
The Paracaspase MALT1 in Cancer


Abstract
1. Introduction
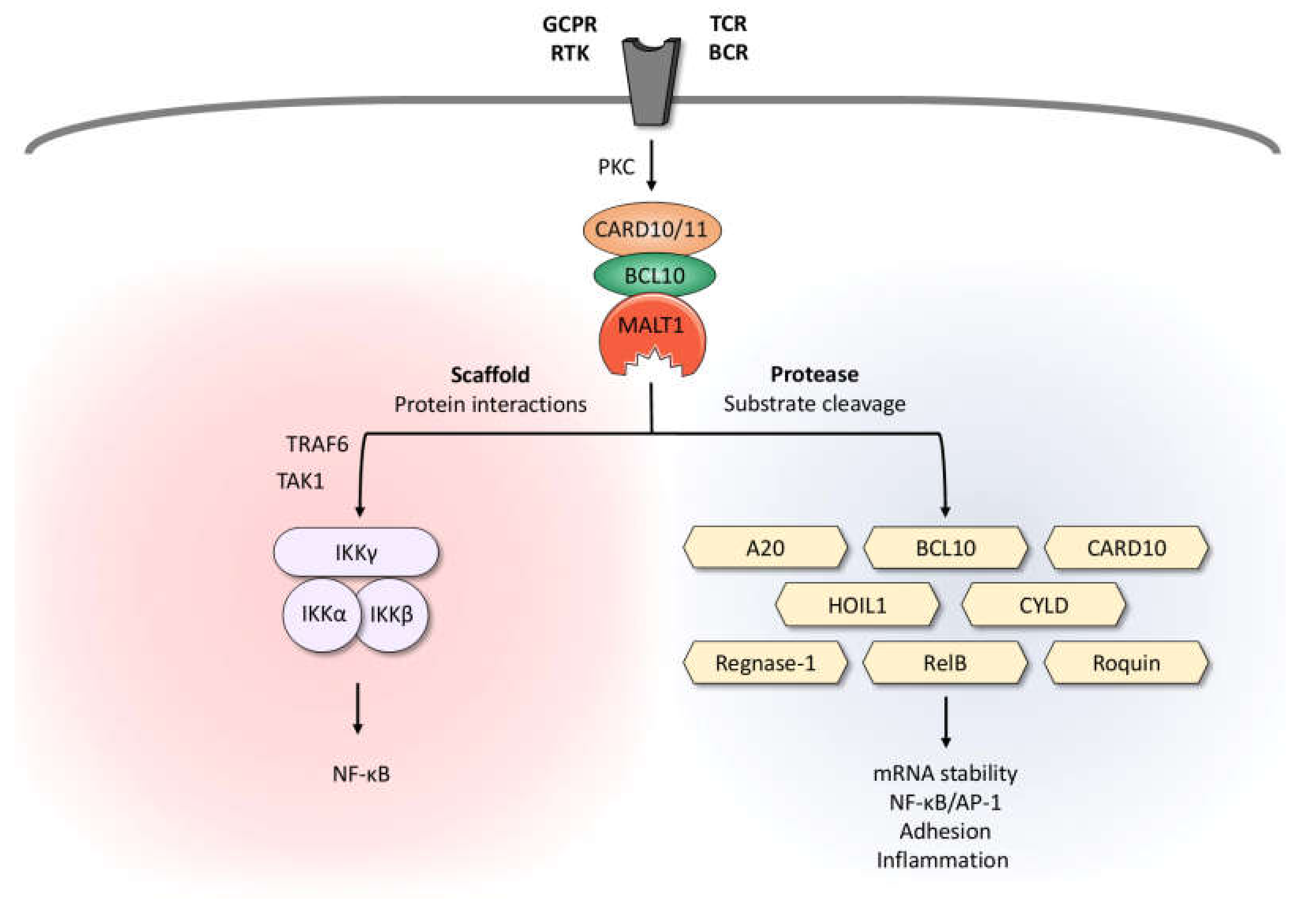
2. MALT1 Is Activated by CARD-CC Proteins
3. MALT1 Protease Function
4. MALT1 in Hematological Malignancies
5. The Role of the CBM Complex in Solid Tumors
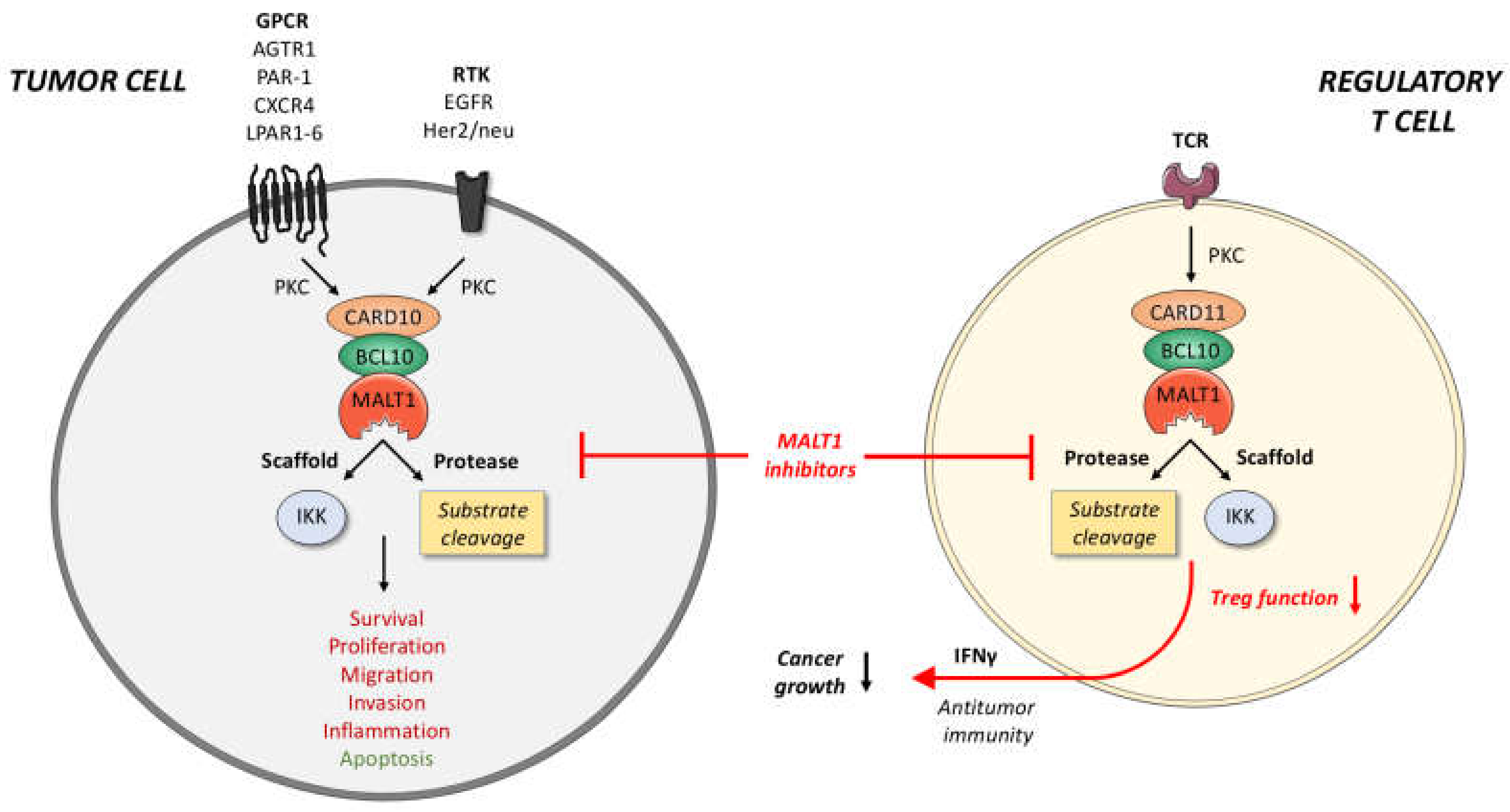
6. MALT1 as a Potential Therapeutic Target in Solid Tumors
6.1. Effects of MALT1 Inhibition on Tumor Cells
6.2. Priming of Solid Tumors via MALT1 Inhibition
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taniguchi, K.; Karin, M. NF-kappaB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the Complexities of the NF-KappaB Signalling Pathway Using Mouse Knockout and Transgenic Models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-kappaB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Ruland, J.; Hartjes, L. CARD-BCL-10-MALT1 Signalling in Protective and Pathological Immunity. Nat. Rev. Immunol. 2019, 19, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Bertin, J.; Guo, Y.; Wang, L.; Srinivasula, S.M.; Jacobson, M.D.; Poyet, J.L.; Merriam, S.; Du, M.Q.; Dyer, M.J.; Robison, K.E.; et al. CARD9 Is a Novel Caspase Recruitment Domain-Containing Protein that Interacts with BCL10/CLAP and Activates NF-Kappa, B. J. Biol. Chem. 2000, 275, 41082–41086. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Gewies, A.; Finger, K.; Schafer, M.; Sparwasser, T.; Peschel, C.; Forster, I.; Ruland, J. Card9 Controls a Non-TLR Signalling Pathway for Innate Anti-Fungal Immunity. Nature 2006, 442, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Gaide, O.; Martinon, F.; Micheau, O.; Bonnet, D.; Thome, M.; Tschopp, J. Carma1, a CARD-Containing Binding Partner of Bcl10, Induces Bcl10 Phosphorylation and NF-KappaB Activation. FEBS Lett. 2001, 496, 121–127. [Google Scholar] [CrossRef]
- Wang, D.; You, Y.; Case, S.M.; McAllister-Lucas, L.M.; Wang, L.; DiStefano, P.S.; Nunez, G.; Bertin, J.; Lin, X. A Requirement for CARMA1 in TCR-Induced NF-Kappa B Activation. Nat. Immunol. 2002, 3, 830–835. [Google Scholar] [CrossRef]
- Gaide, O.; Favier, B.; Legler, D.F.; Bonnet, D.; Brissoni, B.; Valitutti, S.; Bron, C.; Tschopp, J.; Thome, M. CARMA1 Is a Critical Lipid Raft-Associated Regulator of TCR-Induced NF-Kappa B Activation. Nat. Immunol. 2002, 3, 836–843. [Google Scholar] [CrossRef]
- McAllister-Lucas, L.M.; Inohara, N.; Lucas, P.C.; Ruland, J.; Benito, A.; Li, Q.; Chen, S.; Chen, F.F.; Yamaoka, S.; Verma, I.M.; et al. Bimp1, a MAGUK Family Member Linking Protein Kinase C Activation to Bcl10-Mediated NF-KappaB Induction. J. Biol. Chem. 2001, 276, 30589–30597. [Google Scholar] [CrossRef]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 Is due to Mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef]
- Schmitt, A.; Grondona, P.; Maier, T.; Brandle, M.; Schonfeld, C.; Jager, G.; Kosnopfel, C.; Eberle, F.C.; Schittek, B.; Schulze-Osthoff, K.; et al. MALT1 Protease Activity Controls the Expression of Inflammatory Genes in Keratinocytes upon Zymosan Stimulation. J. Invest. Dermatol. 2016, 136, 788–797. [Google Scholar] [CrossRef]
- McAllister-Lucas, L.M.; Ruland, J.; Siu, K.; Jin, X.; Gu, S.; Kim, D.S.; Kuffa, P.; Kohrt, D.; Mak, T.W.; Nunez, G.; et al. CARMA3/Bcl10/MALT1-Dependent NF-kappaB Activation Mediates Angiotensin II-Responsive Inflammatory Signaling in Nonimmune Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 139–144. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Blonska, M.; Lin, P.-C.; You, Y.; Wang, D.; Sun, J.; Darnay, B.G.; Dong, C.; Lin, X. CARMA3 Deficiency Abrogates G Protein-Coupled Receptor-Induced NF-κB Activation. Genes Dev. 2007, 21, 984–996. [Google Scholar] [CrossRef]
- Jiang, T.; Grabiner, B.; Zhu, Y.; Jiang, C.; Li, H.; You, Y.; Lang, J.; Hung, M.-C.; Lin, X. CARMA3 Is Crucial for EGFR-Induced Activation of NF-κB and Tumor Progression. Cancer Res. 2011, 71, 2183–2192. [Google Scholar] [CrossRef]
- Staal, J.; Driege, Y.; Haegman, M.; Kreike, M.; Iliaki, S.; Vanneste, D.; Lork, M.; Afonina, I.S.; Braun, H.; Beyaert, R. Defining the Combinatorial Space of PKC:CARD-CC Signal Transduction Nodes. FEBS J. 2020, 288, 1630–1647. [Google Scholar] [CrossRef]
- Holliday, M.J.; Witt, A.; Gama, A.R.; Walters, B.T.; Arthur, C.P.; Halfmann, R.; Rohou, A.; Dueber, E.C.; Fairbrother, W.J. Structures of Autoinhibited and Polymerized Forms of CARD9 Reveal Mechanisms of CARD9 and CARD11 Activation. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Sommer, K.; Moreno-García, M.E. The CARMA1 Signalosome Links the Signalling Machinery of Adaptive and Innate Immunity in lymphocytes. Nat. Rev. Immunol. 2006, 6, 799–812. [Google Scholar] [CrossRef]
- Sommer, K.; Guo, B.; Pomerantz, J.L.; Bandaranayake, A.D.; Moreno-García, M.E.; Ovechkina, Y.L.; Rawlings, D.J. Phosphorylation of the CARMA1 Linker Controls NF-κB Activation. Immunity 2005, 23, 561–574. [Google Scholar] [CrossRef]
- Matsumoto, R.; Wang, D.; Blonska, M.; Li, H.; Kobayashi, M.; Pappu, B.; Chen, Y.; Wang, D.; Lin, X. Phosphorylation of CARMA1 Plays a Critical Role in T Cell Receptor-Mediated NF-κB Activation. Immunity 2005, 23, 575–585. [Google Scholar] [CrossRef]
- Tanner, M.; Hanel, W.; Gaffen, S.L.; Lin, X. CARMA1 Coiled-Coil Domain Is Involved in the Oligomerization and Subcellular Localization of CARMA1 and Is Required for T Cell Receptor-induced NF-κB Activation. J. Biol. Chem. 2007, 282, 17141–17147. [Google Scholar] [CrossRef]
- Bertin, J.; Wang, L.; Guo, Y.; Jacobson, M.D.; Poyet, J.-L.; Srinivasula, S.M.; Merriam, S.; DiStefano, P.S.; Alnemri, E.S. CARD11 and CARD14 Are Novel Caspase Recruitment Domain (CARD)/Membrane-associated Guanylate Kinase (MAGUK) Family Members that Interact with BCL10 and Activate NF-κB. J. Biol. Chem. 2001, 276, 11877–11882. [Google Scholar] [CrossRef]
- Schlauderer, F.; Seeholzer, T.; Desfosses, A.; Gehring, T.; Strauss, M.; Hopfner, K.-P.; Gutsche, I.; Krappmann, D.; Lammens, K. Molecular Architecture and Regulation of BCL10-MALT1 Filaments. Nat. Commun. 2018, 9, 4041. [Google Scholar] [CrossRef]
- David, L.; Li, Y.; Ma, J.; Garner, E.; Zhang, X.; Wu, H. Assembly Mechanism of the CARMA1–BCL10–MALT1–TRAF6 SignaloSome. Proc. Natl. Acad. Sci. USA 2018, 115, 1499–1504. [Google Scholar] [CrossRef]
- Sun, L.; Deng, L.; Ea, C.-K.; Xia, Z.-P.; Chen, Z.J. The TRAF6 Ubiquitin Ligase and TAK1 Kinase Mediate IKK Activation by BCL10 and MALT1 in T Lymphocytes. Mol. Cell 2004, 14, 289–301. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Wegener, E.; Welteke, V.; Ferch, U.; Arslan, S.; Ruland, J.; Scheidereit, C.; Krappmann, D. Malt1 Ubiquitination Triggers NF-κB Signaling upon T-Cell Activation. EMBO J. 2007, 26, 4634–4645. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB Kinase Complex by TRAF6 Requires a Dimeric Ubiquitin-Conjugating Enzyme Complex and a Unique Polyubiquitin Chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Yang, Y.; Schmitz, R.; Mitala, J.; Whiting, A.L.; Xiao, W.; Ceribelli, M.; Wright, G.W.; Zhao, H.; Yang, Y.; Xu, W.; et al. Essential Role of the Linear Ubiquitin Chain Assembly Complex in Lymphoma Revealed by Rare Germline Polymorphisms. Cancer Discov. 2014, 4, 480–493. [Google Scholar] [CrossRef]
- Wu, C.-J.; Ashwell, J.D. NEMO Recognition of Ubiquitinated Bcl10 is Required for T Cell Receptor-Mediated NF-B Activation. Proc. Natl. Acad. Sci. USA 2008, 105, 3023–3028. [Google Scholar] [CrossRef]
- Zhou, H.; Wertz, I.; O’Rourke, K.; Ultsch, M.; Seshagiri, S.; Eby, M.; Xiao, W.; Dixit, V.M. Bcl10 Activates the NF-κB Pathway through Ubiquitination of NEMO. Nature 2003, 427, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.-I.; Chen, Z.J. TAK1 is a Ubiquitin-Dependent Kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.C.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the Canonical IKK Complex by K63/M1-Linked Hybrid Ubiquitin Chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.; O’Rourke, K.; Aravind, L.; Pisabarro, M.; Seshagiri, S.; Koonin, E.V.; Dixit, V.M. Identification of Paracaspases and Metacaspases: Two Ancient Families of Caspase-like Proteins, One of which Plays a Key Role in MALT Lymphoma. Mol. Cell 2000, 6, 961–967. [Google Scholar] [CrossRef]
- Cheng, J.; Klei, L.R.; Hubel, N.E.; Zhang, M.; Schairer, R.; Maurer, L.M.; Klei, H.B.; Kang, H.; Concel, V.J.; Delekta, P.C.; et al. GRK2 Suppresses Lymphomagenesis by Inhibiting the MALT1 Proto-Oncoprotein. J. Clin. Investig. 2019, 130, 1036–1051. [Google Scholar] [CrossRef]
- O’Neill, T.J.; Seeholzer, T.; Gewies, A.; Gehring, T.; Giesert, F.; Hamp, I.; Graß, C.; Schmidt, H.; Kriegsmann, K.; Tofaute, M.J.; et al. TRAF6 Prevents Fatal Inflammation by Homeostatic Suppression of MALT1 Protease. Sci. Immunol. 2021, 6, eabh2095. [Google Scholar] [CrossRef]
- Pelzer, C.; Cabalzar, K.; Wolf, A.; Gonzalez, M.; Lenz, G.; Thome, M. The Protease Activity of the Paracaspase MALT1 Is Controlled by Monoubiquitination. Nat. Immunol. 2013, 14, 337–345. [Google Scholar] [CrossRef]
- Yu, J.W.; Jeffrey, P.D.; Ha, J.Y.; Yang, X.; Shi, Y. Crystal Structure of the Mucosa-Associated Lymphoid Tissue Lymphoma Translocation 1 (MALT1) Paracaspase Region. Proc. Natl. Acad. Sci. USA 2011, 108, 21004–21009. [Google Scholar] [CrossRef]
- Rebeaud, F.; Hailfinger, S.; Posevitz-Fejfar, A.; Tapernoux, M.; Moser, R.; Rueda, D.; Gaide, O.; Guzzardi, M.; Iancu, E.M.; Rufer, N.; et al. The Proteolytic Activity of the Paracaspase MALT1 is Key in T Cell Activation. Nat. Immunol. 2008, 9, 272–281. [Google Scholar] [CrossRef]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T Cell Antigen Receptor Stimulation Induces MALT1 Paracaspase–Mediated Cleavage of the NF-κB inhibitor A20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef]
- Staal, J.; Driege, Y.; Bekaert, T.; Demeyer, A.; Muyllaert, D.; Van Damme, P.; Gevaert, K.; Beyaert, R. T-Cell Receptor-Induced JNK Activation Requires Proteolytic Inactivation of CYLD by MALT1. EMBO J. 2011, 30, 1742–1752. [Google Scholar] [CrossRef]
- Hailfinger, S.; Nogai, H.; Pelzer, C.; Jaworski, M.; Cabalzar, K.; Charton, J.-E.; Guzzardi, M.; Décaillet, C.; Grau, M.; Dörken, B.; et al. Malt1-Dependent RelB Cleavage Promotes Canonical NF-B Activation in Lymphocytes and Lymphoma Cell Lines. Proc. Natl. Acad. Sci. USA 2011, 108, 14596–14601. [Google Scholar] [CrossRef]
- Douanne, T.; Gavard, J.; Bidère, N. The Paracaspase MALT1 Cleaves the LUBAC Subunit HOIL1 during Antigen Receptor Signaling. J. Cell Sci. 2016, 129, 1775–1780. [Google Scholar] [CrossRef]
- Elton, L.; Carpentier, I.; Staal, J.; Driege, Y.; Haegman, M.; Beyaert, R. MALT1 Cleaves the E3 Ubiquitin Ligase HOIL-1 in Activated T Cells, Generating a Dominant Negative Inhibitor of LUBAC-Induced NF-κB Signaling. FEBS J. 2015, 283, 403–412. [Google Scholar] [CrossRef]
- Klein, T.; Fung, S.-Y.; Renner, F.; Blank, M.A.; Dufour, A.; Kang, S.; Bolger-Munro, M.; Scurll, J.M.; Priatel, J.; Schweigler, P.; et al. The Paracaspase MALT1 Cleaves HOIL1 Reducing Linear Ubiquitination by LUBAC to Dampen Lymphocyte NF-κB Signalling. Nat. Commun. 2015, 6, 8777. [Google Scholar] [CrossRef]
- Uehata, T.; Iwasaki, H.; Vandenbon, A.; Matsushita, K.; Hernandez-Cuellar, E.; Kuniyoshi, K.; Satoh, T.; Mino, T.; Suzuki, Y.; Standley, D.M.; et al. Malt1-Induced Cleavage of Regnase-1 in CD4+ Helper T Cells Regulates Immune Activation. Cell 2013, 153, 1036–1049. [Google Scholar] [CrossRef]
- Jeltsch, K.M.; Hu, D.; Brenner, S.; Zöller, J.; Heinz, G.A.; Nagel, D.; Vogel, K.U.; Rehage, N.; Warth, S.C.; Edelmann, S.L.; et al. Cleavage of Roquin and Regnase-1 by the Paracaspase MALT1 Releases their Cooperatively Repressed Targets to Promote TH17 Differentiation. Nat. Immunol. 2014, 15, 1079–1089. [Google Scholar] [CrossRef]
- Baens, M.; Bonsignore, L.; Somers, R.; Vanderheydt, C.; Weeks, S.; Gunnarsson, J.; Nilsson, E.; Roth, R.G.; Thome, M.; Marynen, P. MALT1 Auto-Proteolysis Is Essential for NF-κB-Dependent Gene Transcription in Activated Lymphocytes. PLoS ONE 2014, 9, e103774. [Google Scholar] [CrossRef]
- Xia, X.; Cao, G.; Sun, G.; Zhu, L.; Tian, Y.; Song, Y.; Guo, C.; Wang, X.; Zhong, J.; Zhou, W.; et al. GLS1-Mediated Glutaminolysis Unbridled by MALT1 Protease Promotes Psoriasis Pathogenesis. J. Clin. Investig. 2020, 130, 5180–5196. [Google Scholar] [CrossRef]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef]
- Xia, X.; Zhou, W.; Guo, C.; Fu, Z.; Zhu, L.; Li, P.; Xu, Y.; Zheng, L.; Zhang, H.; Shan, C.; et al. Glutaminolysis Mediated by MALT1 Protease Activity Facilitates PD-L1 Expression on ABC-DLBCL Cells and Contributes to Their Immune Evasion. Front. Oncol. 2018, 8, 632. [Google Scholar] [CrossRef]
- Juilland, M.; Thome, M. Role of the CARMA1/BCL10/MALT1 Complex in Lymphoid Malignancies. Curr. Opin. Hematol. 2016, 23, 402–409. [Google Scholar] [CrossRef]
- Hailfinger, S.; Lenz, G.; Thome, M. Targeting B-Cell Lymphomas with Inhibitors of the MALT1 Paracaspase. Curr. Opin. Chem. Biol. 2014, 23, 47–55. [Google Scholar] [CrossRef]
- Grondona, P.; Bucher, P.; Schulze-Osthoff, K.; Hailfinger, S.; Schmitt, A. NF-κB Activation in Lymphoid Malignancies: Genetics, Signaling, and Targeted Therapy. Biomedicines 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Motegi, M.; Tamura, A.; Suzuki, R.; Hosokawa, Y.; Suzuki, H.; Ota, H.; Nakamura, S.; Morishima, Y.; Taniwaki, M.; et al. A Novel Gene, MALT1 at 18q21, is Involved in t(11;18) (q21;q21) Found in Low-Grade B-Cell Lymphoma of Mucosa-Associated Lymphoid Tissue. Oncogene 1999, 18, 5785–5794. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, J.; Wang, X.; Xuan, L.; Lai, J.; Xu, L.; Chen, S.; Yang, L.; Luo, G.; Zhu, K.; et al. Overexpression of MALT1-A20-NF-κB in Adult B-Cell Acute Lymphoblastic Leukemia. Cancer Cell Int. 2015, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Izquierdo, D.; Buchonnet, G.; Siebert, R.; Gascoyne, R.D.; Climent, J.; Karran, L.; Marin, M.; Blesa, D.; Horsman, D.; Rosenwald, A.; et al. MALT1 is Deregulated by Both Chromosomal Translocation and Amplification in B-Cell Non-Hodgkin Lymphoma. Blood 2003, 101, 4539–4546. [Google Scholar] [CrossRef]
- Kuper-Hommel, M.J.J.; Schreuder, M.I.; Gemmink, A.H.; Van Krieken, J.H.J.M. T(14;18)(q32;q21) Involving MALT1 and IGH Genes Occurs in Extranodal Diffuse Large B-Cell Lymphomas of the Breast and Testis. Mod. Pathol. 2012, 26, 421–427. [Google Scholar] [CrossRef][Green Version]
- Streubel, B.; Lamprecht, A.; Dierlamm, J.; Cerroni, L.; Stolte, M.; Ott, G.; Raderer, M.; Chott, A. T(14;18)(q32;q21) Involving IGH and MALT1 Is a Frequent Chromosomal Aberration in MALT Lymphoma. Blood 2003, 101, 2335–2339. [Google Scholar] [CrossRef]
- Cook, J.R.; Sherer, M.; Craig, F.E.; Shekhter-Levin, S.; Swerdlow, S.H. T(14;18)(q32;q21) Involving MALT1 and IGH Genes in an Extranodal Diffuse Large B-Cell Lymphoma. Hum. Pathol. 2003, 34, 1212–1215. [Google Scholar] [CrossRef]
- Vicente-Dueñas, C.; Fontan, L.; Gonzalez-Herrero, I.; Romero-Camarero, I.; Segura, V.; Aznar, M.A.; Alonso-Escudero, E.; Campos-Sanchez, E.; Ruiz, L.; Barajas-Diego, M.; et al. Expression of MALT1 Oncogene in Hematopoietic Stem/Progenitor Cells RecapituLates the Pathogenesis of Human Lymphoma in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10534–10539. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Xue, L.; Shin, N.-M.; Roopenian, D.; Xu, W.; Qi, C.-F.; Sangster, M.; Orihuela, C.J.; Tuomanen, E.; et al. Eμ-BCL10 Mice Exhibit Constitutive Activation of Both Canonical and Noncanonical NF-κB Pathways Generating Marginal Zone (MZ) B-Cell Expansion as a Precursor to Splenic MZ Lymphoma. Blood 2009, 114, 4158–4168. [Google Scholar] [CrossRef]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef]
- Almeida, A.C.D.S.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.; Vermeer, M.; Rabadan, R.; Ferrando, A.; Palomero, T. The Mutational Landscape of Cutaneous T Cell Lymphoma and Sézary Syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef]
- Wang, L.; Ni, X.; Covington, K.R.; Yang, B.Y.; Shiu, J.; Zhang, X.; Xi, L.; Meng, Q.; Langridge, T.; Drummond, J.; et al. Genomic Profiling of Sézary Syndrome Identifies Alterations of Key T Cell Signaling and Differentiation Genes. Nat. Genet. 2015, 47, 1426–1434. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.D.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating Mutations in Genes Related to TCR Signaling in Angioimmunoblastic and Other Follicular Helper T-Cell–Derived Lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef]
- Dierlamm, J.; Baens, M.; Wlodarska, I.; Stefanova-Ouzounova, M.; Hernandez, J.M.; Hossfeld, D.K.; De Wolf-Peeters, C.; Hagemeijer, A.; Van den Berghe, H.; Marynen, P. The Apoptosis Inhibitor Gene API2 and a Novel 18q Gene, MLT, are Recurrently Rearranged in the t(11;18)(q21;q21) Associated with Mucosa-Associated Lymphoid Tissue Lymphomas. Blood 1999, 93, 3601–3609. [Google Scholar] [CrossRef]
- Lucas, P.; Kuffa, P.; Gu, S.; Kohrt, D.; Kim, D.S.L.; Siu, K.; Jin, X.; Swenson, J.; McAllister-Lucas, L.M. A Dual Role for the API2 Moiety in API2-MALT1-Dependent NF-κB Activation: Heterotypic Oligomerization and TRAF2 Recruitment. Oncogene 2007, 26, 5643–5654. [Google Scholar] [CrossRef]
- Noels, H.; van Loo, G.; Hagens, S.; Broeckx, V.; Beyaert, R.; Marynen, P.; Baens, M. A Novel TRAF6 Binding Site in MALT1 Defines Distinct Mechanisms of NF-κB Activation by API2·MALT1 Fusions. J. Biol. Chem. 2007, 282, 10180–10189. [Google Scholar] [CrossRef]
- Rosebeck, S.; Madden, L.; Jin, X.; Gu, S.; Apel, I.J.; Appert, A.; Hamoudi, R.A.; Noels, H.; Sagaert, X.; Van Loo, P.; et al. Cleavage of NIK by the API2-MALT1 Fusion Oncoprotein Leads to Noncanonical NF-κB Activation. Science 2011, 331, 468–472. [Google Scholar] [CrossRef]
- Nie, Z.; Du, M.-Q.; McAllister-Lucas, L.M.; Lucas, P.; Bailey, N.; Hogaboam, C.M.; Lim, M.; Elenitoba-Johnson, K. Conversion of the LIMA1 Tumour Suppressor into an Oncogenic LMO-Like Protein by API2–MALT1 in MALT Lymphoma. Nat. Commun. 2015, 6, 5908. [Google Scholar] [CrossRef]
- Young, R.M.; Wu, T.; Schmitz, R.; Dawood, M.; Xiao, W.; Phelan, J.D.; Xu, W.; Menard, L.; Meffre, E.; Chan, W.-C.C.; et al. Survival of Human Lymphoma Cells Requires B-Cell Receptor Engagement by Self-Antigens. Proc. Natl. Acad. Sci. USA 2015, 112, 13447–13454. [Google Scholar] [CrossRef] [PubMed]
- Minden, M.D.-V.; Übelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Köhler, F.; et al. Chronic Lymphocytic Leukaemia is Driven by Antigen-Independent Cell-Autonomous Signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Quinn, E.R.; Chan, C.H.; Hadlock, K.G.; Foung, S.K.H.; Flint, M.; Levy, S. The B-Cell Receptor of a Hepatitis C Virus (HCV)–Associated Non-Hodgkin Lymphoma Binds the Viral E2 Envelope Protein, Implicating HCV in Lymphomagenesis. Blood 2001, 98, 3745–3749. [Google Scholar] [CrossRef] [PubMed]
- Suarez, F.; Lortholary, O.; Hermine, O.; Lecuit, M. Infection-Associated Lymphomas Derived from Marginal Zone B Cells: A Model of Antigen-Driven Lymphoproliferation. Blood 2006, 107, 3034–3044. [Google Scholar] [CrossRef]
- Bonsignore, L.; Passelli, K.; Pelzer, C.; Perroud, M.; Konrad, A.; Thurau, M.; Stürzl, M.; Dai, L.; Trillo-Tinoco, J.; Del Valle, L.; et al. A Role for MALT1 Activity in Kaposi’s Sarcoma-Associated Herpes Virus Latency and Growth of Primary Effusion Lymphoma. Leukemia 2016, 31, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Grau, M.; Juilland, M.; Klener, P.; Höring, E.; Molinsky, J.; Schimmack, G.; Aukema, S.M.; Hoster, E.; Vogt, N.; et al. B-Cell Receptor–Driven MALT1 Activity Regulates MYC Signaling in Mantle Cell Lymphoma. Blood 2017, 129, 333–346. [Google Scholar] [CrossRef]
- Hailfinger, S.; Lenz, G.; Ngo, V.; Posvitz-Fejfar, A.; Rebeaud, F.; Guzzardi, M.; Penas, E.-M.M.; Dierlamm, J.; Chan, W.C.; Staudt, L.M.; et al. Essential Role of MALT1 Protease Activity in Activated B Cell-Like Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 19946–19951. [Google Scholar] [CrossRef]
- Ferch, U.; Kloo, B.; Gewies, A.; Pfänder, V.; Düwel, M.; Peschel, C.; Krappmann, D.; Ruland, J. Inhibition of MALT1 Protease Activity is Selectively Toxic for Activated B Cell–Like Diffuse Large B Cell Lymphoma Cells. J. Exp. Med. 2009, 206, 2313–2320. [Google Scholar] [CrossRef]
- Saba, N.S.; Wong, D.; Tanios, G.; Iyer, J.R.; Lobelle-Rich, P.; Dadashian, E.L.; Liu, D.; Fontan, L.; Flemington, E.K.; Nichols, C.M.; et al. MALT1 Inhibition Is Efficacious in Both Naïve and Ibrutinib-Resistant Chronic Lymphocytic Leukemia. Cancer Res. 2017, 77, 7038–7048. [Google Scholar] [CrossRef]
- Ishikawa, C.; Mori, N. MALT-1 as a Novel Therapeutic Target for Adult T-Cell Leukemia. Eur. J. Haematol. 2020, 105, 460–467. [Google Scholar] [CrossRef]
- Ekambaram, P.; Lee, J.-Y.; Hubel, N.E.; Hu, N.; Yerneni, S.; Campbell, P.G.; Pollock, N.; Klei, L.R.; Concel, V.J.; Delekta, P.C.; et al. The CARMA3-Bcl10-MALT1 Signalosome Drives NFκB Activation and Promotes Aggressiveness in Angiotensin II Receptor-Positive Breast Cancer. Cancer Res. 2017, 78, 1225–1240. [Google Scholar] [CrossRef]
- McAuley, J.R.; Bailey, K.M.; Ekambaram, P.; Klei, L.R.; Kang, H.; Hu, D.; Freeman, T.J.; Concel, V.J.; Hubel, N.E.; Lee, J.-Y.; et al. MALT1 Is a Critical Mediator of PAR1-Driven NF-κB Activation and Metastasis in Multiple Tumor Types. Oncogene 2019, 38, 7384–7398. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Ekambaram, P.; Carleton, N.M.; Hu, D.; Klei, L.R.; Cai, Z.; Myers, M.I.; Hubel, N.E.; Covic, L.; Agnihotri, S.; et al. MALT1 Is a Targetable Driver of Epithelial-to-Mesenchymal Transition in Claudin-Low, Triple-Negative Breast Cancer. Mol. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Mahanivong, C.; Chen, H.M.; Yee, S.W.; Pan, Z.K.; Dong, Z.; Huang, S. Protein Kinase Cα-CARMA3 Signaling Axis Links Ras to NF-κB for Lysophosphatidic Acid-Induced Urokinase Plasminogen Activator Expression in Ovarian Cancer Cells. Oncogene 2007, 27, 1273–1280. [Google Scholar] [CrossRef]
- Liu, X.; Yue, C.; Shi, L.; Liu, G.; Cao, Q.; Shan, Q.; Wang, Y.; Chen, X.; Li, H.; Wang, J.; et al. MALT1 Is a Potential Therapeutic Target in Glioblastoma and Plays a Crucial Role in EGFR-Induced NF-κB Activation. J. Cell Mol. Med. 2020, 24, 7550–7562. [Google Scholar] [CrossRef]
- Pan, D.; Jiang, C.; Ma, Z.-L.; Blonska, M.; You, M.J.; Lin, X. MALT1 Is Required for EGFR-Induced NF-κB Activation and Contributes to EGFR-Driven Lung Cancer Progression. Oncogene 2015, 35, 919–928. [Google Scholar] [CrossRef]
- Pan, D.; Zhu, Y.; Zhou, Z.; Wang, T.; You, H.; Jiang, C.; Lin, X. The CBM Complex Underwrites NF-κB Activation to Promote HER2-Associated Tumor Malignancy. Mol. Cancer Res. 2015, 14, 93–102. [Google Scholar] [CrossRef]
- Konczalla, L.; Perez, D.R.; Wenzel, N.; Wolters-Eisfeld, G.; Klemp, C.; Lüddeke, J.; Wolski, A.; Landschulze, D.; Meier, C.; Buchholz, A.; et al. Biperiden and Mepazine Effectively Inhibit MALT1 Activity and Tumor Growth in Pancreatic Cancer. Int. J. Cancer 2019, 146, 1618–1630. [Google Scholar] [CrossRef]
- Tsui, K.-H.; Chang, K.-S.; Sung, H.-C.; Hsu, S.-Y.; Lin, Y.-H.; Hou, C.-P.; Yang, P.-S.; Chen, C.-L.; Feng, T.-H.; Juang, H.-H. Mucosa-Associated Lymphoid Tissue 1 Is an Oncogene Inducing Cell Proliferation, Invasion, and Tumor Growth via the Upregulation of NF-κB Activity in Human Prostate Carcinoma Cells. Biomedicines 2021, 9, 250. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, G.; Jin, J.; Degan, S.; Tameze, Y.; Zhang, J.Y. MALT1 Promotes Melanoma Progression through JNK/c-Jun Signaling. Oncogenesis 2017, 6, e365. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-N.; Chang, Y.-C.; Su, Y.; Hsu, D.S.-S.; Cheng, C.-T.; Wu, R.-C.; Chung, Y.-H.; Chiang, K.-C.; Yeh, T.-S.; Lu, M.-L.; et al. Identification of MALT1 as Both a Prognostic Factor and a Potential Therapeutic Target of Regorafenib in Cholangiocarcinoma Patients. Oncotarget 2017, 8, 113444–113459. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, X.; Liu, Y.; Liu, Y.; Zhang, C.; Wang, Z.; Jiang, T.; Wang, Y. miR-181d/MALT1 Regulatory Axis Attenuates Mesenchymal Phenotype through NF-κB Pathways in Glioblastoma. Cancer Lett. 2017, 396, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Du, R.; Li, J.; Luo, X.; Zhao, S.; Chang, A.; Zhou, W.; Gao, R.; Luo, D.; Wang, J.; et al. TIFA Suppresses Hepatocellular Carcinoma Progression via MALT1-Dependent and-Independent Signaling Pathways. Signal Transduct. Target. Ther. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Israël, L.; Glück, A.; Berger, M.; Coral, M.; Ceci, M.; Unterreiner, A.; Rubert, J.; Bardet, M.; Ginster, S.; Golding-Ochsenbein, A.M.; et al. CARD10 Cleavage by MALT1 Restricts Lung Carcinoma Growth in vivo. Oncogenesis 2021, 10, 32. [Google Scholar] [CrossRef]
- Rosebeck, S.; Rehman, A.O.; Lucas, P.C.; McAllister-Lucas, L.M. From MALT Lymphoma to the CBM Signalosome: Three Decades of Discovery. Cell Cycle 2011, 10, 2485–2496. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, H.; Xu, J.; Zhang, N.; Pan, T.; Zhong, X.; Zhang, H.; Yin, L.; Yao, Y.; Wu, Q.; et al. MALT1 Inhibition as a Therapeutic Strategy in T-Cell Acute Lymphoblastic Leukemia by Blocking Notch1-Induced NF-κB Activation. Front. Oncol. 2020, 10, 558339. [Google Scholar] [CrossRef]
- Hamp, I.; O’Neill, T.J.; Plettenburg, O.; Krappmann, D. A Patent Review of MALT1 inhibitors (2013-present). Expert Opin. Ther. Pat. 2021, 31, 1079–1096. [Google Scholar] [CrossRef]
- Jaworski, M.; Marsland, B.J.; Gehrig, J.; Held, W.; Favre, S.; Luther, S.; Perroud, M.; Golshayan, D.; Gaide, O.; Thome, M. Malt1 Protease Inactivation Efficiently Dampens Immune Responses but Causes Spontaneous Autoimmunity. EMBO J. 2014, 33, 2765–2781. [Google Scholar] [CrossRef]
- Demeyer, A.; Staal, J.; Beyaert, R. Targeting MALT1 Proteolytic Activity in Immunity, Inflammation and Disease: Good or Bad? Trends Mol. Med. 2016, 22, 135–150. [Google Scholar] [CrossRef]
- Gewies, A.; Gorka, O.; Bergmann, H.; Pechloff, K.; Petermann, F.; Jeltsch, K.M.; Rudelius, M.; Kriegsmann, M.; Weichert, W.; Horsch, M.; et al. Uncoupling Malt1 Threshold Function from Paracaspase Activity Results in Destructive Autoimmune Inflammation. Cell Rep. 2014, 9, 1292–1305. [Google Scholar] [CrossRef]
- Brustle, A.; Brenner, D.; Knobbethomsen, C.B.; Cox, M.; Lang, P.A.; Lang, K.; Mak, T.W. MALT1 Is an Intrinsic Regulator of Regulatory T Cells. Cell Death Differ. 2015, 24, 1214–1223. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Gewies, A.; Pechloff, K.; Heuser, C.; Engleitner, T.; Gehring, T.; Hartjes, L.; Krebs, S.; Krappmann, D.; Kriegsmann, M.; et al. Bcl10-Controlled Malt1 Paracaspase Activity Is Key for the Immune Suppressive Function of Regulatory T Cells. Nat. Commun. 2019, 10, 2352. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Schnalzger, T.; Engleitner, T.; Weiß, C.; Mishra, R.; Mibus, C.; Mitterer, T.; Rad, R.; Ruland, J. MALT1 Protease Function in Regulatory T Cells Induces MYC Activity to Promote Mitochondrial Function and Cellular Expansion. Eur. J. Immunol. 2021, 52, 85–95. [Google Scholar] [CrossRef]
- Di Pilato, M.; Kim, E.Y.; Cadilha, B.; Prüßmann, J.N.; Nasrallah, M.N.; Seruggia, D.; Usmani, S.; Misale, S.; Zappulli, V.; Carrizosa, E.; et al. Targeting the CBM Complex Causes Treg Cells to Prime Tumours for Immune Checkpoint Therapy. Nature 2019, 570, 112–116. [Google Scholar] [CrossRef]
- Demeyer, A.; Driege, Y.; Skordos, I.; Coudenys, J.; Lemeire, K.; Elewaut, D.; Staal, J.; Beyaert, R. Long-Term MALT1 Inhibition in Adult Mice Without Severe Systemic Autoimmunity. iScience 2020, 23, 101557. [Google Scholar] [CrossRef]
- Martin, K.; Junker, U.; Tritto, E.; Sutter, E.; Rubic-Schneider, T.; Morgan, H.; Niwa, S.; Li, J.; Schlapbach, A.; Walker, D.; et al. Pharmacological Inhibition of MALT1 Protease Leads to a Progressive IPEX-Like Pathology. Front. Immunol. 2020, 11, 745. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Hematological Malignancies | Role of MALT1 | MALT1 Activation | Reference |
| ATL | Proliferation Survival Anti-apoptotic signaling NF-κB target gene expression | HTLV-1 infection | [81] |
| B-ALL | Proliferation Survival Anti-apoptotic signaling | MALT1 overexpression | [56] |
| CLL | Cellular activation Proliferation Survival Anti-apoptotic signaling NF-κB target gene expression | Autonomous BCR-derived signaling | [80] |
| DLBCL | Proliferation Survival Anti-apoptotic signaling NF-κB target gene expression Pro-inflammatory signaling | Chronic BCR activation CARD11 mutations | [78,79] |
| MCL | Proliferation Survival Regulation of MYC expression | Chronic BCR activation | [77] |
| MALT lymphoma | Proliferation Survival | Chromosomal translocations resulting in MALT1 overexpression or fusion products | [70,96] |
| PEL | Survival | KSHV infection | [76] |
| T-ALL | Proliferation Survival Anti-apoptotic signaling NF-κB target gene expression | CARD11 overexpression | [97] |
| Solid Tumors | Role of MALT1 | MALT1 Activation | Reference |
| BC | Proliferation Migration Invasion Gene reprogramming Angiogenesis NF-κB target gene expression EMT Metastasis | AGTR1 PAR1 HER2 | [82,83,84,88] |
| CCA | Proliferation Growth Survival NF-κB target gene expression | Raf/Erk/Elk-1 pathway | [92] |
| GBM | Proliferation Clonogenicity Migration Invasion NF-κB target gene expression Metastasis | EGFR | [86] |
| NSCLC | Proliferation Survival Migration NF-κB target gene expression Metastasis | EGFR | [87] |
| MM | Proliferation Survival Growth NF-κB target gene expression Metastasis | TRAIL receptor | [91] |
| OS | Proliferation Survival Growth NF-κB target gene expression | PAR1 | [83] |
| OC | Invasion Migration NF-κB target gene expression | LPA receptor | [85] |
| PDAC | Proliferation Invasion NF-κB target gene expression Metastasis | MALT1 overexpression | [89] |
| PCa | Proliferation Invasion NF-κB target gene expression Metastasis | MALT1 overexpression | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez Solsona, B.; Schmitt, A.; Schulze-Osthoff, K.; Hailfinger, S. The Paracaspase MALT1 in Cancer. Biomedicines 2022, 10, 344. https://doi.org/10.3390/biomedicines10020344
Gomez Solsona B, Schmitt A, Schulze-Osthoff K, Hailfinger S. The Paracaspase MALT1 in Cancer. Biomedicines. 2022; 10(2):344. https://doi.org/10.3390/biomedicines10020344
Chicago/Turabian StyleGomez Solsona, Beatriz, Anja Schmitt, Klaus Schulze-Osthoff, and Stephan Hailfinger. 2022. "The Paracaspase MALT1 in Cancer" Biomedicines 10, no. 2: 344. https://doi.org/10.3390/biomedicines10020344
APA StyleGomez Solsona, B., Schmitt, A., Schulze-Osthoff, K., & Hailfinger, S. (2022). The Paracaspase MALT1 in Cancer. Biomedicines, 10(2), 344. https://doi.org/10.3390/biomedicines10020344