in bipolar disorder. Mol. Psychiatry 2011, 16, 407–418. [Google Scholar] [CrossRef]
- Comings, D.E.; Wu, S.; Rostamkhani, M.; McGue, M.; Iacono, W.G.; MacMurray, J.P. Association of the muscarinic cho-linergic 2 receptor (CHRM2) gene with major depression in women. Am. J. Med. Genet. 2002, 114, 527–529. [Google Scholar] [CrossRef]
- Wang, J.C.; Hinrichs, A.L.; Stock, H.; Budde, J.; Allen, R.; Bertelsen, S.; Kwon, J.M.; Wu, W.; Dick, D.M.; Rice, J.; et al. Evidence of common and specific genetic effects: Association of the muscarinic acetylcholine receptor M2 (CHRM2) gene with alcohol dependence and major depressive syndrome. Hum. Mol. Genet. 2004, 13, 1903–1911. [Google Scholar] [CrossRef]
- Riemann, D.; Hohagen, F.; Bahro, M.; Berger, M. Sleep in depression: The influence of age, gender and diagnostic subtype on baseline sleep and the cholinergic REM induction test with RS 86. Eur. Arch. Psychiatry Clin. Neurosci. 1994, 243, 279–290. [Google Scholar] [CrossRef]
- Rubin, R.T.; O’Toole, S.M.; Rhodes, M.E.; Sekula, L.; Czambel, R. Hypothalamo–pituitary–adrenal cortical responses to low-dose physostigmine and arginine vasopressin administration: Sex differences between major depressives and matched control subjects. Psychiatry Res. 1999, 89, 1–20. [Google Scholar] [CrossRef]
- Luo, X.; Kranzler, H.R.; Zuo, L.; Wang, S.; Blumberg, H.P.; Gelernter, J. CHRM2 gene predisposes to alcohol dependence, drug de-pendence and affective disorders: Results from an extended case--control structured association study. Hum. Mol. Genet. 2005, 14, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hattori, E.; Zou, H.; Badner, J.A.; Christian, S.L.; Gershon, E.S.; Liu, C. No evidence for association between 19 cholinergic genes and bipolar disorder. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. 2007, 144B, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Woods, S.; Gaysina, D.; Craddock, N.; Farmer, A.; Gray, J.; Gunasinghe, C.; Hoda, F.; Jones, L.; Knight, J.; Korszun, A.; et al. Depression Case Control (DeCC) Study fails to support involvement of the muscarinic acetylcholine receptor M2 (CHRM2) gene in recurrent major depressive disorder. Hum. Mol. Genet. 2009, 18, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E. Muscarinic receptors in psychiatric disorders-can we mimic ‘health’? Neurosignals 2009, 17, 298–310. [Google Scholar] [CrossRef]
- Gibbons, A.; Scarr, E.; McLean, C.; Sundram, S.; Dean, B. Decreased muscarinic receptor binding in the frontal cortex of bipolar disorder and major depressive disorder subjects. J. Affect. Disord. 2009, 116, 184–191. [Google Scholar] [CrossRef]
- El Yacoubi, M.; Bouali, S.; Popa, D.; Naudon, L.; Leroux-Nicollet, I.; Hamon, M.; Costentin, J.; Adrien, J.; Vaugeois, J. Behavioral, neu-rochemical, and electrophysiological characterization of a genetic mouse model of depression. Proc. Natl. Acad. Sci. USA 2003, 100, 6227–6232. [Google Scholar] [CrossRef]
- Overstreet, D.H.; Russell, R.W.; Hay, D.A.; Crocker, A.D. Selective breeding for increased cholinergic function: Biometrical genetic analysis of muscarinic responses. Neuropsychopharmacology 1992, 7, 197–204. [Google Scholar]
- Dulawa, S.C.; Janowsky, D.S. Cholinergic regulation of mood: From basic and clinical studies to emerging therapeutics. Mol. Psychiatry 2018, 24, 694–709. [Google Scholar] [CrossRef]
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61, 7–11. [Google Scholar]
- Glennon, R.A.; Iversen, L. Burger’s Medicinal Chemistry and Drug Discovery, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- El-Fakahany, E.; Richelson, E. Antagonism by antidepressants of muscarinic acetylcholine receptors of human brain. J. Cereb. Blood Flow Metab. 1983, 78, 97–102. [Google Scholar]
- Siafis, S.; Papazisis, G. Detecting a potential safety signal of antidepressants and type 2 diabetes: A pharmacovigi-lance-pharmacodynamic study. Br. J. Clin. Pharmacol. 2018, 84, 2405–2414. [Google Scholar] [CrossRef]
- Rathbun, R.C.; Slater, I.H. Amitriptyline and nortriptyline as antagonists of central and peripheral cholinergic activation. Psychopharmacology 1963, 4, 114–125. [Google Scholar] [CrossRef]
- Rawls, S.M.; Mcginty, J.F.; Anderson, I.M. SSRIS versus tricyclic antidepressants in depressed inpatients: A meta-analysis of efficacy and tolerability. Depress. Anxiety 1998, 7 (Suppl. 1), 11–17. [Google Scholar]
- Deisenhammer, E.A.; Whitworth, A.B.; Geretsegger, C.; Kurzthaler, I.; Gritsch, S.; Miller, C.H.; Fleischhacker, W.W.; Stuppäck, C.H. Intravenous Versus Oral Administration of Amitriptyline in Patients with Major Depression. J. Clin. Psychopharmacol. 2000, 20, 417–422. [Google Scholar] [CrossRef]
- Davis, K.L.; Berger, P.A.; Hollister, L.E.; Defraites, E. Physostigmine in Mania. Arch. Gen. Psychiatry 1978, 35, 119–122. [Google Scholar] [CrossRef]
- Fava, M. Diagnosis and definition of treatment-resistant depression. Biol. Psychiatry 2003, 53, 649–659. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Rush, A.J.; Wisniewski, S.R.; Nierenberg, A.A.; Warden, D.; Ritz, L.; Norquist, G.; Howland, R.H.; Lebowitz, B.; McGrath, P.J.; et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: Implications for clinical practice. Am. J. Psychiatry 2006, 163, 28–40. [Google Scholar] [CrossRef]
- Warden, D.; Rush, A.; Trivedi, M.H.; Fava, M.; Wisniewski, S. The STAR*D project results: A comprehensive review of findings. Curr. Psychiatry Rep. 2007, 9, 449–459. [Google Scholar] [CrossRef]
- Blair, D.T.; Dauner, A. Extrapyramidal Symptoms Are Serious Side-effects of Antipsychotic and Other Drugs. Nurse Pr. 1992, 17, 56, 62–64, 67. [Google Scholar] [CrossRef]
- Sayyah, M.; Eslami, K.; AlaiShehni, S.; Kouti, L. Cognitive Function before and during Treatment with Selective Serotonin Reuptake Inhibitors in Patients with Depression or Obsessive-Compulsive Disorder Cognitive Function before and during Treatment with Selective Serotonin Reuptake Inhibitors in Patients with Depression or Obsessive-Compulsive Disorder. Psychiatry J. 2016, 2016, 5480391. [Google Scholar]
- Fava, M.; Graves, L.M.; Benazzi, F.; Scalia, M.J.; Iosifescu, D.V.; Alpert, J.; Papakostas, G.I. A Cross-Sectional Study of the Prevalence of Cognitive and Physical Symptoms During Long-Term Antidepressant Treatment. J. Clin. Psychiatry 2006, 67, 1754–1759. [Google Scholar] [CrossRef]
- Schatzberg, A.F.; Blier, P.; Delgado, P.L.; Fava, M.; Haddad, P.; Shelton, R.C. Antidepressant discontinuation syndrome: Consensus panel recommendations for clinical management and additional research. J. Clin. Psychiatry 2006, 67, 27–30. [Google Scholar]
- Shelton, R.C. The nature of the discontinuation syndrome associated with antidepressant drugs. J. Clin. Psychiatry 2006, 67, 3–7. [Google Scholar]
- Gray, S.L.; Anderson, M.L.; Dublin, S.; Hanlon, J.T.; Hubbard, R.; Walker, R.; Yu, O.; Crane, P.K.; Larson, E.B. Cumulative use of strong anticholinergics and incident dementia: A prospective cohort study. JAMA Intern Med. 2015, 175, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Jessen, F.; Kaduszkiewicz, H.; Daerr, M.; Bickel, H.; Pentzek, M.; Riedel-Heller, S.; Wagner, M.; Weyerer, S.; Wiese, B.; van den Bussche, H.; et al. Anticholinergic drug use and risk for de-mentia: Target for dementia prevention. Eur. Arch. Psychiatry Clin. Neurosci. 2010, 260 (Suppl. 2), S111–S115. [Google Scholar] [CrossRef] [PubMed]
- Coupland, C.A.C.; Hill, T.; Dening, T.; Morriss, R.; Moore, M.; Hippisley-Cox, J. Anticholinergic Drug Exposure and the Risk of Dementia: A Nested Case-Control Study. JAMA Intern. Med. 2019, 179, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, O.F.; Dinan, T.; Cryan, J.F. Faster, better, stronger: Towards new antidepressant therapeutic strategies. Eur. J. Pharmacol. 2015, 753, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Han, C.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Pae, C.-U. Five potential therapeutic agents as antidepressants: A brief review and future directions. Expert Rev. Neurother. 2015, 15, 1015–1029. [Google Scholar] [CrossRef]
- Anderson, I.M. Selective serotonin reuptake inhibitors versus tricyclic antidepressants: A meta-analysis of efficacy and toler-ability. J. Affect. Disord. 2000, 58, 19–36. [Google Scholar] [CrossRef]
- Souery, D.; Papakostas, G.I.; Trivedi, M.H. Treatment-resistant depression. J. Clin. Psychiatry 2006, 67 (Suppl. 6), 16–22. [Google Scholar]
- Trivedi, M.H.; Daly, E.J. Treatment strategies to improve and sustain remission in major depressive disorder. Dialogues Clin. Neurosci. 2008, 10, 377–384. [Google Scholar]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Zarate, C.A., Jr.; Mathews, D.; Ibrahim, L.; Chaves, J.F.; Marquardt, C.; Ukoh, I.; Jolkovsky, L.; Brutsche, N.E.; Smith, M.A.; Luckenbaugh, D.A. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol. Psychiatry 2013, 74, 257–264. [Google Scholar] [CrossRef]
- Trullas, R.; Skolnick, P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol. 1990, 185, 1–10. [Google Scholar] [CrossRef]
- Daly, E.J.; Trivedi, M.H.; Janik, A.; Li, H.; Zhang, Y.; Li, X.; Lane, R.; Lim, P.; Duca, A.R.; Hough, D.; et al. Efficacy of Esketamine Nasal Spray Plus Oral Antidepressant Treatment for Relapse Prevention in Patients with Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry 2019, 76, 893–903. [Google Scholar] [CrossRef]
- Nijs, M.; Wajs, E.; Aluisio, L.; Turkoz, I.; Daly, E.; Janik, A.; Borentain, S.; Singh, J.B.; DiBernardo, A.; Wiegand, F. Managing esketamine treatment frequency toward successful outcomes: Analysis of phase 3 data. Int. J. Neuropsychopharmacol. 2020, 23, 426–433. [Google Scholar] [CrossRef]
- Kaur, U.; Pathak, B.K.; Singh, A.; Chakrabarti, S.S. Esketamine: A glimmer of hope in treatment-resistant depression. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 271, 417–429. [Google Scholar] [CrossRef]
- Ballard, E.D.; Zarate, C.A. The role of dissociation in ketamine’s antidepressant effects. Nat. Commun. 2020, 11, 6431. [Google Scholar] [CrossRef]
- Phillips, J.; Norris, S.; Talbot, J.; Birmingham, M.; Hatchard, T.; Ortiz, A.; Owoeye, O.; Batten, L.A.; Blier, P. Single, Repeated, and Maintenance Ketamine Infusions for Treatment-Resistant Depression: A Randomized Controlled Trial. Am. J. Psychiatry 2019, 176, 401–409. [Google Scholar] [CrossRef]
- Phillips, J.L.; Norris, S.; Talbot, J.; Hatchard, T.; Ortiz, A.; Birmingham, M.; Owoeye, O.; Batten, L.A.; Blier, P. Single and repeated ketamine infusions for reduction of suicidal ideation in treatment-resistant depression. Neuropsychopharmacology 2019, 45, 606–612. [Google Scholar] [CrossRef]
- Furey, M.L.; Drevets, W.C. Antidepressant efficacy of the antimuscarinic drug scopolamine: A randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry. 2006, 63, 1121–1129. [Google Scholar] [CrossRef]
- Drevets, W.C.; Bhattacharya, A.; Furey, M.L. The antidepressant efficacy of the muscarinic antagonist scopolamine: Past findings and future directions. Adv. Pharmacol. 2020, 89, 357–386. [Google Scholar] [CrossRef]
- Furey, M.L.; Pietrini, P.; Haxby, J.V.; Drevets, W.C. Selective Effects of Cholinergic Modulation on Task Performance during Selective Attention. Neuropsychopharmacology 2007, 33, 913–923. [Google Scholar] [CrossRef]
- Lakstygal, A.M.; Kolesnikova, T.O.; Khatsko, S.L.; Zabegalov, K.N.; Volgin, A.D.; Demin, K.A.; Shevyrin, V.A.; Wappler-Guzzetta, E.A.; Kalueff, A.V. Dark classics in chemical neuroscience: Atropine, scopolamine, and other anticholinergic deliriant hallucinogens. ACS Chem. Neurosci. 2019, 10, 2144–2159. [Google Scholar] [CrossRef]
- Clark, L.; Chamberlain, S.R.; Sahakian, B.J. Neurocognitive mechanisms in depression: Implications for treatment. Annu. Rev. Neurosci. 2009, 32, 57–74. [Google Scholar] [CrossRef]
- Porter, R.J.; Gallagher, P.; Thompson, J.M.; Young, A.H. Neurocognitive impairment in drug-free patients with major depressive disorder. Br. J. Psychiatry 2003, 182, 214–220. [Google Scholar] [CrossRef]
- Drevets, W.C.; Zarate, C.A.; Furey, M.L. Antidepressant Effects of the Muscarinic Cholinergic Receptor Antagonist Scopoloamine: A Review. Biol. Psychiatry. 2013, 73, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Voleti, B.; Navarria, A.; Liu, R.J.; Banasr, M.; Li, N.; Terwilliger, R.; Sanacora, G.; Eid, T.; Aghajanian, G.; Duman, R.S. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol. Psychiatry 2013, 74, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.Y.; Liu, R.J.; Banasr, M.; Dwyer, J.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTORC1-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Navarria, A.; Wohleb, E.S.; Voleti, B.; Ota, K.T.; Dutheil, S.; Lepack, A.E.; Dwyer, J.M.; Fuchikami, M.; Becker, A.; Drago, F.; et al. Rapid antidepressant actions of scopolamine: Role of medial prefrontal cortex and M1-subtype muscarinic acetylcholine receptors. Neurobiol. Dis. 2015, 82, 254–261. [Google Scholar] [CrossRef]
- Witkin, J.M.; Overshiner, C.; Li, X.; Catlow, J.T.; Wishart, G.N.; Schober, D.A.; Heinz, B.A.; Nikolayev, A.; Tolstikov, V.V.; Anderson, W.H.; et al. M1 and M2 muscarinic receptor subtypes regulate antidepressant-like effects of the rapidly acting antidepressant scopolamine. J. Pharmacol. Exp. Ther. 2014, 351, 448–456. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Gerhard, D.; Thomas, A.; Duman, R.S. Molecular and Cellular Mechanisms of Rapid-Acting Antidepressants Ketamine and Scopolamine. Curr. Neuropharmacol. 2016, 15, 11–20. [Google Scholar] [CrossRef]
- Duman, R.S. Ketamine and rapid-acting antidepressants: A new era in the battle against depression and suicide. F1000Research 2018, 7, 659. [Google Scholar] [CrossRef]
- Nenasheva, T.A.; Neary, M.; Mashanov, G.I.; Birdsall, N.J.; Breckenridge, R.A.; Molloy, J.E. Abundance, distribution, mobility and oligomeric state of M2; muscarinic acetylcholine receptors in live cardiac muscle. J. Mol. Cell. Cardiol. 2013, 57, 129–136. [Google Scholar] [CrossRef]
- Levey, A.I. Muscarinic acetylcholine receptor expression in memory circuits: Implications for treatment of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1996, 93, 13541–13546. [Google Scholar] [CrossRef]
- Yamada, M.; Miyakawa, T.; Duttaroy, A.; Yamanaka, A.; Moriguchi, T.; Makita, R.; Ogawa, M.; Chou, C.J.; Xia, B.; Crawley, J.N.; et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature 2001, 410, 207–212. [Google Scholar] [CrossRef]
- Zhang, W.; Yamada, M.; Gomeza, J.; Basile, A.S.; Wess, J. Multiple Muscarinic Acetylcholine Receptor Subtypes Modulate Striatal Dopamine Release, as Studied with M1–M5Muscarinic Receptor Knock-Out Mice. J. Neurosci. 2002, 22, 6347–6352. [Google Scholar] [CrossRef]
- Vilaro, M.T.; Palacios, J.M.; Mengod, G. Localization of M5 muscarinic receptor mRNA in rat brain examined by in situ hybrid-ization histochemistry. Neurosci. Lett. 1990, 114, 154–159. [Google Scholar] [CrossRef]
- Basile, A.S.; Fedorova, I.; Zapata, A.; Liu, X.; Shippenberg, T.; Duttaroy, A.; Yamada, M.; Wess, J. Deletion of the M5 muscarinic ace-tylcholine receptor attenuates morphine reinforcement and withdrawal but not morphine analgesia. Proc. Natl. Acad. Sci. USA 2002, 99, 11452–11457. [Google Scholar] [CrossRef]
- Thomsen, M.; Woldbye, D.P.D.; Wortwein, G.; Fink-Jensen, A.; Wess, J.; Caine, S.B. Reduced Cocaine Self-Administration in Musarinic M5 Acetylcholine Receptor-Deficient Mice. J. Neurosci. 2005, 25, 8141–8149. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, D.; Walsh, J.J.; Friedman, A.K.; Juarez, B.; Ku, S.M.; Koo, J.W.; Ferguson, D.; Tsai, H.C.; Pomeranz, L.; Christoffel, D.J.; et al. Rapid regulation of depression-related behaviors by control of midbrain dopamine neurons. Nature 2013, 493, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.R.; Kangas, B.D.; Jutkiewicz, E.M.; Winger, G.; Bergman, J.; Coop, A.; Woods, J.H. Novel Antimuscarinic Antidepressant-like Compounds with Reduced Effects on Cognition. J. Pharmacol. Exp. Ther. 2021, 377, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.; Showell, G.A.; Street, L.J.; Saunders, J.; Hoogsteen, K.; Freedman, S.B.; Hargreaves, R.J. Synthesis, physicochemical and conformational properties of (3R, 4R)-3-(3-cyclopropyl-1,2,4-oxadiazol-5-yl)-1-azabicyclo[2.2.1]heptane, a novel M1 selective muscarinic partial agonist. J. Chem. Soc. Chem. Comm. 1992, 11, 817–819. [Google Scholar] [CrossRef]
- Freedman, S.B.; Dawson, G.R.; Iversen, L.L.; Baker, R.; Hargreaves, R.J. The design of novel muscarinic partial agonists that have functional selectivity in pharmacological preparations in vitro and reduced side-effect profile in vivo. Life Sci. 1993, 52, 489–495. [Google Scholar] [CrossRef]
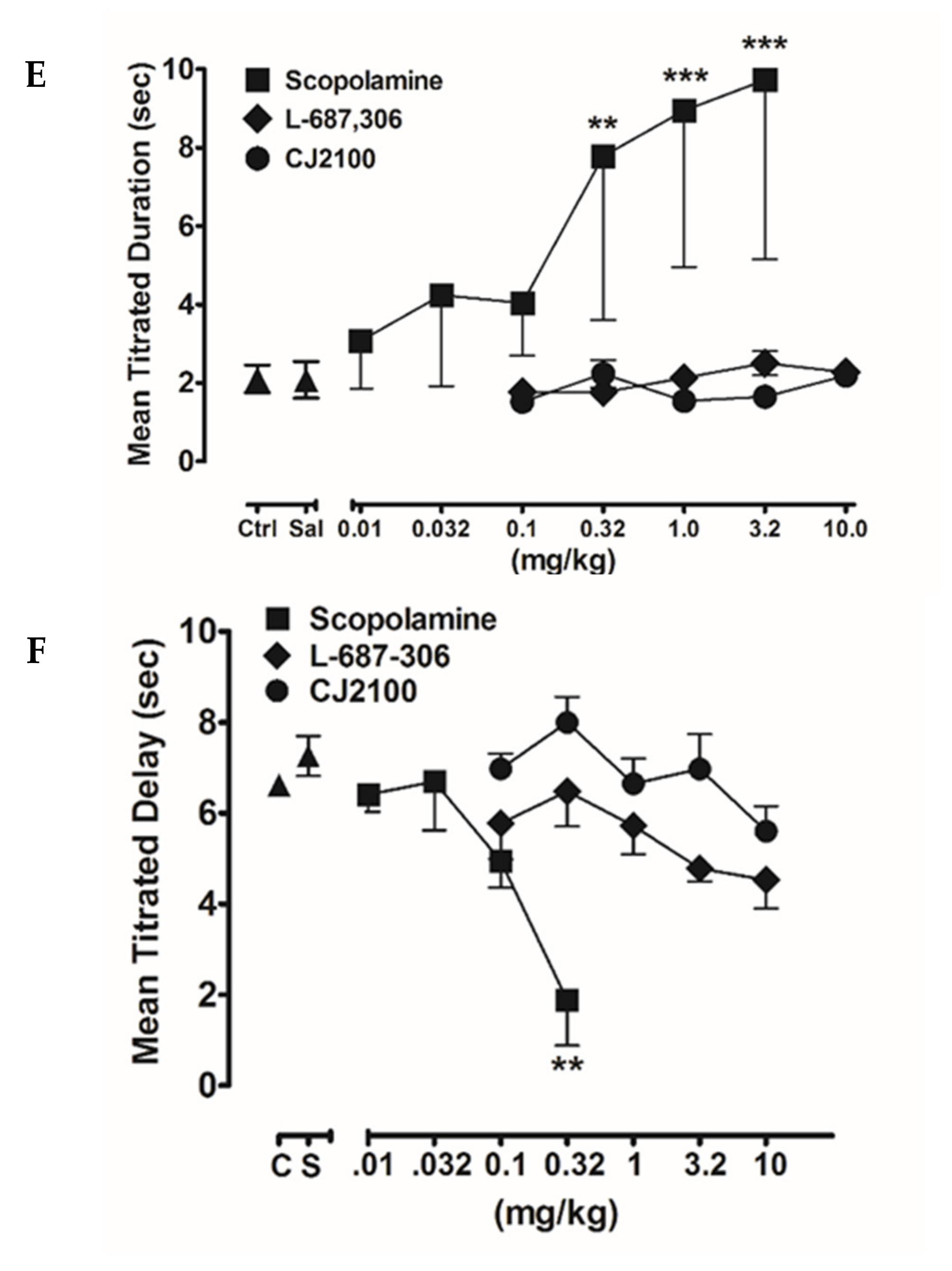
- Freedman, S.B.; Patel, S.; Harley, E.A.; Iversen, L.L.; Baker, R.; Showell, G.A.; Saunders, J.; McKnight, A.; Newberry, N.; Scholey, K.; et al. L-687,306: A functionally selective and potent muscarinic M1 receptor agonist. Eur. J. Pharmacol. 1992, 215, 135–136. [Google Scholar] [CrossRef]
- Nunes, E.J.; Rupprecht, L.E.; Foster, D.; Lindsley, C.W.; Conn, P.J.; Addy, N.A. Examining the role of muscarinic M5 receptors in VTA cholinergic modulation of depressive-like and anxiety-related behaviors in rats. Neuropharmacology 2020, 171, 108089. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, C.R.; Kangas, B.D.; Jutkiewicz, E.M.; Bergman, J.; Coop, A. Drug Design Targeting the Muscarinic Receptors and the Implications in Central Nervous System Disorders. Biomedicines 2022, 10, 398. https://doi.org/10.3390/biomedicines10020398
Johnson CR, Kangas BD, Jutkiewicz EM, Bergman J, Coop A. Drug Design Targeting the Muscarinic Receptors and the Implications in Central Nervous System Disorders. Biomedicines. 2022; 10(2):398. https://doi.org/10.3390/biomedicines10020398
Chicago/Turabian StyleJohnson, Chad R., Brian D. Kangas, Emily M. Jutkiewicz, Jack Bergman, and Andrew Coop. 2022. "Drug Design Targeting the Muscarinic Receptors and the Implications in Central Nervous System Disorders" Biomedicines 10, no. 2: 398. https://doi.org/10.3390/biomedicines10020398
APA StyleJohnson, C. R., Kangas, B. D., Jutkiewicz, E. M., Bergman, J., & Coop, A. (2022). Drug Design Targeting the Muscarinic Receptors and the Implications in Central Nervous System Disorders. Biomedicines, 10(2), 398. https://doi.org/10.3390/biomedicines10020398