
Neuroimaging of Mouse Models of Alzheimer’s Disease


Abstract
1. Introduction
2. Human Imaging of Alzheimer’s Disease: Brief Overview
2.1. Positron Emission Tomography (PET)
2.2. Magnetic Resonance Imaging (MRI)
3. Mouse Models of AD
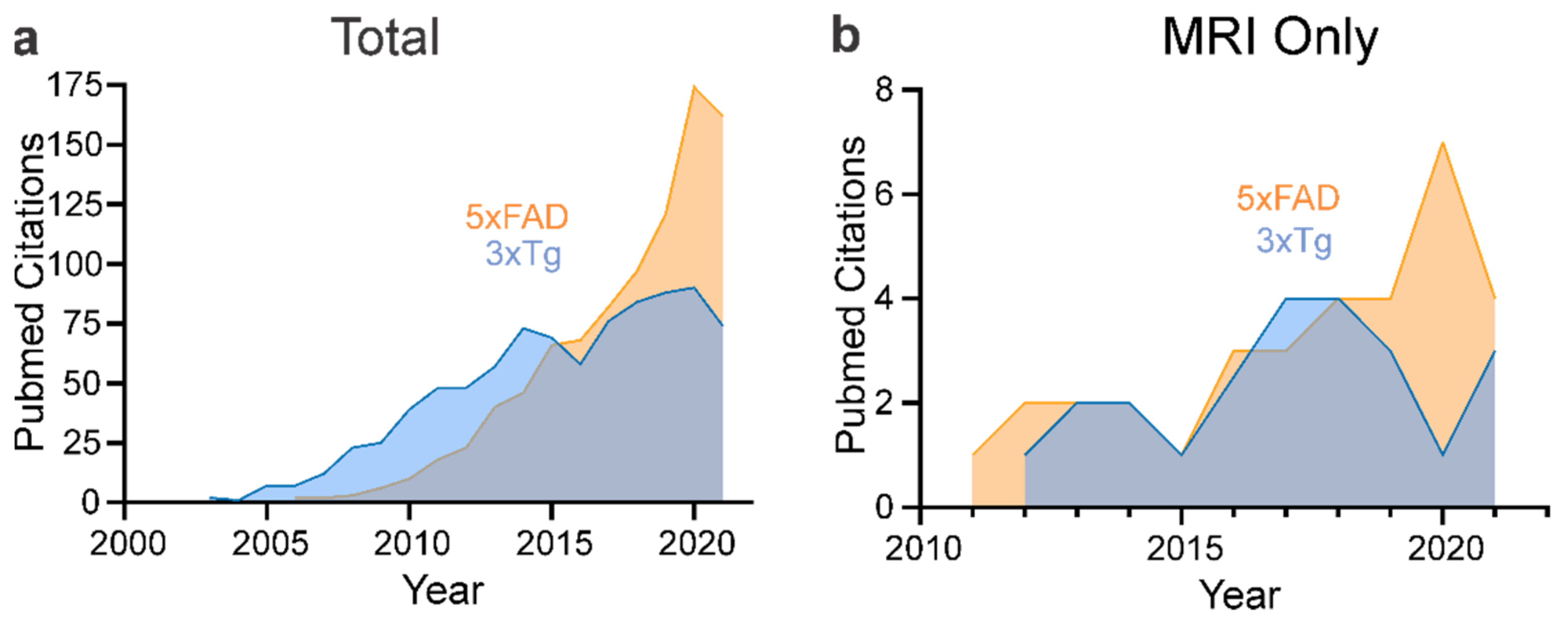
4. Neuroimaging of Mouse Models of AD
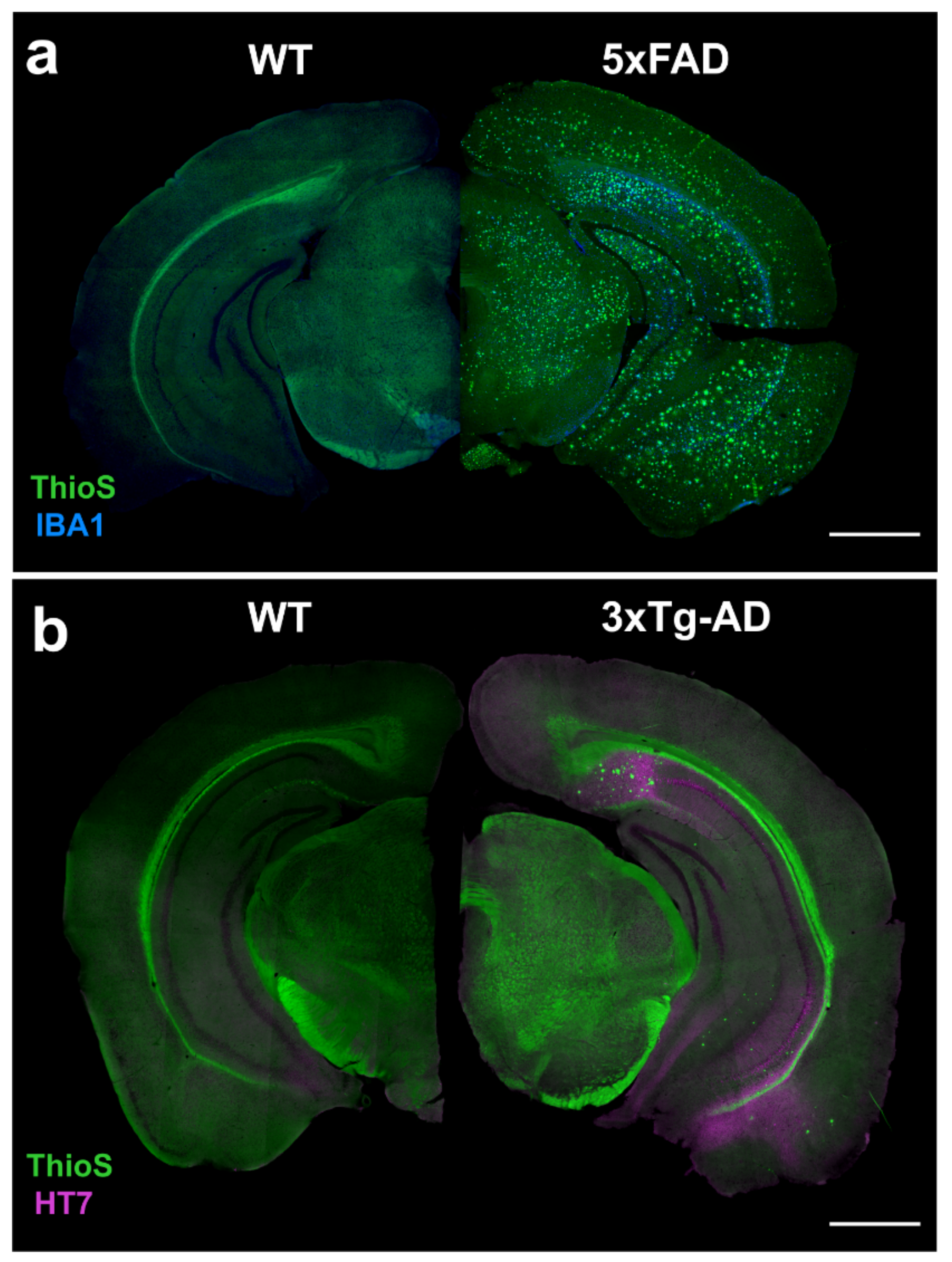
4.1. 5xFAD
4.1.1. MRI: Volumetric
4.1.2. MRI: Morphologic
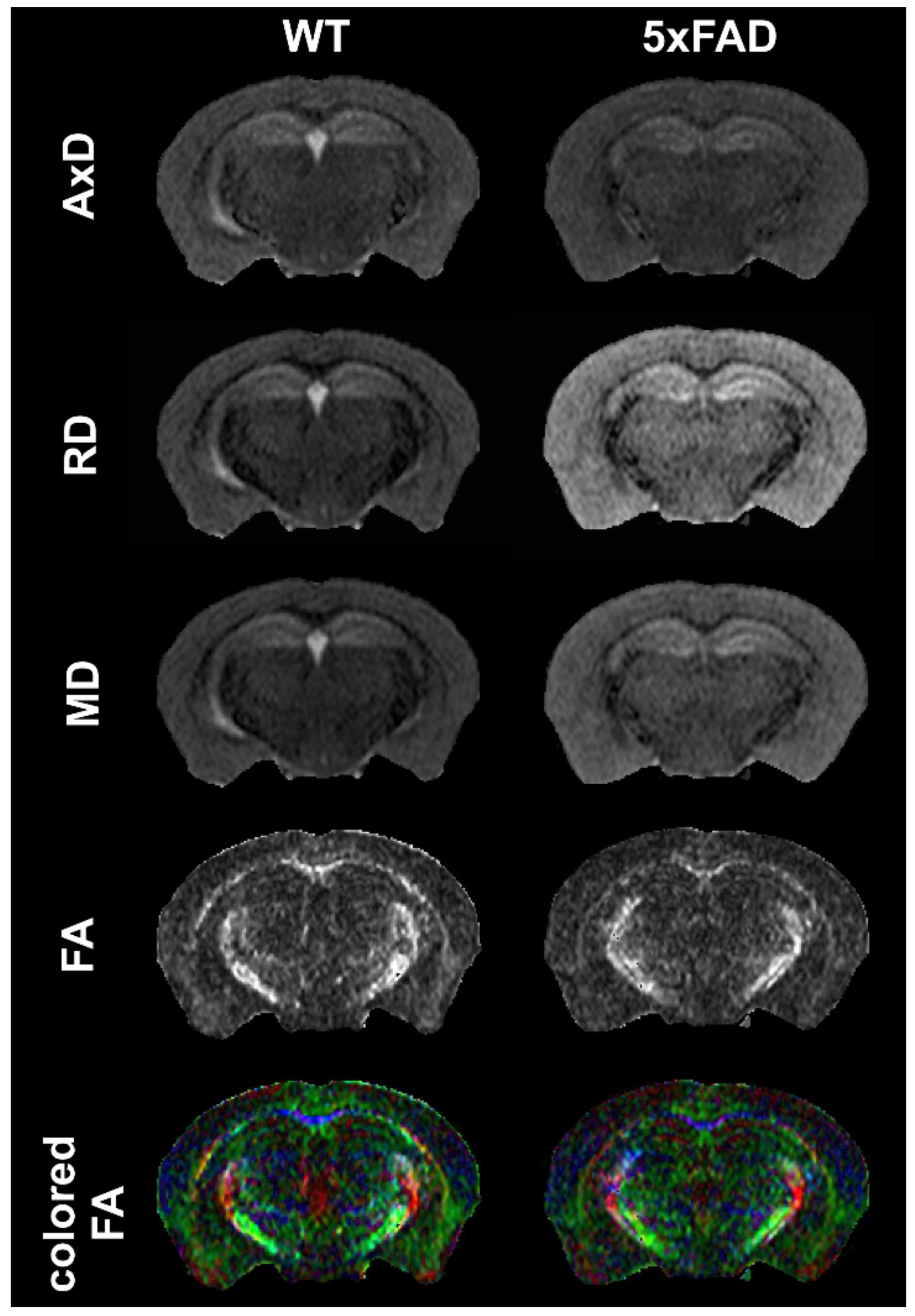
4.1.3. MRI: Diffusion
4.1.4. MRI: Functional MRI and Connectivity Studies
4.1.5. MRI: Spectroscopy
4.1.6. PET: Fluorodeoxyglucose
4.1.7. PET: Amyloid Imaging
4.1.8. PET: Other Tracers (TSPO, mGluR5, D2R, BChE…)
4.2. 3xTg-AD
4.2.1. MRI: Volumetric
4.2.2. MRI: Blood–Brain Barrier Permeability
4.2.3. MRI: Morphological
4.2.4. MRI: Diffusion
4.2.5. MRI: Functional MRI
4.2.6. MRI: Spectroscopy
4.2.7. PET: Fluorodeoxyglucose
4.2.8. PET: Amyloid Imaging
4.2.9. PET: Other Tracers (TSPO, HDAC)
5. Magnetic Resonance Findings in Other AD Mouse Models
5.1. Volumetric MRI
5.2. Magnetic Resonance Spectroscopy (MRS)
5.3. Vascular MR Imaging
5.4. Diffusion MRI (dMRI)
5.5. Relaxometry Imaging
5.6. fMRI
5.7. Manganese-Enhanced MRI (MEMRI)
5.8. Susceptibility-Weighted Imaging (SWI) and Quantitative Susceptibility Mapping (QSM)
6. Comments on Rigor and Reproducibility in AD Preclinical Neuroimaging
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heron, M. Deaths: Leading Causes for 2013. Natl. Vital Stat. Rep. 2016, 65, 1–95. [Google Scholar]
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged >/=65 years. Alzheimer’s Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Badhwar, A.; McFall, G.P.; Sapkota, S.; Black, S.E.; Chertkow, H.; Duchesne, S.; Masellis, M.; Li, L.; Dixon, R.A.; Bellec, P. A multiomics approach to heterogeneity in Alzheimer’s disease: Focused review and roadmap. Brain 2020, 143, 1315–1331. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, N.; Tosto, G. Genetics of Alzheimer’s Disease: The Importance of Polygenic and Epistatic Components. Curr. Neurol. Neurosci. Rep. 2017, 17, 78. [Google Scholar] [CrossRef]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.Y.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial Alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef]
- Nardini, E.; Hogan, R.; Flamier, A.; Bernier, G. Alzheimer’s disease: A tale of two diseases? Neural Regen. Res. 2021, 16, 1958–1964. [Google Scholar] [CrossRef]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.; Sorbi, S. The complexity of Alzheimer’s disease: An evolving puzzle. Physiol. Rev. 2021, 101, 1047–1081. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Leuzy, A.; Karikari, T.K.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; et al. The validation status of blood biomarkers of amyloid and phospho-tau assessed with the 5-phase development framework for AD biomarkers. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2140–2156. [Google Scholar] [CrossRef] [PubMed]
- D’Abramo, C.; D’Adamio, L.; Giliberto, L. Significance of Blood and Cerebrospinal Fluid Biomarkers for Alzheimer’s Disease: Sensitivity, Specificity and Potential for Clinical Use. J. Pers. Med. 2020, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Piersson, A.D.; Mohamad, M.; Rajab, F.; Suppiah, S. Cerebrospinal Fluid Amyloid Beta, Tau Levels, Apolipoprotein, and (1)H-MRS Brain Metabolites in Alzheimer’s Disease: A Systematic Review. Acad. Radiol. 2021, 28, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Araujo, J.A.; Fossel, M.; Greenberg, B.D.; Howell, G.R.; Rizzo, S.J.S.; Seyfried, N.T.; Tenner, A.J.; Territo, P.R.; Windisch, M.; et al. Translational animal models for Alzheimer’s disease: An Alzheimer’s Association Business Consortium Think Tank. Alzheimer’s Dement. 2020, 6, e12114. [Google Scholar] [CrossRef]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimer’s Dement. 2020, 6, e12110. [Google Scholar] [CrossRef]
- Hogervorst, E.; Bandelow, S.; Combrinck, M.; Irani, S.R.; Smith, A.D. The validity and reliability of 6 sets of clinical criteria to classify Alzheimer’s disease and vascular dementia in cases confirmed post-mortem: Added value of a decision tree approach. Dement. Geriatr. Cogn. Disord. 2003, 16, 170–180. [Google Scholar] [CrossRef]
- Holmes, C.; Cairns, N.; Lantos, P.; Mann, A. Validity of current clinical criteria for Alzheimer’s disease, vascular dementia and dementia with Lewy bodies. Br. J. Psychiatry 1999, 174, 45–50. [Google Scholar] [CrossRef]
- Pan, G.; King, A.; Wu, F.; Simpson-Yap, S.; Woodhouse, A.; Phipps, A.; Vickers, J.C. The potential roles of genetic factors in predicting ageing-related cognitive change and Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101402. [Google Scholar] [CrossRef]
- Buckley, R.F. Recent Advances in Imaging of Preclinical, Sporadic, and Autosomal Dominant Alzheimer’s Disease. Neurotherapeutics 2021, 18, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.G. Uses of Human MR and PET Imaging in Research of Neurodegenerative Brain Diseases. Neurotherapeutics 2021, 18, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergstrom, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Sintini, I.; Graff-Radford, J.; Senjem, M.L.; Schwarz, C.G.; Machulda, M.M.; Martin, P.R.; Jones, D.T.; Boeve, B.F.; Knopman, D.S.; Kantarci, K.; et al. Longitudinal neuroimaging biomarkers differ across Alzheimer’s disease phenotypes. Brain 2020, 143, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Panegyres, P.K.; Rogers, J.M.; McCarthy, M.; Campbell, A.; Wu, J.S. Fluorodeoxyglucose-positron emission tomography in the differential diagnosis of early-onset dementia: A prospective, community-based study. BMC Neurol. 2009, 9, 41. [Google Scholar] [CrossRef]
- Schoemaker, D.; Charidimou, A.; Zanon Zotin, M.C.; Raposo, N.; Johnson, K.A.; Sanchez, J.S.; Greenberg, S.M.; Viswanathan, A. Association of Memory Impairment With Concomitant Tau Pathology in Patients with Cerebral Amyloid Angiopathy. Neurology 2021, 96, e1975–e1986. [Google Scholar] [CrossRef]
- Cho, H.; Baek, M.S.; Lee, H.S.; Lee, J.H.; Ryu, Y.H.; Lyoo, C.H. Principal components of tau positron emission tomography and longitudinal tau accumulation in Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 114. [Google Scholar] [CrossRef]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef]
- Lagarde, J.; Sarazin, M.; Bottlaender, M. In vivo PET imaging of neuroinflammation in Alzheimer’s disease. J. Neural Transm. 2018, 125, 847–867. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, K.; Shao, T.; Hou, L.; Zhang, S.; Ye, W.; Josephson, L.; Meyer, J.H.; Zhang, M.R.; Vasdev, N.; et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta Pharm. Sin. B 2021, 11, 373–393. [Google Scholar] [CrossRef]
- Kumar, A.; Fontana, I.C.; Nordberg, A. Reactive astrogliosis: A friend or foe in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, H.; Qin, X.; Tian, M.; Zhang, H. PET imaging of reactive astrocytes in neurological disorders. Eur. J. Nucl. Med. Mol. Imaging, 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Donat, C.K.; Sastre, M. In vivo Imaging of Glial Activation in Alzheimer’s Disease. Front. Neurol. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed]
- Bastin, C.; Bahri, M.A.; Meyer, F.; Manard, M.; Delhaye, E.; Plenevaux, A.; Becker, G.; Seret, A.; Mella, C.; Giacomelli, F.; et al. In vivo imaging of synaptic loss in Alzheimer’s disease with [18F]UCB-H positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 390–402. [Google Scholar] [CrossRef]
- Chen, M.K.; Mecca, A.P.; Naganawa, M.; Finnema, S.J.; Toyonaga, T.; Lin, S.F.; Najafzadeh, S.; Ropchan, J.; Lu, Y.; McDonald, J.W.; et al. Assessing Synaptic Density in Alzheimer Disease with Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 2018, 75, 1215–1224. [Google Scholar] [CrossRef]
- McCluskey, S.P.; Plisson, C.; Rabiner, E.A.; Howes, O. Advances in CNS PET: The state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 451–489. [Google Scholar] [CrossRef]
- Carli, G.; Tondo, G.; Boccalini, C.; Perani, D. Brain Molecular Connectivity in Neurodegenerative Conditions. Brain Sci. 2021, 11, 433. [Google Scholar] [CrossRef]
- Ferreira, L.K.; Busatto, G.F. Neuroimaging in Alzheimer’s disease: Current role in clinical practice and potential future applications. Clinics 2011, 66 (Suppl. 1), 19–24. [Google Scholar] [CrossRef]
- Schroeter, M.L.; Stein, T.; Maslowski, N.; Neumann, J. Neural correlates of Alzheimer’s disease and mild cognitive impairment: A systematic and quantitative meta-analysis involving 1351 patients. Neuroimage 2009, 47, 1196–1206. [Google Scholar] [CrossRef]
- Jobson, D.D.; Hase, Y.; Clarkson, A.N.; Kalaria, R.N. The role of the medial prefrontal cortex in cognition, ageing and dementia. Brain Commun. 2021, 3, fcab125. [Google Scholar] [CrossRef]
- Montero-Crespo, M.; Dominguez-Alvaro, M.; Alonso-Nanclares, L.; DeFelipe, J.; Blazquez-Llorca, L. Three-dimensional analysis of synaptic organization in the hippocampal CA1 field in Alzheimer’s disease. Brain 2021, 144, 553–573. [Google Scholar] [CrossRef] [PubMed]
- Veldsman, M.; Nobis, L.; Alfaro-Almagro, F.; Manohar, S.; Husain, M. The human hippocampus and its subfield volumes across age, sex and APOE e4 status. Brain Commun. 2021, 3, fcaa219. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.B.; Ryman, S.G.; Hartman, A.P.; Wertz, C.J.; Vakhtin, A.A.; Alzheimer’s Disease Neuroimaging Initiative. Specific White Matter Tracts and Diffusion Properties Predict Conversion From Mild Cognitive Impairment to Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 711579. [Google Scholar] [CrossRef] [PubMed]
- Kamagata, K.; Andica, C.; Kato, A.; Saito, Y.; Uchida, W.; Hatano, T.; Lukies, M.; Ogawa, T.; Takeshige-Amano, H.; Akashi, T.; et al. Diffusion Magnetic Resonance Imaging-Based Biomarkers for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 5216. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, S.; Jiaerken, Y.; Li, K.; Zeng, Q.; Zhang, R.; Wang, C.; Xu, X.; Wu, D.; Huang, P.; et al. Distinct fiber-specific white matter reductions pattern in early- and late-onset Alzheimer’s disease. Aging 2021, 13, 12410–12430. [Google Scholar] [CrossRef]
- Nir, T.M.; Jahanshad, N.; Villalon-Reina, J.E.; Toga, A.W.; Jack, C.R.; Weiner, M.W.; Thompson, P.M.; Alzheimer’s Disease Neuroimaging Initiative. Effectiveness of regional DTI measures in distinguishing Alzheimer’s disease, MCI, and normal aging. Neuroimage Clin. 2013, 3, 180–195. [Google Scholar] [CrossRef]
- Wen, Q.; Mustafi, S.M.; Li, J.; Risacher, S.L.; Tallman, E.; Brown, S.A.; West, J.D.; Harezlak, J.; Farlow, M.R.; Unverzagt, F.W.; et al. White matter alterations in early-stage Alzheimer’s disease: A tract-specific study. Alzheimer’s Dement. 2019, 11, 576–587. [Google Scholar] [CrossRef]
- Fu, X.; Shrestha, S.; Sun, M.; Wu, Q.; Luo, Y.; Zhang, X.; Yin, J.; Ni, H. Microstructural White Matter Alterations in Mild Cognitive Impairment and Alzheimer’s Disease: Study Based on Neurite Orientation Dispersion and Density Imaging (NODDI). Clin. Neuroradiol. 2020, 30, 569–579. [Google Scholar] [CrossRef]
- Vogt, N.M.; Hunt, J.F.; Adluru, N.; Dean, D.C.; Johnson, S.C.; Asthana, S.; Yu, J.J.; Alexander, A.L.; Bendlin, B.B. Cortical Microstructural Alterations in Mild Cognitive Impairment and Alzheimer’s Disease Dementia. Cereb. Cortex 2020, 30, 2948–2960. [Google Scholar] [CrossRef]
- Chen, Q.; Turnbull, A.; Baran, T.M.; Lin, F.V. Longitudinal stability of medial temporal lobe connectivity is associated with tau-related memory decline. eLife 2020, 9, e62114. [Google Scholar] [CrossRef]
- Yan, T.; Wang, W.; Yang, L.; Chen, K.; Chen, R.; Han, Y. Rich club disturbances of the human connectome from subjective cognitive decline to Alzheimer’s disease. Theranostics 2018, 8, 3237–3255. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, A.; Dalboni da Rocha, J.L.; Nagaraju, D.B.; Tovar-Moll, F.; Bramati, I.; Coutinho, G.; Sitaram, R.; Rashidi, P. Ensemble Classification of Alzheimer’s Disease and Mild Cognitive Impairment Based on Complex Graph Measures from Diffusion Tensor Images. Front. Neurosci. 2017, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, B.; Suppiah, S.; Ibrahim, N.; Mohamad, M.; Hassan, H.A.; Nasser, N.S.; Saripan, M.I. Diagnostic power of resting-state fMRI for detection of network connectivity in Alzheimer’s disease and mild cognitive impairment: A systematic review. Hum. Brain Mapp. 2021, 42, 2941–2968. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.K.; Leung, M.K.; Lee, T.M.; Law, A.C. Resting-state abnormalities in amnestic mild cognitive impairment: A meta-analysis. Transl. Psychiatry 2016, 6, e790. [Google Scholar] [CrossRef] [PubMed]
- Schwindt, G.C.; Black, S.E. Functional imaging studies of episodic memory in Alzheimer’s disease: A quantitative meta-analysis. Neuroimage 2009, 45, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Nellessen, N.; Rottschy, C.; Eickhoff, S.B.; Ketteler, S.T.; Kuhn, H.; Shah, N.J.; Schulz, J.B.; Reske, M.; Reetz, K. Specific and disease stage-dependent episodic memory-related brain activation patterns in Alzheimer’s disease: A coordinate-based meta-analysis. Brain Struct. Funct. 2015, 220, 1555–1571. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, M.; Desouky, A.; Sirry, W. Imaging Transcriptomics in Neurodegenerative Diseases. J. Neuroimaging 2021, 31, 244–250. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prevot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Mlynarik, V.; Cacquevel, M.; Sun-Reimer, L.; Janssens, S.; Cudalbu, C.; Lei, H.; Schneider, B.L.; Aebischer, P.; Gruetter, R. Proton and phosphorus magnetic resonance spectroscopy of a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31 (Suppl. 3), S87–S99. [Google Scholar] [CrossRef] [PubMed]
- Aytan, N.; Choi, J.K.; Carreras, I.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Combination therapy in a transgenic model of Alzheimer’s disease. Exp. Neurol. 2013, 250, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.G.; Bridges, L.R.; Elderfield, K.; Amir, K.; Austen, B.; Howe, F.A. Quantitative evaluation of MRI and histological characteristics of the 5xFAD Alzheimer mouse brain. Neuroimage 2013, 76, 108–115. [Google Scholar] [CrossRef]
- Girard, S.D.; Baranger, K.; Gauthier, C.; Jacquet, M.; Bernard, A.; Escoffier, G.; Marchetti, E.; Khrestchatisky, M.; Rivera, S.; Roman, F.S. Evidence for early cognitive impairment related to frontal cortex in the 5XFAD mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33, 781–796. [Google Scholar] [CrossRef]
- Rojas, S.; Herance, J.R.; Gispert, J.D.; Abad, S.; Torrent, E.; Jimenez, X.; Pareto, D.; Perpina, U.; Sarroca, S.; Rodriguez, E.; et al. In vivo evaluation of amyloid deposition and brain glucose metabolism of 5XFAD mice using positron emission tomography. Neurobiol. Aging 2013, 34, 1790–1798. [Google Scholar] [CrossRef]
- Girard, S.D.; Jacquet, M.; Baranger, K.; Migliorati, M.; Escoffier, G.; Bernard, A.; Khrestchatisky, M.; Feron, F.; Rivera, S.; Roman, F.S.; et al. Onset of hippocampus-dependent memory impairments in 5XFAD transgenic mouse model of Alzheimer’s disease. Hippocampus 2014, 24, 762–772. [Google Scholar] [CrossRef]
- Macdonald, I.R.; DeBay, D.R.; Reid, G.A.; O’Leary, T.P.; Jollymore, C.T.; Mawko, G.; Burrell, S.; Martin, E.; Bowen, C.V.; Brown, R.E.; et al. Early detection of cerebral glucose uptake changes in the 5XFAD mouse. Curr. Alzheimer Res. 2014, 11, 450–460. [Google Scholar] [CrossRef]
- Tang, X.; Wu, D.; Gu, L.H.; Nie, B.B.; Qi, X.Y.; Wang, Y.J.; Wu, F.F.; Li, X.L.; Bai, F.; Chen, X.C.; et al. Spatial learning and memory impairments are associated with increased neuronal activity in 5XFAD mouse as measured by manganese-enhanced magnetic resonance imaging. Oncotarget 2016, 7, 57556–57570. [Google Scholar] [CrossRef]
- Aytan, N.; Choi, J.K.; Carreras, I.; Brinkmann, V.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 24939. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, N.; Tang, S.P.; Ashworth, S.; Coello, C.; Plisson, C.; Passchier, J.; Selvaraj, V.; Tyacke, R.J.; Nutt, D.J.; Sastre, M. In vivo imaging of microglial activation by positron emission tomography with [(11)C]PBR28 in the 5XFAD model of Alzheimer’s disease. Glia 2016, 64, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.G.; Lovell, D.P.; Elderfield, K.; Austen, B.; Howe, F.A. Can MRI T1 be used to detect early changes in 5xFAD Alzheimer’s mouse brain? Magn. Reson. Mater. Phys. Biol. Med. 2017, 30, 153–163. [Google Scholar] [CrossRef] [PubMed]
- DeBay, D.R.; Reid, G.A.; Macdonald, I.R.; Mawko, G.; Burrell, S.; Martin, E.; Bowen, C.V.; Darvesh, S. Butyrylcholinesterase-knockout reduces fibrillar beta-amyloid and conserves (18)FDG retention in 5XFAD mouse model of Alzheimer’s disease. Brain Res. 2017, 1671, 102–110. [Google Scholar] [CrossRef]
- Kesler, S.R.; Acton, P.; Rao, V.; Ray, W.J. Functional and structural connectome properties in the 5XFAD transgenic mouse model of Alzheimer’s disease. Netw. Neurosci. 2018, 2, 241–258. [Google Scholar] [CrossRef]
- Lee, M.; Lee, H.J.; Park, I.S.; Park, J.A.; Kwon, Y.J.; Ryu, Y.H.; Kim, C.H.; Kang, J.H.; Hyun, I.Y.; Lee, K.C.; et al. Abeta pathology downregulates brain mGluR5 density in a mouse model of Alzheimer. Neuropharmacology 2018, 133, 512–517. [Google Scholar] [CrossRef]
- Son, Y.; Kim, J.S.; Jeong, Y.J.; Jeong, Y.K.; Kwon, J.H.; Choi, H.D.; Pack, J.K.; Kim, N.; Lee, Y.S.; Lee, H.J. Long-term RF exposure on behavior and cerebral glucose metabolism in 5xFAD mice. Neurosci. Lett. 2018, 666, 64–69. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, H.J.; Kang, K.J.; Han, S.J.; Lee, Y.J.; Lee, K.C.; Lim, S.M.; Chi, D.Y.; Kim, K.M.; Park, J.A.; et al. Early Detection of Abeta Deposition in the 5xFAD Mouse by Amyloid PET. Contrast Media Mol. Imaging 2018, 2018, 5272014. [Google Scholar] [CrossRef]
- Nie, X.; Falangola, M.F.; Ward, R.; McKinnon, E.T.; Helpern, J.A.; Nietert, P.J.; Jensen, J.H. Diffusion MRI detects longitudinal white matter changes in the 3xTg-AD mouse model of Alzheimer’s disease. Magn. Reson. Imaging 2019, 57, 235–242. [Google Scholar] [CrossRef]
- Son, Y.; Jeong, Y.J.; Shin, N.R.; Oh, S.J.; Nam, K.R.; Choi, H.D.; Choi, J.Y.; Lee, H.J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. Int. J. Mol. Sci. 2020, 21, 5553. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, H.J.; Jeong, Y.J.; Nam, K.R.; Kang, K.J.; Han, S.J.; Lee, K.C.; Lee, Y.J.; Choi, J.Y. Evaluation of the neuroprotective effect of taurine in Alzheimer’s disease using functional molecular imaging. Sci. Rep. 2020, 10, 15551. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.R.; Longo, V.; Li, T.; Jonas, L.A.; Judenhofer, M.; Cherry, S.; Koutcher, J.; Lekaye, C.; Zanzonico, P.; Li, Y.M. Hybrid PET/MRI enables high-spatial resolution, quantitative imaging of amyloid plaques in an Alzheimer’s disease mouse model. Sci. Rep. 2020, 10, 10379. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.N.; Irwin, C.; Bayer, T.A.; Brenner, W.; Beindorff, N.; Bouter, C.; Bouter, Y. In vivo Imaging with (18)F-FDG- and (18)F-Florbetaben-PET/MRI Detects Pathological Changes in the Brain of the Commonly Used 5XFAD Mouse Model of Alzheimer’s Disease. Front. Med. 2020, 7, 529. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Huynh, T.T.; Rogers, B.E.; Mirica, L.M. Design of a multivalent bifunctional chelator for diagnostic (64)Cu PET imaging in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 30928–30933. [Google Scholar] [CrossRef]
- Rejc, L.; Gomez-Vallejo, V.; Joya, A.; Moreno, O.; Egimendia, A.; Castellnou, P.; Rios-Anglada, X.; Cossio, U.; Baz, Z.; Passannante, R.; et al. Longitudinal evaluation of a novel BChE PET tracer as an early in vivo biomarker in the brain of a mouse model for Alzheimer disease. Theranostics 2021, 11, 6542–6559. [Google Scholar] [CrossRef]
- Kim, E.; Di Censo, D.; Baraldo, M.; Simmons, C.; Rosa, I.; Randall, K.; Ballard, C.; Dickie, B.R.; Williams, S.C.R.; Killick, R.; et al. In vivo multi-parametric manganese-enhanced MRI for detecting amyloid plaques in rodent models of Alzheimer’s disease. Sci. Rep. 2021, 11, 12419. [Google Scholar] [CrossRef]
- Chang, S.K.; Kim, J.; Lee, D.; Yoo, C.H.; Jin, S.; Rhee, H.Y.; Ryu, C.W.; Lee, J.K.; Cho, H.; Jahng, G.H. Mapping of microvascular architecture in the brain of an Alzheimer’s disease mouse model using MRI. NMR Biomed. 2021, 34, e4481. [Google Scholar] [CrossRef]
- Tataryn, N.M.; Singh, V.; Dyke, J.P.; Berk-Rauch, H.E.; Clausen, D.M.; Aronowitz, E.; Norris, E.H.; Strickland, S.; Ahn, H.J. Vascular endothelial growth factor associated dissimilar cerebrovascular phenotypes in two different mouse models of Alzheimer’s Disease. Neurobiol. Aging 2021, 107, 96–108. [Google Scholar] [CrossRef]
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C.; et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front. Aging Neurosci. 2021, 13, 713726. [Google Scholar] [CrossRef]
- Haller, S.; Haacke, E.M.; Thurnher, M.M.; Barkhof, F. Susceptibility-weighted Imaging: Technical Essentials and Clinical Neurologic Applications. Radiology 2021, 299, 3–26. [Google Scholar] [CrossRef]
- Petrushina, I.; Hovakimyan, A.; Harahap-Carrillo, I.S.; Davtyan, H.; Antonyan, T.; Chailyan, G.; Kazarian, K.; Antonenko, M.; Jullienne, A.; Hamer, M.M.; et al. Characterization and preclinical evaluation of the cGMP grade DNA based vaccine, AV-1959D to enter the first-in-human clinical trials. Neurobiol. Dis. 2020, 139, 104823. [Google Scholar] [CrossRef] [PubMed]
- Tropres, I.; Pannetier, N.; Grand, S.; Lemasson, B.; Moisan, A.; Peoc’h, M.; Remy, C.; Barbier, E.L. Imaging the microvessel caliber and density: Principles and applications of microvascular MRI. Magn. Reson. Med. 2015, 73, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Nie, B.; Wu, D.; Liang, S.; Liu, H.; Sun, X.; Li, P.; Huang, Q.; Zhang, T.; Feng, T.; Ye, S.; et al. A stereotaxic MRI template set of mouse brain with fine sub-anatomical delineations: Application to MEMRI studies of 5XFAD mice. Magn. Reson. Imaging 2019, 57, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Bassett, D.S.; Bullmore, E. Small-world brain networks. Neuroscientist 2006, 12, 512–523. [Google Scholar] [CrossRef]
- Ishibashi, K.; Onishi, A.; Fujiwara, Y.; Ishiwata, K.; Ishii, K. Effects of glucose, insulin, and insulin resistance on cerebral 18F-FDG distribution in cognitively normal older subjects. PLoS ONE 2017, 12, e0181400. [Google Scholar] [CrossRef]
- Fueger, B.J.; Czernin, J.; Hildebrandt, I.; Tran, C.; Halpern, B.S.; Stout, D.; Phelps, M.E.; Weber, W.A. Impact of animal handling on the results of 18F-FDG PET studies in mice. J. Nucl. Med. 2006, 47, 999–1006. [Google Scholar]
- Byun, B.H.; Kim, B.I.; Park, S.Y.; Ko, I.O.; Lee, K.C.; Kim, K.M.; Kim, Y.K.; Lee, J.Y.; Bu, S.H.; Kim, J.H.; et al. Head-to-head comparison of 11C-PiB and 18F-FC119S for Abeta imaging in healthy subjects, mild cognitive impairment patients, and Alzheimer’s disease patients. Medicine 2017, 96, e6441. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Andersson, E.; Janelidze, S.; Ossenkoppele, R.; Insel, P.; Strandberg, O.; Zetterberg, H.; Rosen, H.J.; Rabinovici, G.; Chai, X.; et al. Abeta deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer’s disease. Sci. Adv. 2020, 6, eaaz2387. [Google Scholar] [CrossRef]
- Roussakis, A.A.; Piccini, P. PET Imaging in Huntington’s Disease. J. Huntington’s Dis. 2015, 4, 287–296. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Perry, E.K.; Perry, R.H.; Blessed, G.; Tomlinson, B.E. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol. Appl. Neurobiol. 1978, 4, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Algarzae, N.; Hebron, M.; Miessau, M.; Moussa, C.E. Parkin prevents cortical atrophy and Abeta-induced alterations of brain metabolism: (1)(3)C NMR and magnetic resonance imaging studies in AD models. Neuroscience 2012, 225, 22–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ishihara, Y.; Itoh, K.; Mitsuda, Y.; Shimada, T.; Kubota, T.; Kato, C.; Song, S.Y.; Kobayashi, Y.; Mori-Yasumoto, K.; Sekita, S.; et al. Involvement of brain oxidation in the cognitive impairment in a triple transgenic mouse model of Alzheimer’s disease: Noninvasive measurement of the brain redox state by magnetic resonance imaging. Free Radic. Res. 2013, 47, 731–739. [Google Scholar] [CrossRef]
- Kastyak-Ibrahim, M.Z.; Di Curzio, D.L.; Buist, R.; Herrera, S.L.; Albensi, B.C.; Del Bigio, M.R.; Martin, M. Neurofibrillary tangles and plaques are not accompanied by white matter pathology in aged triple transgenic-Alzheimer disease mice. Magn. Reson. Imaging 2013, 31, 1515–1521. [Google Scholar] [CrossRef]
- Sancheti, H.; Akopian, G.; Yin, F.; Brinton, R.D.; Walsh, J.P.; Cadenas, E. Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e69830. [Google Scholar] [CrossRef]
- Sancheti, H.; Patil, I.; Kanamori, K.; Diaz Brinton, R.; Zhang, W.; Lin, A.L.; Cadenas, E. Hypermetabolic state in the 7-month-old triple transgenic mouse model of Alzheimer’s disease and the effect of lipoic acid: A 13C-NMR study. J. Cereb. Blood Flow Metab. 2014, 34, 1749–1760. [Google Scholar] [CrossRef]
- Hohsfield, L.A.; Daschil, N.; Oradd, G.; Stromberg, I.; Humpel, C. Vascular pathology of 20-month-old hypercholesterolemia mice in comparison to triple-transgenic and APPSwDI Alzheimer’s disease mouse models. Mol. Cell. Neurosci. 2014, 63, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, B.; Liu, C.; Liang, G.; Eckenhoff, M.F.; Liu, W.; Pickup, S.; Meng, Q.; Tian, Y.; Li, S.; et al. Long-term dantrolene treatment reduced intraneuronal amyloid in aged Alzheimer triple transgenic mice. Alzheimer Dis. Assoc. Disord. 2015, 29, 184–191. [Google Scholar] [CrossRef]
- Ye, M.; Chung, H.S.; An, Y.H.; Lim, S.J.; Choi, W.; Yu, A.R.; Kim, J.S.; Kang, M.; Cho, S.; Shim, I.; et al. Standardized Herbal Formula PM012 Decreases Cognitive Impairment and Promotes Neurogenesis in the 3xTg AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 5401–5412. [Google Scholar] [CrossRef]
- Ye, M.; Chung, H.S.; Lee, C.; Yoon, M.S.; Yu, A.R.; Kim, J.S.; Hwang, D.S.; Shim, I.; Bae, H. Neuroprotective effects of bee venom phospholipase A2 in the 3xTg AD mouse model of Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 10. [Google Scholar] [CrossRef]
- Baek, H.; Ye, M.; Kang, G.H.; Lee, C.; Lee, G.; Choi, D.B.; Jung, J.; Kim, H.; Lee, S.; Kim, J.S.; et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget 2016, 7, 69347–69357. [Google Scholar] [CrossRef] [PubMed]
- Snow, W.M.; Dale, R.; O’Brien-Moran, Z.; Buist, R.; Peirson, D.; Martin, M.; Albensi, B.C. In Vivo Detection of Gray Matter Neuropathology in the 3xTg Mouse Model of Alzheimer’s Disease with Diffusion Tensor Imaging. J. Alzheimer’s Dis. 2017, 58, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Dudeffant, C.; Vandesquille, M.; Herbert, K.; Garin, C.M.; Alves, S.; Blanchard, V.; Comoy, E.E.; Petit, F.; Dhenain, M. Contrast-enhanced MR microscopy of amyloid plaques in five mouse models of amyloidosis and in human Alzheimer’s disease brains. Sci. Rep. 2017, 7, 4955. [Google Scholar] [CrossRef] [PubMed]
- Montoliu-Gaya, L.; Guell-Bosch, J.; Esquerda-Canals, G.; Roda, A.R.; Serra-Mir, G.; Lope-Piedrafita, S.; Sanchez-Quesada, J.L.; Villegas, S. Differential effects of apoE and apoJ mimetic peptides on the action of an anti-Abeta scFv in 3xTg-AD mice. Biochem. Pharmacol. 2018, 155, 380–392. [Google Scholar] [CrossRef]
- Kong, V.; Devenyi, G.A.; Gallino, D.; Ayranci, G.; Germann, J.; Rollins, C.; Chakravarty, M.M. Early-in-life neuroanatomical and behavioural trajectories in a triple transgenic model of Alzheimer’s disease. Brain Struct. Funct. 2018, 223, 3365–3382. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Ko, J.H.; Albensi, B.C. Regional hypometabolism in the 3xTg mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef]
- Chiquita, S.; Ribeiro, M.; Castelhano, J.; Oliveira, F.; Sereno, J.; Batista, M.; Abrunhosa, A.; Rodrigues-Neves, A.C.; Carecho, R.; Baptista, F.; et al. A longitudinal multimodal in vivo molecular imaging study of the 3xTg-AD mouse model shows progressive early hippocampal and taurine loss. Hum. Mol. Genet. 2019, 28, 2174–2188. [Google Scholar] [CrossRef]
- Manno, F.A.M.; Isla, A.G.; Manno, S.H.C.; Ahmed, I.; Cheng, S.H.; Barrios, F.A.; Lau, C. Early Stage Alterations in White Matter and Decreased Functional Interhemispheric Hippocampal Connectivity in the 3xTg Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 39. [Google Scholar] [CrossRef]
- Rollins, C.P.E.; Gallino, D.; Kong, V.; Ayranci, G.; Devenyi, G.A.; Germann, J.; Chakravarty, M.M. Contributions of a high-fat diet to Alzheimer’s disease-related decline: A longitudinal behavioural and structural neuroimaging study in mouse models. Neuroimage Clin. 2019, 21, 101606. [Google Scholar] [CrossRef]
- Guell-Bosch, J.; Lope-Piedrafita, S.; Esquerda-Canals, G.; Montoliu-Gaya, L.; Villegas, S. Progression of Alzheimer’s disease and effect of scFv-h3D6 immunotherapy in the 3xTg-AD mouse model: An in vivo longitudinal study using Magnetic Resonance Imaging and Spectroscopy. NMR Biomed. 2020, 33, e4263. [Google Scholar] [CrossRef]
- Falangola, M.F.; Nie, X.; Ward, R.; Dhiman, S.; Voltin, J.; Nietert, P.J.; Jensen, J.H. Diffusion MRI detects basal forebrain cholinergic abnormalities in the 3xTg-AD mouse model of Alzheimer’s disease. Magn. Reson. Imaging 2021, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stojakovic, A.; Chang, S.Y.; Nesbitt, J.; Pichurin, N.P.; Ostroot, M.A.; Aikawa, T.; Kanekiyo, T.; Trushina, E. Partial Inhibition of Mitochondrial Complex I Reduces Tau Pathology and Improves Energy Homeostasis and Synaptic Function in 3xTg-AD Mice. J. Alzheimer’s Dis. 2021, 79, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lu, C.H.; Ke, C.C.; Chiu, S.J.; Chang, C.W.; Yang, B.H.; Gelovani, J.G.; Liu, R.S. Evaluation of Class IIa Histone Deacetylases Expression and In Vivo Epigenetic Imaging in a Transgenic Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8633. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.F. Genetic drift in mammals. An. Acad. Bras. Cienc. 2019, 91, e20190339. [Google Scholar] [CrossRef]
- Carreras, I.; McKee, A.C.; Choi, J.K.; Aytan, N.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. R-flurbiprofen improves tau, but not Ass pathology in a triple transgenic model of Alzheimer’s disease. Brain Res. 2013, 1541, 115–127. [Google Scholar] [CrossRef][Green Version]
- Janelsins, M.C.; Mastrangelo, M.A.; Oddo, S.; LaFerla, F.M.; Federoff, H.J.; Bowers, W.J. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J. Neuroinflamm. 2005, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Redwine, J.M.; Kosofsky, B.; Jacobs, R.E.; Games, D.; Reilly, J.F.; Morrison, J.H.; Young, W.G.; Bloom, F.E. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: A magnetic resonance microscopy and stereologic analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 1381–1386. [Google Scholar] [CrossRef]
- Weiss, C.; Venkatasubramanian, P.N.; Aguado, A.S.; Power, J.M.; Tom, B.C.; Li, L.; Chen, K.S.; Disterhoft, J.F.; Wyrwicz, A.M. Impaired eyeblink conditioning and decreased hippocampal volume in PDAPP V717F mice. Neurobiol. Dis. 2002, 11, 425–433. [Google Scholar] [CrossRef][Green Version]
- Van Broeck, B.; Vanhoutte, G.; Pirici, D.; Van Dam, D.; Wils, H.; Cuijt, I.; Vennekens, K.; Zabielski, M.; Michalik, A.; Theuns, J.; et al. Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer’s disease. Neurobiol. Aging 2008, 29, 241–252. [Google Scholar] [CrossRef]
- Lau, J.C.; Lerch, J.P.; Sled, J.G.; Henkelman, R.M.; Evans, A.C.; Bedell, B.J. Longitudinal neuroanatomical changes determined by deformation-based morphometry in a mouse model of Alzheimer’s disease. Neuroimage 2008, 42, 19–27. [Google Scholar] [CrossRef]
- Grand’maison, M.; Zehntner, S.P.; Ho, M.K.; Hebert, F.; Wood, A.; Carbonell, F.; Zijdenbos, A.P.; Hamel, E.; Bedell, B.J. Early cortical thickness changes predict beta-amyloid deposition in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2013, 54, 59–67. [Google Scholar] [CrossRef]
- Hebert, F.; Grand’maison, M.; Ho, M.K.; Lerch, J.P.; Hamel, E.; Bedell, B.J. Cortical atrophy and hypoperfusion in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1644–1652. [Google Scholar] [CrossRef]
- Liang, S.; Huang, J.; Liu, W.; Jin, H.; Li, L.; Zhang, X.; Nie, B.; Lin, R.; Tao, J.; Zhao, S.; et al. Magnetic resonance spectroscopy analysis of neurochemical changes in the atrophic hippocampus of APP/PS1 transgenic mice. Behav. Brain Res. 2017, 335, 26–31. [Google Scholar] [CrossRef]
- Badea, A.; Johnson, G.A.; Jankowsky, J.L. Remote sites of structural atrophy predict later amyloid formation in a mouse model of Alzheimer’s disease. Neuroimage 2010, 50, 416–427. [Google Scholar] [CrossRef]
- Maheswaran, S.; Barjat, H.; Rueckert, D.; Bate, S.T.; Howlett, D.R.; Tilling, L.; Smart, S.C.; Pohlmann, A.; Richardson, J.C.; Hartkens, T.; et al. Longitudinal regional brain volume changes quantified in normal aging and Alzheimer’s APP x PS1 mice using MRI. Brain Res. 2009, 1270, 19–32. [Google Scholar] [CrossRef]
- Micotti, E.; Paladini, A.; Balducci, C.; Tolomeo, D.; Frasca, A.; Marizzoni, M.; Filibian, M.; Caroli, A.; Valbusa, G.; Dix, S.; et al. Striatum and entorhinal cortex atrophy in AD mouse models: MRI comprehensive analysis. Neurobiol. Aging 2015, 36, 776–788. [Google Scholar] [CrossRef]
- Xie, Z.; Yang, D.; Stephenson, D.; Morton, D.; Hicks, C.; Brown, T.; Bocan, T. Characterizing the regional structural difference of the brain between tau transgenic (rTg4510) and wild-type mice using MRI. Med. Image Comput. Comput. Assist. Interv. 2010, 13, 308–315. [Google Scholar] [CrossRef]
- Yang, D.; Xie, Z.; Stephenson, D.; Morton, D.; Hicks, C.D.; Brown, T.M.; Sriram, R.; O’Neill, S.; Raunig, D.; Bocan, T. Volumetric MRI and MRS provide sensitive measures of Alzheimer’s disease neuropathology in inducible Tau transgenic mice (rTg4510). Neuroimage 2011, 54, 2652–2658. [Google Scholar] [CrossRef]
- Hinteregger, B.; Loeffler, T.; Flunkert, S.; Neddens, J.; Bayer, T.A.; Madl, T.; Hutter-Paier, B. Metabolic, Phenotypic, and Neuropathological Characterization of the Tg4-42 Mouse Model for Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 1151–1168. [Google Scholar] [CrossRef]
- Wei, Z.; Xu, J.; Chen, L.; Hirschler, L.; Barbier, E.L.; Li, T.; Wong, P.C.; Lu, H. Brain metabolism in tau and amyloid mouse models of Alzheimer’s disease: An MRI study. NMR Biomed. 2021, 34, e4568. [Google Scholar] [CrossRef]
- Yin, J.; Turner, G.H.; Coons, S.W.; Maalouf, M.; Reiman, E.M.; Shi, J. Association of amyloid burden, brain atrophy and memory deficits in aged apolipoprotein epsilon4 mice. Curr. Alzheimer Res. 2014, 11, 283–290. [Google Scholar] [CrossRef]
- Allemang-Grand, R.; Scholz, J.; Ellegood, J.; Cahill, L.S.; Laliberte, C.; Fraser, P.E.; Josselyn, S.A.; Sled, J.G.; Lerch, J.P. Altered brain development in an early-onset murine model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 638–647. [Google Scholar] [CrossRef]
- Borg, J.; Chereul, E. Differential MRI patterns of brain atrophy in double or single transgenic mice for APP and/or SOD. J. Neurosci. Res. 2008, 86, 3275–3284. [Google Scholar] [CrossRef]
- Marjanska, M.; Curran, G.L.; Wengenack, T.M.; Henry, P.G.; Bliss, R.L.; Poduslo, J.F.; Jack, C.R., Jr.; Ugurbil, K.; Garwood, M. Monitoring disease progression in transgenic mouse models of Alzheimer’s disease with proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 11906–11910. [Google Scholar] [CrossRef]
- Chen, S.Q.; Wang, P.J.; Ten, G.J.; Zhan, W.; Li, M.H.; Zang, F.C. Role of myo-inositol by magnetic resonance spectroscopy in early diagnosis of Alzheimer’s disease in APP/PS1 transgenic mice. Dement. Geriatr. Cogn. Disord. 2009, 28, 558–566. [Google Scholar] [CrossRef]
- Chen, S.Q.; Cai, Q.; Shen, Y.Y.; Wang, P.J.; Teng, G.J.; Zhang, W.; Zang, F.C. Age-related changes in brain metabolites and cognitive function in APP/PS1 transgenic mice. Behav. Brain Res. 2012, 235, 1–6. [Google Scholar] [CrossRef]
- Woo, D.C.; Lee, S.H.; Lee, D.W.; Kim, S.Y.; Kim, G.Y.; Rhim, H.S.; Choi, C.B.; Kim, H.Y.; Lee, C.U.; Choe, B.Y. Regional metabolic alteration of Alzheimer’s disease in mouse brain expressing mutant human APP-PS1 by 1H HR-MAS. Behav. Brain Res. 2010, 211, 125–131. [Google Scholar] [CrossRef]
- Oberg, J.; Spenger, C.; Wang, F.H.; Andersson, A.; Westman, E.; Skoglund, P.; Sunnemark, D.; Norinder, U.; Klason, T.; Wahlund, L.O.; et al. Age related changes in brain metabolites observed by 1H MRS in APP/PS1 mice. Neurobiol. Aging 2008, 29, 1423–1433. [Google Scholar] [CrossRef]
- Haris, M.; Nath, K.; Cai, K.; Singh, A.; Crescenzi, R.; Kogan, F.; Verma, G.; Reddy, S.; Hariharan, H.; Melhem, E.R.; et al. Imaging of glutamate neurotransmitter alterations in Alzheimer’s disease. NMR Biomed. 2013, 26, 386–391. [Google Scholar] [CrossRef]
- Haris, M.; Singh, A.; Cai, K.; Nath, K.; Crescenzi, R.; Kogan, F.; Hariharan, H.; Reddy, R. MICEST: A potential tool for non-invasive detection of molecular changes in Alzheimer’s disease. J. Neurosci. Methods 2013, 212, 87–93. [Google Scholar] [CrossRef]
- Xu, W.; Zhan, Y.; Huang, W.; Wang, X.; Zhang, S.; Lei, H. Reduction of hippocampal N-acetyl aspartate level in aged APP(Swe)/PS1(dE9) transgenic mice is associated with degeneration of CA3 pyramidal neurons. J. Neurosci. Res. 2010, 88, 3155–3160. [Google Scholar] [CrossRef]
- Kuhla, A.; Ruhlmann, C.; Lindner, T.; Polei, S.; Hadlich, S.; Krause, B.J.; Vollmar, B.; Teipel, S.J. APPswe/PS1dE9 mice with cortical amyloid pathology show a reduced NAA/Cr ratio without apparent brain atrophy: A MRS and MRI study. Neuroimage Clin. 2017, 15, 581–586. [Google Scholar] [CrossRef]
- Dedeoglu, A.; Choi, J.K.; Cormier, K.; Kowall, N.W.; Jenkins, B.G. Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 2004, 1012, 60–65. [Google Scholar] [CrossRef]
- Doert, A.; Pilatus, U.; Zanella, F.; Muller, W.E.; Eckert, G.P. (1)H- and (1)(3)C-NMR spectroscopy of Thy-1-APPSL mice brain extracts indicates metabolic changes in Alzheimer’s disease. J. Neural Transm. 2015, 122, 541–550. [Google Scholar] [CrossRef]
- Lalande, J.; Halley, H.; Balayssac, S.; Gilard, V.; Dejean, S.; Martino, R.; Frances, B.; Lassalle, J.M.; Malet-Martino, M. 1H NMR metabolomic signatures in five brain regions of the AbetaPPswe Tg2576 mouse model of Alzheimer’s disease at four ages. J. Alzheimer’s Dis. 2014, 39, 121–143. [Google Scholar] [CrossRef]
- Van Duijn, S.; Nabuurs, R.J.; van Duinen, S.G.; Natte, R.; van Buchem, M.A.; Alia, A. Longitudinal monitoring of sex-related in vivo metabolic changes in the brain of Alzheimer’s disease transgenic mouse using magnetic resonance spectroscopy. J. Alzheimer’s Dis. 2013, 34, 1051–1059. [Google Scholar] [CrossRef]
- Jansen, D.; Zerbi, V.; Janssen, C.I.; Dederen, P.J.; Mutsaers, M.P.; Hafkemeijer, A.; Janssen, A.L.; Nobelen, C.L.; Veltien, A.; Asten, J.J.; et al. A longitudinal study of cognition, proton MR spectroscopy and synaptic and neuronal pathology in aging wild-type and AbetaPPswe-PS1dE9 mice. PLoS ONE 2013, 8, e63643. [Google Scholar] [CrossRef]
- Forster, D.; Davies, K.; Williams, S. Magnetic resonance spectroscopy in vivo of neurochemicals in a transgenic model of Alzheimer’s disease: A longitudinal study of metabolites, relaxation time, and behavioral analysis in TASTPM and wild-type mice. Magn. Reson. Med. 2013, 69, 944–955. [Google Scholar] [CrossRef]
- Roy, U.; Stute, L.; Hofling, C.; Hartlage-Rubsamen, M.; Matysik, J.; Robetaner, S.; Alia, A. Sex- and age-specific modulation of brain GABA levels in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 168–179. [Google Scholar] [CrossRef]
- Beckmann, N.; Schuler, A.; Mueggler, T.; Meyer, E.P.; Wiederhold, K.H.; Staufenbiel, M.; Krucker, T. Age-dependent cerebrovascular abnormalities and blood flow disturbances in APP23 mice modeling Alzheimer’s disease. J. Neurosci. 2003, 23, 8453–8459. [Google Scholar] [CrossRef]
- Krucker, T.; Schuler, A.; Meyer, E.P.; Staufenbiel, M.; Beckmann, N. Magnetic resonance angiography and vascular corrosion casting as tools in biomedical research: Application to transgenic mice modeling Alzheimer’s disease. Neurol. Res. 2004, 26, 507–516. [Google Scholar] [CrossRef]
- Mueggler, T.; Baumann, D.; Rausch, M.; Staufenbiel, M.; Rudin, M. Age-dependent impairment of somatosensory response in the amyloid precursor protein 23 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2003, 23, 8231–8236. [Google Scholar] [CrossRef]
- Thal, D.R.; Capetillo-Zarate, E.; Larionov, S.; Staufenbiel, M.; Zurbruegg, S.; Beckmann, N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 2009, 30, 1936–1948. [Google Scholar] [CrossRef]
- Beckmann, N.; Gerard, C.; Abramowski, D.; Cannet, C.; Staufenbiel, M. Noninvasive magnetic resonance imaging detection of cerebral amyloid angiopathy-related microvascular alterations using superparamagnetic iron oxide particles in APP transgenic mouse models of Alzheimer’s disease: Application to passive Abeta immunotherapy. J. Neurosci. 2011, 31, 1023–1031. [Google Scholar] [CrossRef][Green Version]
- Xu, X.; Meng, T.; Wen, Q.; Tao, M.; Wang, P.; Zhong, K.; Shen, Y. Dynamic changes in vascular size and density in transgenic mice with Alzheimer’s disease. Aging 2020, 12, 17224–17234. [Google Scholar] [CrossRef]
- Shen, Z.; Lei, J.; Li, X.; Wang, Z.; Bao, X.; Wang, R. Multifaceted assessment of the APP/PS1 mouse model for Alzheimer’s disease: Applying MRS, DTI, and ASL. Brain Res. 2018, 1698, 114–120. [Google Scholar] [CrossRef]
- Faure, A.; Verret, L.; Bozon, B.; El Tannir El Tayara, N.; Ly, M.; Kober, F.; Dhenain, M.; Rampon, C.; Delatour, B. Impaired neurogenesis, neuronal loss, and brain functional deficits in the APPxPS1-Ki mouse model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 407–418. [Google Scholar] [CrossRef]
- El Tayara Nel, T.; Delatour, B.; Volk, A.; Dhenain, M. Detection of vascular alterations by in vivo magnetic resonance angiography and histology in APP/PS1 mouse model of Alzheimer’s disease. Magn. Reson. Mater. Phys. Biol. Med. 2010, 23, 53–64. [Google Scholar] [CrossRef]
- Zazulia, A.R.; Videen, T.O.; Morris, J.C.; Powers, W.J. Autoregulation of cerebral blood flow to changes in arterial pressure in mild Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2010, 30, 1883–1889. [Google Scholar] [CrossRef]
- Govaerts, K.; Dresselaers, T.; Van Leuven, F.; Himmelreich, U. Quantitative Assessment of Age-Associated Alterations in Brain Vasculature in Wild-Type Mice and in Bigenic Mice that Model Alzheimer’s Disease. Mol. Imaging Biol. 2020, 22, 578–586. [Google Scholar] [CrossRef]
- Weidensteiner, C.; Metzger, F.; Bruns, A.; Bohrmann, B.; Kuennecke, B.; von Kienlin, M. Cortical hypoperfusion in the B6.PS2APP mouse model for Alzheimer’s disease: Comprehensive phenotyping of vascular and tissular parameters by MRI. Magn. Reson. Med. 2009, 62, 35–45. [Google Scholar] [CrossRef]
- Wiesmann, M.; Zerbi, V.; Jansen, D.; Lutjohann, D.; Veltien, A.; Heerschap, A.; Kiliaan, A.J. Hypertension, cerebrovascular impairment, and cognitive decline in aged AbetaPP/PS1 mice. Theranostics 2017, 7, 1277–1289. [Google Scholar] [CrossRef]
- Guo, Y.; Li, X.; Zhang, M.; Chen, N.; Wu, S.; Lei, J.; Wang, Z.; Wang, R.; Wang, J.; Liu, H. Age and brain regionassociated alterations of cerebral blood flow in early Alzheimer’s disease assessed in AbetaPPSWE/PS1DeltaE9 transgenic mice using arterial spin labeling. Mol. Med. Rep. 2019, 19, 3045–3052. [Google Scholar] [CrossRef]
- Zerbi, V.; Jansen, D.; Dederen, P.J.; Veltien, A.; Hamans, B.; Liu, Y.; Heerschap, A.; Kiliaan, A.J. Microvascular cerebral blood volume changes in aging APP(swe)/PS1(dE9) AD mouse model: A voxel-wise approach. Brain Struct. Funct. 2013, 218, 1085–1098. [Google Scholar] [CrossRef]
- Luo, F.; Rustay, N.R.; Ebert, U.; Hradil, V.P.; Cole, T.B.; Llano, D.A.; Mudd, S.R.; Zhang, Y.; Fox, G.B.; Day, M. Characterization of 7- and 19-month-old Tg2576 mice using multimodal in vivo imaging: Limitations as a translatable model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 933–944. [Google Scholar] [CrossRef]
- Kara, F.; Dongen, E.S.; Schliebs, R.; Buchem, M.A.; Groot, H.J.; Alia, A. Monitoring blood flow alterations in the Tg2576 mouse model of Alzheimer’s disease by in vivo magnetic resonance angiography at 17.6 T. Neuroimage 2012, 60, 958–966. [Google Scholar] [CrossRef]
- Wells, J.A.; Holmes, H.E.; O’Callaghan, J.M.; Colgan, N.; Ismail, O.; Fisher, E.M.; Siow, B.; Murray, T.K.; Schwarz, A.J.; O’Neill, M.J.; et al. Increased cerebral vascular reactivity in the tau expressing rTg4510 mouse: Evidence against the role of tau pathology to impair vascular health in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 359–362. [Google Scholar] [CrossRef]
- Wells, J.A.; O’Callaghan, J.M.; Holmes, H.E.; Powell, N.M.; Johnson, R.A.; Siow, B.; Torrealdea, F.; Ismail, O.; Walker-Samuel, S.; Golay, X.; et al. In vivo imaging of tau pathology using multi-parametric quantitative MRI. Neuroimage 2015, 111, 369–378. [Google Scholar] [CrossRef]
- Decker, Y.; Muller, A.; Nemeth, E.; Schulz-Schaeffer, W.J.; Fatar, M.; Menger, M.D.; Liu, Y.; Fassbender, K. Analysis of the vasculature by immunohistochemistry in paraffin-embedded brains. Brain Struct. Funct. 2018, 223, 1001–1015. [Google Scholar] [CrossRef]
- Mueggler, T.; Meyer-Luehmann, M.; Rausch, M.; Staufenbiel, M.; Jucker, M.; Rudin, M. Restricted diffusion in the brain of transgenic mice with cerebral amyloidosis. Eur. J. Neurosci. 2004, 20, 811–817. [Google Scholar] [CrossRef]
- Sykova, E.; Vorisek, I.; Antonova, T.; Mazel, T.; Meyer-Luehmann, M.; Jucker, M.; Hajek, M.; Ort, M.; Bures, J. Changes in extracellular space size and geometry in APP23 transgenic mice: A model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Y.; Li, M.W.; Zhang, S.; Zhang, Y.; Zhao, L.Y.; Lei, H.; Oishi, K.; Zhu, W.Z. In vivo quantitative whole-brain diffusion tensor imaging analysis of APP/PS1 transgenic mice using voxel-based and atlas-based methods. Neuroradiology 2013, 55, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Qin, Y.Y.; Zhang, S.; Jiang, J.J.; Zhang, Y.; Zhao, L.Y.; Shan, D.; Zhu, W.Z. Voxel-based diffusion tensor imaging of an APP/PS1 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, G.; Pereson, S.; Delgado, Y.P.R.; Guns, P.J.; Asselbergh, B.; Veraart, J.; Sijbers, J.; Verhoye, M.; Van Broeckhoven, C.; Van der Linden, A. Diffusion kurtosis imaging to detect amyloidosis in an APP/PS1 mouse model for Alzheimer’s disease. Magn. Reson. Med. 2013, 69, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Zerbi, V.; Kleinnijenhuis, M.; Fang, X.; Jansen, D.; Veltien, A.; Van Asten, J.; Timmer, N.; Dederen, P.J.; Kiliaan, A.J.; Heerschap, A. Gray and white matter degeneration revealed by diffusion in an Alzheimer mouse model. Neurobiol. Aging 2013, 34, 1440–1450. [Google Scholar] [CrossRef]
- Sun, S.W.; Song, S.K.; Harms, M.P.; Lin, S.J.; Holtzman, D.M.; Merchant, K.M.; Kotyk, J.J. Detection of age-dependent brain injury in a mouse model of brain amyloidosis associated with Alzheimer’s disease using magnetic resonance diffusion tensor imaging. Exp. Neurol. 2005, 191, 77–85. [Google Scholar] [CrossRef]
- Harms, M.P.; Kotyk, J.J.; Merchant, K.M. Evaluation of white matter integrity in ex vivo brains of amyloid plaque-bearing APPsw transgenic mice using magnetic resonance diffusion tensor imaging. Exp. Neurol. 2006, 199, 408–415. [Google Scholar] [CrossRef]
- Sahara, N.; Perez, P.D.; Lin, W.L.; Dickson, D.W.; Ren, Y.; Zeng, H.; Lewis, J.; Febo, M. Age-related decline in white matter integrity in a mouse model of tauopathy: An in vivo diffusion tensor magnetic resonance imaging study. Neurobiol. Aging 2014, 35, 1364–1374. [Google Scholar] [CrossRef]
- Colgan, N.; Siow, B.; O’Callaghan, J.M.; Harrison, I.F.; Wells, J.A.; Holmes, H.E.; Ismail, O.; Richardson, S.; Alexander, D.C.; Collins, E.C.; et al. Application of neurite orientation dispersion and density imaging (NODDI) to a tau pathology model of Alzheimer’s disease. Neuroimage 2016, 125, 739–744. [Google Scholar] [CrossRef]
- Thiessen, J.D.; Glazner, K.A.; Nafez, S.; Schellenberg, A.E.; Buist, R.; Martin, M.; Albensi, B.C. Histochemical visualization and diffusion MRI at 7 Tesla in the TgCRND8 transgenic model of Alzheimer’s disease. Brain Struct. Funct. 2010, 215, 29–36. [Google Scholar] [CrossRef]
- Colon-Perez, L.M.; Ibanez, K.R.; Suarez, M.; Torroella, K.; Acuna, K.; Ofori, E.; Levites, Y.; Vaillancourt, D.E.; Golde, T.E.; Chakrabarty, P.; et al. Neurite orientation dispersion and density imaging reveals white matter and hippocampal microstructure changes produced by Interleukin-6 in the TgCRND8 mouse model of amyloidosis. Neuroimage 2019, 202, 116138. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Mishra, A.; Wang, T.; Wang, Y.; Desai, M.; Chen, S.; Mao, Z.; Do, L.; Bernstein, A.S.; Trouard, T.P.; et al. Evidence in support of chromosomal sex influencing plasma based metabolome vs APOE genotype influencing brain metabolome profile in humanized APOE male and female mice. PLoS ONE 2020, 15, e0225392. [Google Scholar] [CrossRef] [PubMed]
- Badea, A.; Kane, L.; Anderson, R.J.; Qi, Y.; Foster, M.; Cofer, G.P.; Medvitz, N.; Buckley, A.F.; Badea, A.K.; Wetsel, W.C.; et al. The fornix provides multiple biomarkers to characterize circuit disruption in a mouse model of Alzheimer’s disease. Neuroimage 2016, 142, 498–511. [Google Scholar] [CrossRef]
- Helpern, J.A.; Lee, S.P.; Falangola, M.F.; Dyakin, V.V.; Bogart, A.; Ardekani, B.; Duff, K.; Branch, C.; Wisniewski, T.; de Leon, M.J.; et al. MRI assessment of neuropathology in a transgenic mouse model of Alzheimer’s disease. Magn. Reson. Med. 2004, 51, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Falangola, M.F.; Dyakin, V.V.; Lee, S.P.; Bogart, A.; Babb, J.S.; Duff, K.; Nixon, R.; Helpern, J.A. Quantitative MRI reveals aging-associated T2 changes in mouse models of Alzheimer’s disease. NMR Biomed. 2007, 20, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, X.Y.; Gao, F.B.; Wang, L.; Xia, R.; Li, Z.X.; Xing, W.; Tang, B.S.; Zeng, Y.; Zhou, G.F.; et al. Magnetic resonance T2 relaxation time at 7 Tesla associated with amyloid beta pathology and age in a double-transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2016, 610, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Alquezar, C.; Bartolome, F.; Antequera, D.; Barrios, L.; Carro, E.; Cerdan, S.; Martin-Requero, A. Systematic evaluation of magnetic resonance imaging and spectroscopy techniques for imaging a transgenic model of Alzheimer’s disease (AbetaPP/PS1). J. Alzheimer’s Dis. 2012, 30, 337–353. [Google Scholar] [CrossRef]
- Teipel, S.J.; Kaza, E.; Hadlich, S.; Bauer, A.; Bruning, T.; Plath, A.S.; Krohn, M.; Scheffler, K.; Walker, L.C.; Lotze, M.; et al. Automated detection of amyloid-beta-related cortical and subcortical signal changes in a transgenic model of Alzheimer’s disease using high-field MRI. J. Alzheimer’s Dis. 2011, 23, 221–237. [Google Scholar] [CrossRef]
- Kara, F.; Hofling, C.; Rossner, S.; Schliebs, R.; Van der Linden, A.; Groot, H.J.; Alia, A. In Vivo Longitudinal Monitoring of Changes in the Corpus Callosum Integrity During Disease Progression in a Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 941–950. [Google Scholar] [CrossRef]
- Roy, U.; Heredia-Munoz, M.T.; Stute, L.; Hofling, C.; Matysik, J.; Meijer, J.H.; Rossner, S.; Alia, A. Degeneration of the Suprachiasmatic Nucleus in an Alzheimer’s Disease Mouse Model Monitored by in vivo Magnetic Resonance Relaxation Measurements and Immunohistochemistry. J. Alzheimer’s Dis. 2019, 69, 363–375. [Google Scholar] [CrossRef]
- Grandjean, J.; Derungs, R.; Kulic, L.; Welt, T.; Henkelman, M.; Nitsch, R.M.; Rudin, M. Complex interplay between brain function and structure during cerebral amyloidosis in APP transgenic mouse strains revealed by multi-parametric MRI comparison. Neuroimage 2016, 134, 1–11. [Google Scholar] [CrossRef]
- Grandjean, J.; Schroeter, A.; He, P.; Tanadini, M.; Keist, R.; Krstic, D.; Konietzko, U.; Klohs, J.; Nitsch, R.M.; Rudin, M. Early alterations in functional connectivity and white matter structure in a transgenic mouse model of cerebral amyloidosis. J. Neurosci. 2014, 34, 13780–13789. [Google Scholar] [CrossRef]
- Latif-Hernandez, A.; Shah, D.; Craessaerts, K.; Saido, T.; Saito, T.; De Strooper, B.; Van der Linden, A.; D’Hooge, R. Subtle behavioral changes and increased prefrontal-hippocampal network synchronicity in APP(NL-G-F) mice before prominent plaque deposition. Behav. Brain Res. 2019, 364, 431–441. [Google Scholar] [CrossRef]
- Shah, D.; Praet, J.; Latif Hernandez, A.; Hofling, C.; Anckaerts, C.; Bard, F.; Morawski, M.; Detrez, J.R.; Prinsen, E.; Villa, A.; et al. Early pathologic amyloid induces hypersynchrony of BOLD resting-state networks in transgenic mice and provides an early therapeutic window before amyloid plaque deposition. Alzheimer’s Dement. 2016, 12, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Detrez, J.R.; Ben-Nejma, I.R.H.; Van Kolen, K.; Van Dam, D.; De Deyn, P.P.; Fransen, E.; Verhoye, M.; Timmermans, J.P.; Nuydens, R.; Van der Linden, A.; et al. Progressive tau aggregation does not alter functional brain network connectivity in seeded hTau.P301L mice. Neurobiol. Dis. 2020, 143, 105011. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.J.; Zhang, X.; Ziomek, G.J.; Jacobs, R.E.; Bearer, E.L. Deficits in axonal transport in hippocampal-based circuitry and the visual pathway in APP knock-out animals witnessed by manganese enhanced MRI. Neuroimage 2012, 60, 1856–1866. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bearer, E.L.; Manifold-Wheeler, B.C.; Medina, C.S.; Gonzales, A.G.; Chaves, F.L.; Jacobs, R.E. Alterations of functional circuitry in aging brain and the impact of mutated APP expression. Neurobiol. Aging 2018, 70, 276–290. [Google Scholar] [CrossRef]
- Smith, K.D.; Kallhoff, V.; Zheng, H.; Pautler, R.G. In vivo axonal transport rates decrease in a mouse model of Alzheimer’s disease. Neuroimage 2007, 35, 1401–1408. [Google Scholar] [CrossRef][Green Version]
- Androuin, A.; Abada, Y.S.; Ly, M.; Santin, M.; Petiet, A.; Epelbaum, S.; Bertrand, A.; Delatour, B. Activity-induced MEMRI cannot detect functional brain anomalies in the APPxPS1-Ki mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 1140. [Google Scholar] [CrossRef]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef]
- Braakman, N.; Matysik, J.; van Duinen, S.G.; Verbeek, F.; Schliebs, R.; de Groot, H.J.; Alia, A. Longitudinal assessment of Alzheimer’s beta-amyloid plaque development in transgenic mice monitored by in vivo magnetic resonance microimaging. J. Magn. Reson. Imaging 2006, 24, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.J.; Dibb, R.; Bulk, M.; van der Weerd, L.; Liu, C. Imaging beta amyloid aggregation and iron accumulation in Alzheimer’s disease using quantitative susceptibility mapping MRI. Neuroimage 2019, 191, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease—The gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Benavides, F.; Rulicke, T.; Prins, J.B.; Bussell, J.; Scavizzi, F.; Cinelli, P.; Herault, Y.; Wedekind, D. Genetic quality assurance and genetic monitoring of laboratory mice and rats: FELASA Working Group Report. Lab. Anim. 2020, 54, 135–148. [Google Scholar] [CrossRef]
- Jessen, F.; Gur, O.; Block, W.; Ende, G.; Frolich, L.; Hammen, T.; Wiltfang, J.; Kucinski, T.; Jahn, H.; Heun, R.; et al. A multicenter (1)H-MRS study of the medial temporal lobe in AD and MCI. Neurology 2009, 72, 1735–1740. [Google Scholar] [CrossRef]
- Modrego, P.J.; Fayed, N. Longitudinal magnetic resonance spectroscopy as marker of cognitive deterioration in mild cognitive impairment. Am. J. Alzheimer’s Dis. Dement. 2011, 26, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Foy, C.M.; Daly, E.M.; Glover, A.; O’Gorman, R.; Simmons, A.; Murphy, D.G.; Lovestone, S. Hippocampal proton MR spectroscopy in early Alzheimer’s disease and mild cognitive impairment. Brain Topogr. 2011, 24, 316–322. [Google Scholar] [CrossRef]
- Walecki, J.; Barcikowska, M.; Cwikla, J.B.; Gabryelewicz, T. N-acetylaspartate, choline, myoinositol, glutamine and glutamate (glx) concentration changes in proton MR spectroscopy (1H MRS) in patients with mild cognitive impairment (MCI). Med. Sci. Monit. 2011, 17, MT105–MT111. [Google Scholar] [CrossRef]
- Silverman, D.H.; Small, G.W.; Chang, C.Y.; Lu, C.S.; Kung De Aburto, M.A.; Chen, W.; Czernin, J.; Rapoport, S.I.; Pietrini, P.; Alexander, G.E.; et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA 2001, 286, 2120–2127. [Google Scholar] [CrossRef]
- Levin, F.; Ferreira, D.; Lange, C.; Dyrba, M.; Westman, E.; Buchert, R.; Teipel, S.J.; Grothe, M.J.; Alzheimer’s Disease Neuroimaging Initiative. Data-driven FDG-PET subtypes of Alzheimer’s disease-related neurodegeneration. Alzheimer’s Res. Ther. 2021, 13, 49. [Google Scholar] [CrossRef]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative; Swedish BioFinder Study. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef] [PubMed]
- Brendel, M.; Jaworska, A.; Probst, F.; Overhoff, F.; Korzhova, V.; Lindner, S.; Carlsen, J.; Bartenstein, P.; Harada, R.; Kudo, Y.; et al. Small-Animal PET Imaging of Tau Pathology with 18F-THK5117 in 2 Transgenic Mouse Models. J. Nucl. Med. 2016, 57, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Ninan, S.; Atkinson, R.; Fan, Z.; Brooks, D.J.; Edison, P. Does Microglial Activation Influence Hippocampal Volume and Neuronal Function in Alzheimer’s Disease and Parkinson’s Disease Dementia? J. Alzheimer’s Dis. 2016, 51, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, N.; Malpetti, M.; Mak, E.; Williams, G.B.; Bevan-Jones, W.R.; Carter, S.F.; Passamonti, L.; Fryer, T.D.; Hong, Y.T.; Aigbirhio, F.I.; et al. Gray matter changes related to microglial activation in Alzheimer’s disease. Neurobiol. Aging 2020, 94, 236–242. [Google Scholar] [CrossRef]
- Kumar, A.; Koistinen, N.A.; Malarte, M.L.; Nennesmo, I.; Ingelsson, M.; Ghetti, B.; Lemoine, L.; Nordberg, A. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer’s disease brain. Mol. Psychiatry 2021, 26, 5833–5847. [Google Scholar] [CrossRef]
- Rodriguez-Vieitez, E.; Ni, R.; Gulyas, B.; Toth, M.; Haggkvist, J.; Halldin, C.; Voytenko, L.; Marutle, A.; Nordberg, A. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: A correlative positron emission tomography and in vitro imaging study. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1119–1132. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.C.; Jung, K.S.; Kim, J.; Jang, J.S.; Kwon, S.; Byun, M.S.; Yi, D.; Byeon, G.; Jung, G.; et al. The clinical use of blood-test factors for Alzheimer’s disease: Improving the prediction of cerebral amyloid deposition by the QPLEX(TM) Alz plus assay kit. Exp. Mol. Med. 2021, 53, 1046–1054. [Google Scholar] [CrossRef]
- Fabrizio, C.; Termine, A.; Caltagirone, C.; Sancesario, G. Artificial Intelligence for Alzheimer’s Disease: Promise or Challenge? Diagnostics 2021, 11, 1473. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| MR Sequence | Information Provided |
|---|---|
| T2-weighted imaging (T2WI) | Regional volumes and multi-echo regional tissue relaxation for iron and water content. |
| Diffusion tensor imaging (DTI) | Tissue microstructure. Regional axial, radial and mean diffusivity and fractional anisotropy. Multishell DTI enables alternate reconstruction schemes such as NODDI for neurite density (NDI), dispersion (ODI) and isotropic water (ISOVF) indices. |
| Susceptibility-weighted imaging (SWI) | Iron associated with amyloid β deposition, iron content and extravascular blood; useful in cerebral amyloid angiopathy. Newer QSM methods allow for quantification. |
| Perfusion-weighted imaging (PWI) | Cerebrovascular function, cerebral blood volumes and flow. |
| Functional MRI (fMRI) | Resting state (rsfMRI) for brain-wide connectivity and task evoked functional MRI for task-specific connectivity. |
| Spectroscopy (MRS) | Brain metabolites |
| References | Imaging Age (Months) * | Sex | Imaging Modality | Magnet (T) | In Vivo, Ex Vivo, In Vitro | Key Findings |
|---|---|---|---|---|---|---|
| Mlynarik et al., 2012 [63] | 8, 9, 10, 16 | NS | MRS | 14.1 | in vivo | Increased Myo, decreased NAA and GABA at 9 months in dorsal hippocampus |
| Aytan et al., 2013 [64] | 8 | F | MRS | 14 | in vitro | Decreased NAA, GABA and Glu, increased Myo and Gln in the motor cortex |
| Spencer et al., 2013 [65] | 11 | M, F | MRI: T1, T2 | 4.7 | in vivo | Lower T1 and T2 values in 5xFAD mice, T1 more sensitive to change |
| Girard et al., 2013 [66] | 2, 4, 6 | NS | MRI: T2 RARE | 7 | in vivo | No differences in volumes (whole brain, forebrain, cerebral cortex, ventricles, frontal cortex, hippocampus, striatum, olfactory bulbs) |
| Rojas et al., 2013 [67] | 10–16 | NS | PET | in vivo | Detection of Aβ with 11C-PiB and 18F-Florbetapir, increased 18F-FDG uptake in 5xFAD compared to WT | |
| Girard et al., 2014 [68] | 2, 4, 6 | M, F | MRI: T2 RARE | 7 | in vivo | No differences in volumes (whole brain, forebrain, cerebral cortex, ventricles, frontal cortex, hippocampus, striatum, olfactory bulbs) |
| Macdonald et al., 2014 [69] | 2, 5, 13 | M | PET-CT, MRI | 3 | in vivo | Reduced 18F-FDG uptake and 10% decrease in hippocampal volume at 13 months, no other volume differences |
| Tang et al., 2016 [70] | 1, 2, 3, 5 | M | MRI: T1, MEMRI | 7 | in vivo | Increased signal intensity in brain regions involved in spatial cognition |
| Aytan et al., 2016 [71] | 3 | F | MRS | 14 | in vitro | Decreased taurine, NAA, GABA and Glu in the hippocampus |
| Mirzaei et al., 2016 [72] | 6 | F | PET | in vivo | Uptake of 11C-PBR28 is increased in 5xFAD mice | |
| Spencer et al., 2017 [73] | 2.5, 5 | M, F | MRI: T1 | 4.7 | in vivo | T1 is not a sensitive measure to detect disease onset or progression at early stages |
| DeBay et al., 2017 [74] | 5 | M | PET-CT | in vivo | Decreased 18F-FDG uptake in 5xFAD vs. WT | |
| Kesler et al., 2018 [75] | 6 | M | MRI: fMRI, T2RARE, DTI | 7, 9.4 | in vivo, ex vivo | Structural networks exhibited higher path lengths in vivo and ex vivo 5xFAD vs. WT |
| Lee M et al., 2018 [76] | 10 | M | MRI: T2, PET | 9.4 | in vivo | 18F-FPEB shows mGluR5 is decreased in hippocampus and striatum of 5xFAD mice |
| Son et al., 2018 [77] | 9.5 | F | PET | in vivo | Decreased 18F-FDG uptake in 5xFAD vs. WT | |
| Oh et al., 2018 [78] | 5.5 | M | PET-CT | in vivo | Used 11C-FC119S to quantify Aβ in 5xFAD brain | |
| Nie et al., 2019 [79] | 1, 2, 3, 5 | M | MRI: MEMRI | 7 | in vivo | Increased neuronal activity in hippocampus and amygdala at 1, 2, and 5 months |
| Son et al., 2020 [80] | 10 | F | PET, microCT | in vivo | Binding of DR2 tracer (18F-Fallypride) is decreased in 5xFAD mice, no differences/effects seen with mGluR5 tracer (18F-FPEB) | |
| Oh et al., 2020 [81] | 9 | NS | PET-CT | in vivo | Decreased 18F-FPEB uptake in 5xFAD mice | |
| Frost et al., 2020 [82] | 14 | NS | PET-MRI | 7 | in vivo | Increased uptake of 18F-Florbetapir in cortex, hippocampus and thalamus in 5xFAD mice |
| Franke et al., 2020 [83] | 7, 12 | M | PET-MRI | 1 | in vivo | Detection of cerebral hypometabolism and increased plaque load before the onset of severe memory deficits |
| Cho et al., 2020 [84] | NS | NS | PET-CT | in vivo | Testing of new 64Cu tracers to detect Aβ | |
| Rejc et al., 2021 [85] | longitudinal from 2 to 12 | F | PET-CT | in vivo | Increased 18F-Florbetaben uptake in cortex and hippocampus starting at 5 months Possible use of 11C-BChE as biomarker | |
| Kim et al., 2021 [86] | 8–9.5 | M, F | MRI: MEMRI, SWI | 9.4 | in vivo | Manganese improves SNR, but SWI alone is sufficient to detect amyloid plaques |
| Chang et al., 2021 [87] | 6 | M | MRI: microvascular MRI | 7 | in vivo | Microvascular damage in the cortex of 5xFAD mice |
| Tataryn et al., 2021 [88] | 7, 12 | F | MRI: DSC-MRI, PET-CT | 7 | In vivo | No significant differences in whole brain glucose uptake between WT and 5xFAD |
| Oblak et al., 2021 [89] | 4, 12 | M, F | PET-MRI | 3 | In vivo | Increased 18F-FDG retention in cortex of 5xFAD females at 12 months Using 18F-Florbetapir, detection of Aβ at 4 months and significant increase by 12 months |
| References | Imaging Age (Months) * | Sex | Imaging Modality | Magnet (T) | In Vivo, Ex Vivo | Key Findings |
|---|---|---|---|---|---|---|
| Algarzae et al., 2012 [102] | 12, 18 | NS | MRI: T2 | 7 | in vivo | Cortical atrophy and increased ventricular volumes at 18 months |
| Ishihara et al., 2013 [103] | 6 | NS | MRI: T1 w Gd | 1.5 | in vivo | No differences in BBB permeability |
| Kastyak-Ibrahim et al., 2013 [104] | 11, 13, 15, 17 | NS | MRI: T2 and DTI | 7 | in vivo, ex vivo | No detectable white matter changes (volumes, DTI metrics or myelin staining) |
| Sancheti et al., 2013 [105] | 7, 13 | NS | PET-CT | in vivo | Decreased 18F-FDG uptake in 3xTg-AD mice compared to WT | |
| Sancheti et al., 2014 [106] | 7 | M | MRS, PET | ex vivo | 50% increased influx of 13C in 3xTg-AD mice compared to controls, no differences in 18F-FDG uptake in the hippocampus and motor and somatosensory cortex of 3xTg-AD mice compared to WT | |
| Hohsfield et al., 2014 [107] | 7, 14, 20 | M | MRI: T2*FLASH, SWI | 9.4 | ex vivo | No microbleeds found, increased ventricle size in 3xTg-AD mice at 14 and 20 months |
| Wu Z et al., 2015 [108] | 22 | M, F | MRI: T2 | 9.4 | in vivo | No volume differences in cortex or hippocampus between 3xTg-AD and WT mice |
| Ye M et al., 2016a [109] | 6 | NS | PET | in vivo | Decreased 18F-FDG uptake in 3xTg-AD mice compared to WT in hippocampus and prefrontal cortex | |
| Ye M et al., 2016b [110] | 6 | NS | PET | in vivo | Decreased 18F-FDG uptake in 3xTg-AD mice compared to WT in diencephalon | |
| Baek et al., 2016 [111] | 6 | M | PET | in vivo | Decreased 18F-FDG uptake in 3xTg-AD mice compared to WT | |
| Snow et al., 2017 [112] | 12, 14 | M, F | MRI: T2RARE, EPI DTI | 7 | in vivo | Changes of DTI metrics in hippocampus but not in cortex |
| Dudeffant et al., 2017 [113] | from 11 to 24 | M | MRI: 3D GRE w Gd | 7 | in vivo, ex vivo | Amyloid plaques could not be detected with MRI in 3xTg-AD mice |
| Montoliu-Gaya et al., 2018 [114] | 6 | F | MRI: T2RARE, MRS | 7 | in vivo | No significant difference in whole brain volume, increased alanine in cortex and hippocampus of 3xTg-AD mice |
| Kong et al., 2018 [115] | 1.5, 2, 3, 4, 5, 6 | M, F | MRI: 3D Flash T1WI, MEMRI | 7 | in vivo | Decreased brain regions volume |
| Adlimoghaddam et al., 2019 [116] | 11 | M | PET-MRI | 7 | in vivo | Decreased 18F-FDG uptake in the bilateral piriform area and insular cortex of 3xTg-AD mice compared to WT, but no differences in the whole brain |
| Chiquita et al., 2019 [117] | 4, 8, 12, 16 | M | MRI: T2, 2D DCE-FLASH, PET | 9.4 | in vivo | Decreased hippocampal volume at all ages, decreased BBB permeability index at 16 months, decreased taurine levels in hippocampus, no difference in 11C-PiB and 11C-PK11195 uptake |
| Manno et al., 2019 [118] | 2 | F | MRI: T2RARE, DTI, rsfMRI | 7 | in vivo | 4-fold increase in ventricle volume, decreased hippocampal interhemispheric connectivity, increased cortical FA but decreased RD |
| Rollins et al., 2019 [119] | 2, 4, 6 | M, F | MRI: MEMRI | 7 | in vivo | Decreased brain regions volume |
| Guëll-Bosch et al., 2020 [120] | 5, 7, 9, 12 | F | MRI: T2RARE, T2MSME, EPI, MRS | 7 | in vivo | Decreased brain volume, increased Aβ, increased inflammation in hippocampus and cortex |
| Falangola et al., 2021 [121] | 2, 8, 15 | NS | MRI: dMRI | 7 | in vivo | Changes in DTI metrics in 3xTg-AD mice compared to WT at 2 and 8 months |
| Stojakovic et al., 2021 [122] | 16 | M, F | PET-CT | in vivo | Decreased 18F-FDG uptake in males and females 3xTg-AD mice compared to WT | |
| Chen et al., 2021 [123] | 5, 8, 11 | NS | PET-CT | in vivo | Increased 11C-PiB in 8- and 11-month-old 3xTg-AD mice, increased uptake of the HDAC tracer 18F-TFAHA at 8 and 11 months |
| Imaging Modality | Generalized Findings | Human Studies | Mouse Studies |
|---|---|---|---|
| MRI–Structural | Volumetric decreases Brain atrophy in human studies Less robust findings in mouse AD models | Schroeter et al., 2009 [39], Jobson et al., 2021 [40] | Girard et al., 2014 [68], Mcdonald et al., 2014 [69], Hohsfield et al., 2014 [107], Guëll-Bosch et al., 2020 [120], Lau et al., 2008 [130] |
| MRI-dMRI | Increased FA Decreased MD, AxD, RD Reduced connectivity | Nir et al., 2013 [46], Chen et al., 2020 [50] | Manno et al., 2019 [118], Falangola et al., 2021 [121], Qin et al., 2013 [182], Shu et al., 2013 [183] |
| MRI–multishell dMRI | Increased ODI Decreased NDI Increased intracellular volume fraction (ICVF) | Wen et al., 2019 [47], Fu et al., 2020 [48] | Colgan et al., 2016 [189], Colon-Perez et al., 2019 [191] |
| rs-fMRI | Decreased connectivity (temporal lobe) Increased path lengths; increased disconnectivity | Schwindt et al., 2009 [55] | Kesler et al., 2018 [75], Manno et al., 2019 [118], Shah et al., 2016 [204] |
| MRI–MRS | Decreased NAA Decreased NAA/Cr Decreased Glu/Gln (Glx) Increased Myo | Jessen et al., 2009 [215], Modrego and Fayed 2012 [216], Foy et al., 2011 [217], Walecki et al., 2011 [218] | Mlynarik et al., 2012 [63], Guëll-Bosch et al., 2020 [120], Oberg et al., 2008 [148] |
| PET-FDG | Reduced metabolism | Silverman et al., 2001 [219], Levin et al., 2021 [220] | Son et al., 2018 [77], Franke et al., 2020 [83], Adlimoghaddam et al., 2019 [116] |
| PET-Aβ | Increased uptake | Sintini et al., 2020 [24], Panegyres et al., 2009 [25] | Rojas et al., 2013 [67], Frost et al., 2020 [82], Chen et al., 2021 [123] |
| PET–Tau | Increased uptake with advancing AD Tau labeling recapitulates Braak staging | Cho et al., 2020 [27], Johnson et al., 2016 [28], Vogel et al., 2020 [221] | Brendel et al., 2016 [222], Sahara et al., 2017 [188] |
| PET- glial | Increased microglial binding associated with atrophy Increased astrocyte binding | Femminella et al., 2016 [223], Nicastro et al., 2020 [224], Kumar et al., 2021 [225] | Mirzaei et al., 2016 [72], Rodriguez-Vieitez et al., 2015 [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jullienne, A.; Trinh, M.V.; Obenaus, A. Neuroimaging of Mouse Models of Alzheimer’s Disease. Biomedicines 2022, 10, 305. https://doi.org/10.3390/biomedicines10020305
Jullienne A, Trinh MV, Obenaus A. Neuroimaging of Mouse Models of Alzheimer’s Disease. Biomedicines. 2022; 10(2):305. https://doi.org/10.3390/biomedicines10020305
Chicago/Turabian StyleJullienne, Amandine, Michelle V. Trinh, and Andre Obenaus. 2022. "Neuroimaging of Mouse Models of Alzheimer’s Disease" Biomedicines 10, no. 2: 305. https://doi.org/10.3390/biomedicines10020305
APA StyleJullienne, A., Trinh, M. V., & Obenaus, A. (2022). Neuroimaging of Mouse Models of Alzheimer’s Disease. Biomedicines, 10(2), 305. https://doi.org/10.3390/biomedicines10020305