
Identification of Targetable Liabilities in the Dynamic Metabolic Profile of EGFR-Mutant Lung Adenocarcinoma: Thinking beyond Genomics for Overcoming EGFR TKI Resistance

 , , ,
, , ,  ,
,  and
and 
Abstract
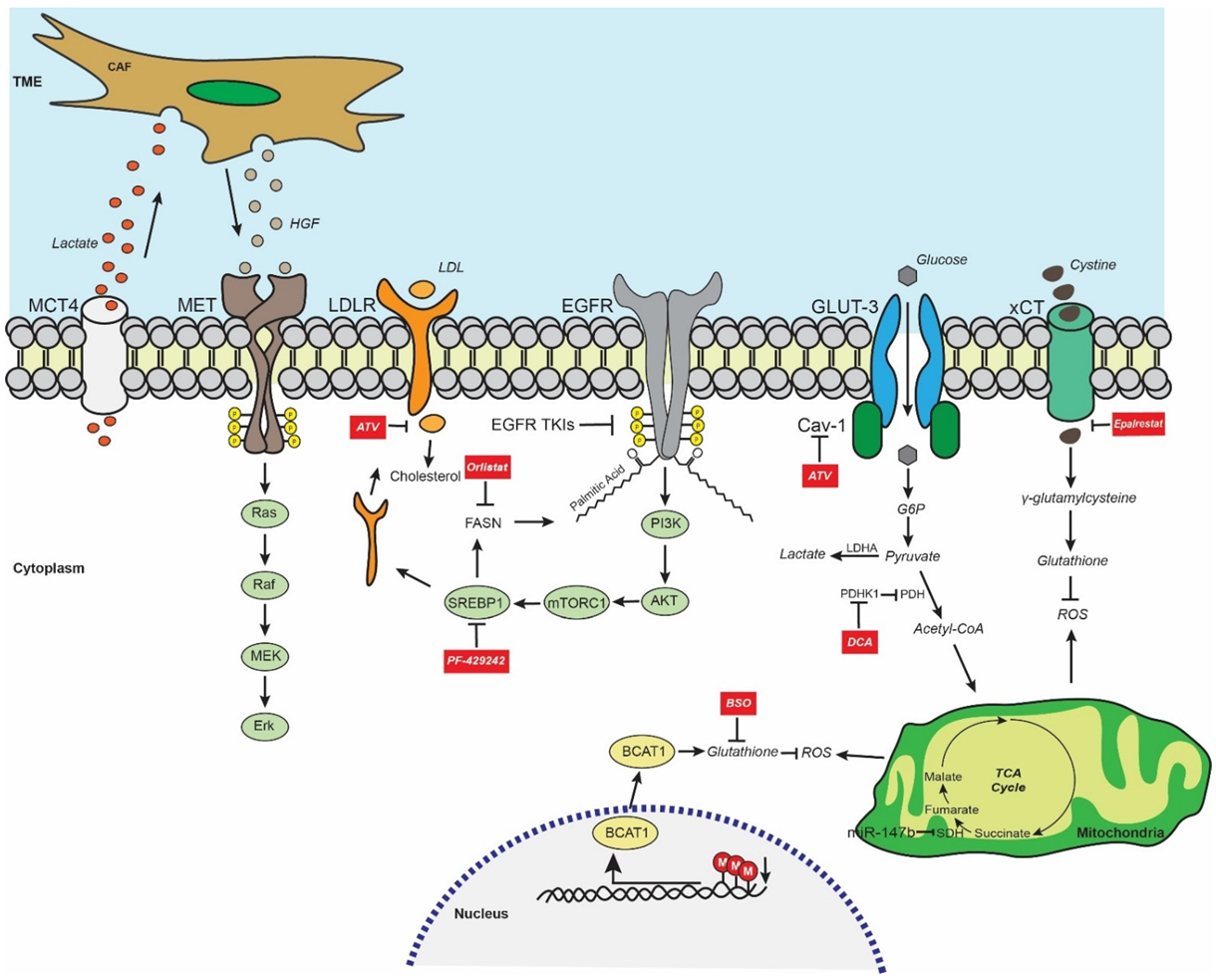
:1. Introduction
2. EGFR-Mutant LUAD Global Metabolic Profile
3. EGFR TKI Resistance and Glycolysis/Lactate Metabolism

{kind=link}
| Compound | Mechanism of Action | Downstream Effect | Reference |
|---|---|---|---|
| Dichloroacetate | PDHK inhibitor | Induction of Acetyl-CoA formation | [26] |
| Atorvastatin | CAV-1/GLUT-3 inhibitor | Inhibition of glucose uptake and cholesterol synthesis | [30] |
| PF-429242 | SREBP1 inhibitor | Inhibition of lipid synthesis | [31] |
| Orlistat | FASN inhibitor | Inhibition of EGFR palmitoylation | [32] |
| Epalrestat | AKR1B1 inhibitor | Inhibition of GSH synthesis | [33] |
| PiperlongumineAuranofin | ROS inducing agent | Induction of oxidative stress | [34] |
| Buthionine sulfoximine | GSH synthesis inhibitor | Inhibition of GSH synthesis | [34] |
| 1,25D | VDR agonist | Inhibition of stemness phenotype | [35] |
| AZ12756122 | FASN inhibitor | Inhibition of stemness phenotype | [36] |
4. EGFR TKI Resistance and Fatty Acid Metabolism
5. EGFR TKI Resistance and Redox Homeostasis
6. EGFR TKI Resistance and Metabolism-Mediated Stemness
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.G.; Tsao, M.S.; Beasley, M.B.; Borczuk, A.C.; Brambilla, E.; Cooper, W.A.; Dacic, S.; Jain, D.; Kerr, K.M.; Lantuejoul, S.; et al. The 2021 WHO Classification of Lung Tumors: Impact of advances since 2015. J. Thorac. Oncol. 2021, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; McCormack, R.; Webster, A.; Milenkova, T. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: A phase-IV, open-label, single-arm study. Br. J. Cancer 2014, 110, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.; John, T.; Okamoto, I.; Yang, J.C.H.; Bulusu, K.; Laus, G.; et al. LBA51Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Schmid, S.; Li, J.J.N.; Leighl, N.B. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer 2020, 147, 123–129. [Google Scholar] [CrossRef]
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation 2021, 2, 100103. [Google Scholar] [CrossRef]
- Wei, N.; Song, Y.; Zhang, F.; Sun, Z.; Zhang, X. Transcriptome Profiling of Acquired Gefitinib Resistant Lung Cancer Cells Reveals Dramatically Changed Transcription Programs and New Treatment Targets. Front. Oncol. 2020, 10, 1424. [Google Scholar] [CrossRef]
- Zhang, X.; Maity, T.K.; Ross, K.E.; Qi, Y.; Cultraro, C.M.; Bahta, M.; Pitts, S.; Keswani, M.; Gao, S.; Nguyen, K.D.P.; et al. Alterations in the Global Proteome and Phosphoproteome in Third Generation EGFR TKI Resistance Reveal Drug Targets to Circumvent Resistance. Cancer Res. 2021, 81, 3051–3066. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyrstad, S.E.; Lotsberg, M.L.; Tan, T.Z.; Pettersen, I.K.N.; Hjellbrekke, S.; Tusubira, D.; Engelsen, A.S.T.; Daubon, T.; Mourier, A.; Thiery, J.P.; et al. Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells. Cancers 2021, 13, 941. [Google Scholar] [CrossRef]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernandez-Perez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C.; Wells, J.M.; Chow, K.H.; Huang, H.; Yuan, M.; Saxena, T.; Melnick, M.A.; Politi, K.; Asara, J.M.; Costa, D.B.; et al. miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat. Metab. 2019, 1, 460–474. [Google Scholar] [CrossRef]
- Ali, A.; Levantini, E.; Fhu, C.W.; Teo, J.T.; Clohessy, J.G.; Goggi, J.L.; Wu, C.S.; Chen, L.; Chin, T.M.; Tenen, D.G. CAV—GLUT3 signaling is important for cellular energy and can be targeted by Atorvastatin in Non-Small Cell Lung Cancer. Theranostics 2019, 9, 6157–6174. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, D.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. Induction of SREBP1 degradation coupled with suppression of SREBP1-mediated lipogenesis impacts the response of EGFR mutant NSCLC cells to osimertinib. Oncogene 2021, 40, 6653–6665. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Levantini, E.; Teo, J.T.; Goggi, J.; Clohessy, J.G.; Wu, C.S.; Chen, L.; Yang, H.; Krishnan, I.; Kocher, O.; et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol. Med. 2018, 10, e8313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.R.; Zhang, Y.F.; Lei, H.M.; Tang, Y.B.; Ma, C.S.; Lv, Q.M.; Wang, S.Y.; Lu, L.M.; Shen, Y.; Chen, H.Z.; et al. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Sci. Transl. Med. 2021, 13, eabg6428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Ren, S.; Sun, D.; Huang, H.Y.; Wang, H.; Jin, Y.; Li, F.; Zheng, C.; Yang, L.; et al. Branched-Chain Amino Acid Metabolic Reprogramming Orchestrates Drug Resistance to EGFR Tyrosine Kinase Inhibitors. Cell Rep. 2019, 28, 512–525.e516. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, Y.; Yan, A.; Wang, M.; Han, Q.; Wang, K.; Wang, J.; Qiao, C.; Pan, Z.; Chen, C.; et al. 1,25-dihydroxyvitamin D3 signaling-induced decreases in IRX4 inhibits NANOG-mediated cancer stem-like properties and gefitinib resistance in NSCLC cells. Cell Death Dis. 2020, 11, 670. [Google Scholar] [CrossRef]
- Polonio, E.; Palomeras, S.; Porta-Balanya, R.; Bosch-Barrera, J.; Vásquez, C.A.; Ciurana, J.; Ruiz-Martínez, S.; Puig, T. 3P AZ12756122, a novel fatty acid synthase (FASN) inhibitor, reduces resistance properties in gefitinib- and osimertinib-resistant EGFR-mutated non-small cell lung cancer models. J. Thorac. Oncol. 2021, 16, S700. [Google Scholar] [CrossRef]
- Han, X.; Luo, R.; Wang, L.; Zhang, L.; Wang, T.; Zhao, Y.; Xiao, S.; Qiao, N.; Xu, C.; Ding, L.; et al. Potential predictive value of serum targeted metabolites and concurrently mutated genes for EGFR-TKI therapeutic efficacy in lung adenocarcinoma patients with EGFR sensitizing mutations. Am. J. Cancer Res. 2020, 10, 4266–4286. [Google Scholar]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Hung, M.S.; Chen, I.C.; Lee, C.P.; Huang, R.J.; Chen, P.C.; Tsai, Y.H.; Yang, Y.H. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS ONE 2017, 12, e0171137. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.A.; Chang, C.C.; Galvin, C.J.; Wang, Y.C.; An, S.Y.; Huang, C.W.; Wang, Y.H.; Hsu, M.H.; Li, Y.J.; Yang, H.C. Statins use and its impact in EGFR-TKIs resistance to prolong the survival of lung cancer patients: A Cancer registry cohort study in Taiwan. Cancer Sci. 2020, 111, 2965–2973. [Google Scholar] [CrossRef]
- Martinez, C.A.; Scafoglio, C. Heterogeneity of Glucose Transport in Lung Cancer. Biomolecules 2020, 10, 868. [Google Scholar] [CrossRef] [PubMed]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Yang, Y.; Peng, P.; Zhan, J.; Wang, Z.; Zhu, Z.; Zhang, Z.; Liu, L.; Fang, W.; Zhang, L. Cholesterol synthesis disruption combined with a molecule-targeted drug is a promising metabolic therapy for EGFR mutant non-small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, L.; Wang, D.; Jiang, S.; Cao, D.; Zhao, Z.; Huang, M.; Jin, J. Lipidomics reveals that sustained SREBP-1-dependent lipogenesis is a key mediator of gefitinib-acquired resistance in EGFR-mutant lung cancer. Cell Death Discov. 2021, 7, 353. [Google Scholar] [CrossRef]
- Kim, L.; Tsao, M.S. Tumour tissue sampling for lung cancer management in the era of personalised therapy: What is good enough for molecular testing? Eur. Respir. J. 2014, 44, 1011–1022. [Google Scholar] [CrossRef]
- Ho, Y.S.; Yip, L.Y.; Basri, N.; Chong, V.S.; Teo, C.C.; Tan, E.; Lim, K.L.; Tan, G.S.; Yang, X.; Yeo, S.Y.; et al. Lipidomic Profiling of Lung Pleural Effusion Identifies Unique Metabotype for EGFR Mutants in Non-Small Cell Lung Cancer. Sci. Rep. 2016, 6, 35110. [Google Scholar] [CrossRef] [Green Version]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Bi, A.; Lan, X.; Xu, J.; Wang, X.; Liu, Y.; Wang, T.; Tang, S.; Zeng, H.; Chen, Z.; et al. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat. Commun. 2019, 10, 2701. [Google Scholar] [CrossRef] [Green Version]
- Thiagarajan, P.S.; Wu, X.; Zhang, W.; Shi, I.; Bagai, R.; Leahy, P.; Feng, Y.; Veigl, M.; Lindner, D.; Danielpour, D.; et al. Transcriptomic-metabolomic reprogramming in EGFR-mutant NSCLC early adaptive drug escape linking TGFbeta2-bioenergetics-mitochondrial priming. Oncotarget 2016, 7, 82013–82027. [Google Scholar] [CrossRef] [Green Version]
- Lei, H.M.; Zhang, K.R.; Wang, C.H.; Wang, Y.; Zhuang, G.L.; Lu, L.M.; Zhang, J.; Shen, Y.; Chen, H.Z.; Zhu, L. Aldehyde dehydrogenase 1A1 confers erlotinib resistance via facilitating the reactive oxygen species-reactive carbonyl species metabolic pathway in lung adenocarcinomas. Theranostics 2019, 9, 7122–7139. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Lv, Q.M.; Zhang, K.R.; Tang, Y.B.; Zhang, Y.F.; Shen, Y.; Lei, H.M.; Zhu, L. NRF2-GPX4/SOD2 axis imparts resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer cells. Acta Pharmacol. Sin. 2021, 42, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Mao, W.; Gao, Y.; Wang, D.; Song, Z.; Chen, Z. Liquid chromatography-mass spectrometry based metabolic characterization of pleural effusion in patients with acquired EGFR-TKI resistance. J. Pharm. Biomed. Anal. 2021, 202, 114147. [Google Scholar] [CrossRef] [PubMed]
- Hashida, S.; Yamamoto, H.; Shien, K.; Miyoshi, Y.; Ohtsuka, T.; Suzawa, K.; Watanabe, M.; Maki, Y.; Soh, J.; Asano, H.; et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015, 106, 1377–1384. [Google Scholar] [CrossRef]
- Ji, M.; Liu, L.; Hou, Y.; Li, B. 1alpha,25Dihydroxyvitamin D3 restrains stem celllike properties of ovarian cancer cells by enhancing vitamin D receptor and suppressing CD44. Oncol. Rep. 2019, 41, 3393–3403. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkountakos, A.; Centonze, G.; Vita, E.; Belluomini, L.; Milella, M.; Bria, E.; Milione, M.; Scarpa, A.; Simbolo, M. Identification of Targetable Liabilities in the Dynamic Metabolic Profile of EGFR-Mutant Lung Adenocarcinoma: Thinking beyond Genomics for Overcoming EGFR TKI Resistance. Biomedicines 2022, 10, 277. https://doi.org/10.3390/biomedicines10020277
Gkountakos A, Centonze G, Vita E, Belluomini L, Milella M, Bria E, Milione M, Scarpa A, Simbolo M. Identification of Targetable Liabilities in the Dynamic Metabolic Profile of EGFR-Mutant Lung Adenocarcinoma: Thinking beyond Genomics for Overcoming EGFR TKI Resistance. Biomedicines. 2022; 10(2):277. https://doi.org/10.3390/biomedicines10020277
Chicago/Turabian StyleGkountakos, Anastasios, Giovanni Centonze, Emanuele Vita, Lorenzo Belluomini, Michele Milella, Emilio Bria, Massimo Milione, Aldo Scarpa, and Michele Simbolo. 2022. "Identification of Targetable Liabilities in the Dynamic Metabolic Profile of EGFR-Mutant Lung Adenocarcinoma: Thinking beyond Genomics for Overcoming EGFR TKI Resistance" Biomedicines 10, no. 2: 277. https://doi.org/10.3390/biomedicines10020277
APA StyleGkountakos, A., Centonze, G., Vita, E., Belluomini, L., Milella, M., Bria, E., Milione, M., Scarpa, A., & Simbolo, M. (2022). Identification of Targetable Liabilities in the Dynamic Metabolic Profile of EGFR-Mutant Lung Adenocarcinoma: Thinking beyond Genomics for Overcoming EGFR TKI Resistance. Biomedicines, 10(2), 277. https://doi.org/10.3390/biomedicines10020277