
Inhibition of SGLT2 Preserves Function and Promotes Proliferation of Human Islets Cells In Vivo in Diabetic Mice

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Human Islets, Islet Transplantation, and Diabetes Induction
2.3. Dapagliflozin Treatment
2.4. Pharmacokinetic Analysis of Dapagliflozin Administered in the Drinking Water
2.5. Bioanalysis of Dapagliflozin
2.6. Intravenous Arginine/Glucose Tolerance Test (IVArgGTT)
2.7. Tissue and Blood Sampling and Biochemical Analysis
2.8. Histology and Immunohistochemistry
2.9. Human Islets Culture and Dapagliflozin Treatment
2.10. Statistical Analysis
3. Results
3.1. SGLT2 Inhibition Maintained Long-Term Glycemic Control in Diabetic Mice
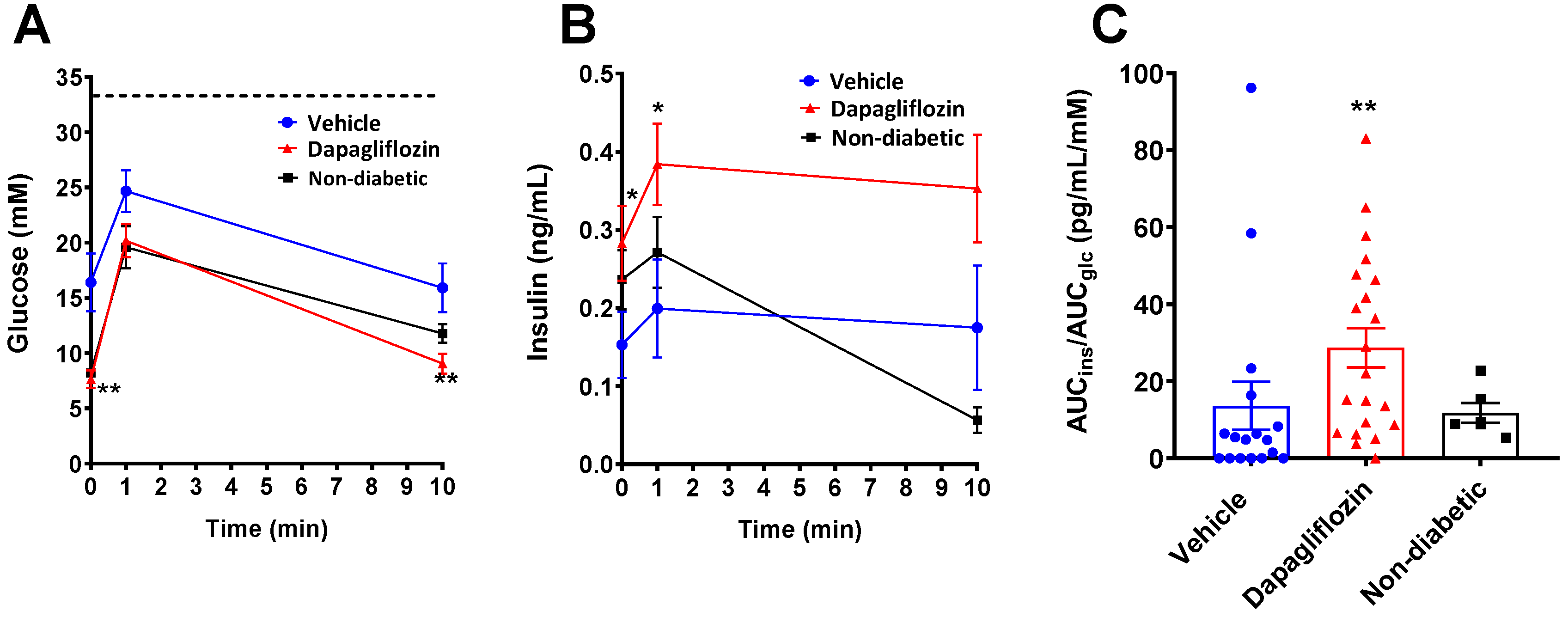
3.2. SGLT2 Inhibition Preserved Human Insulin Levels in Diabetic Mice
3.3. SGLT2 Inhibition Preserved Human Beta Cell Function
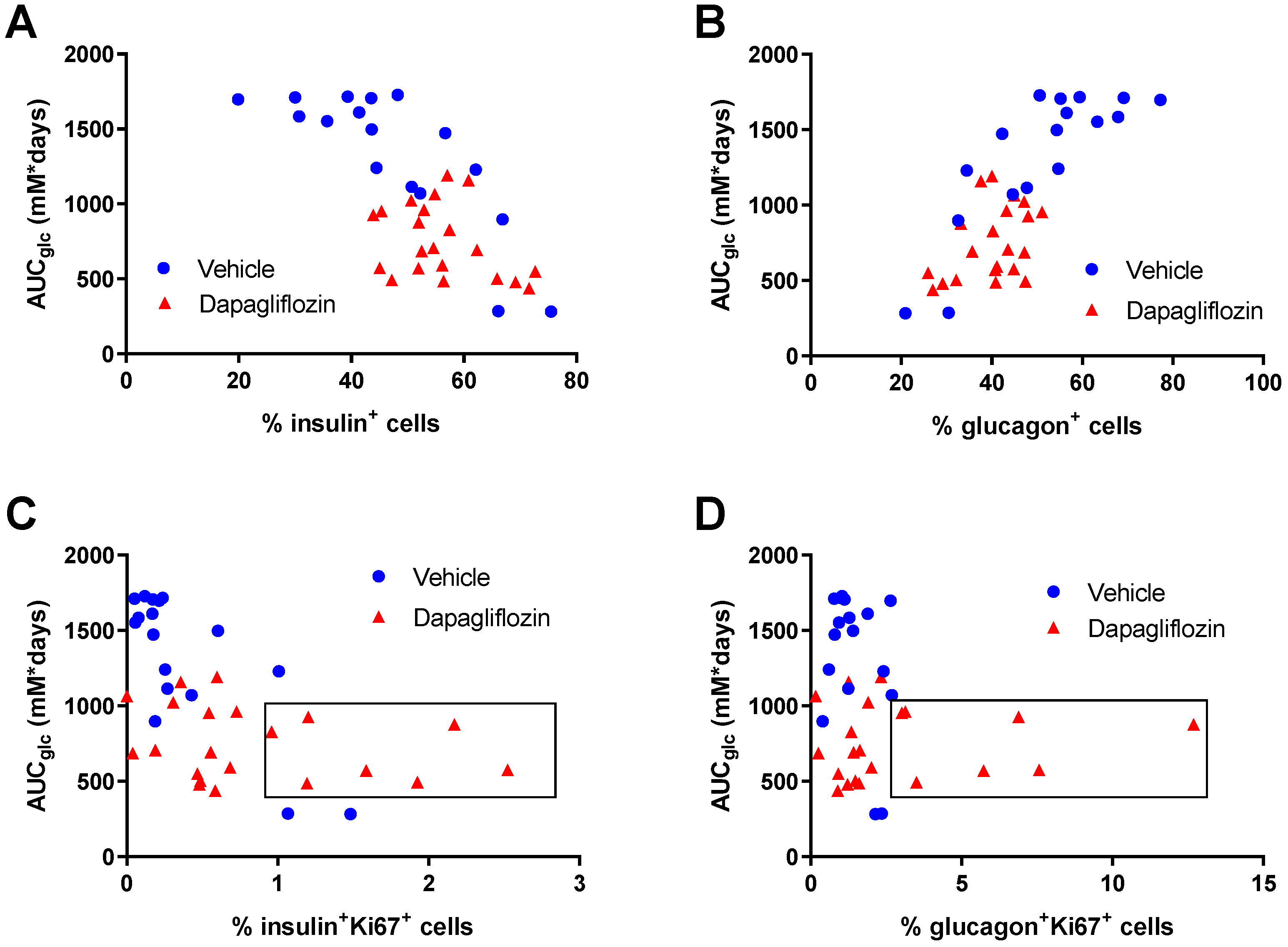
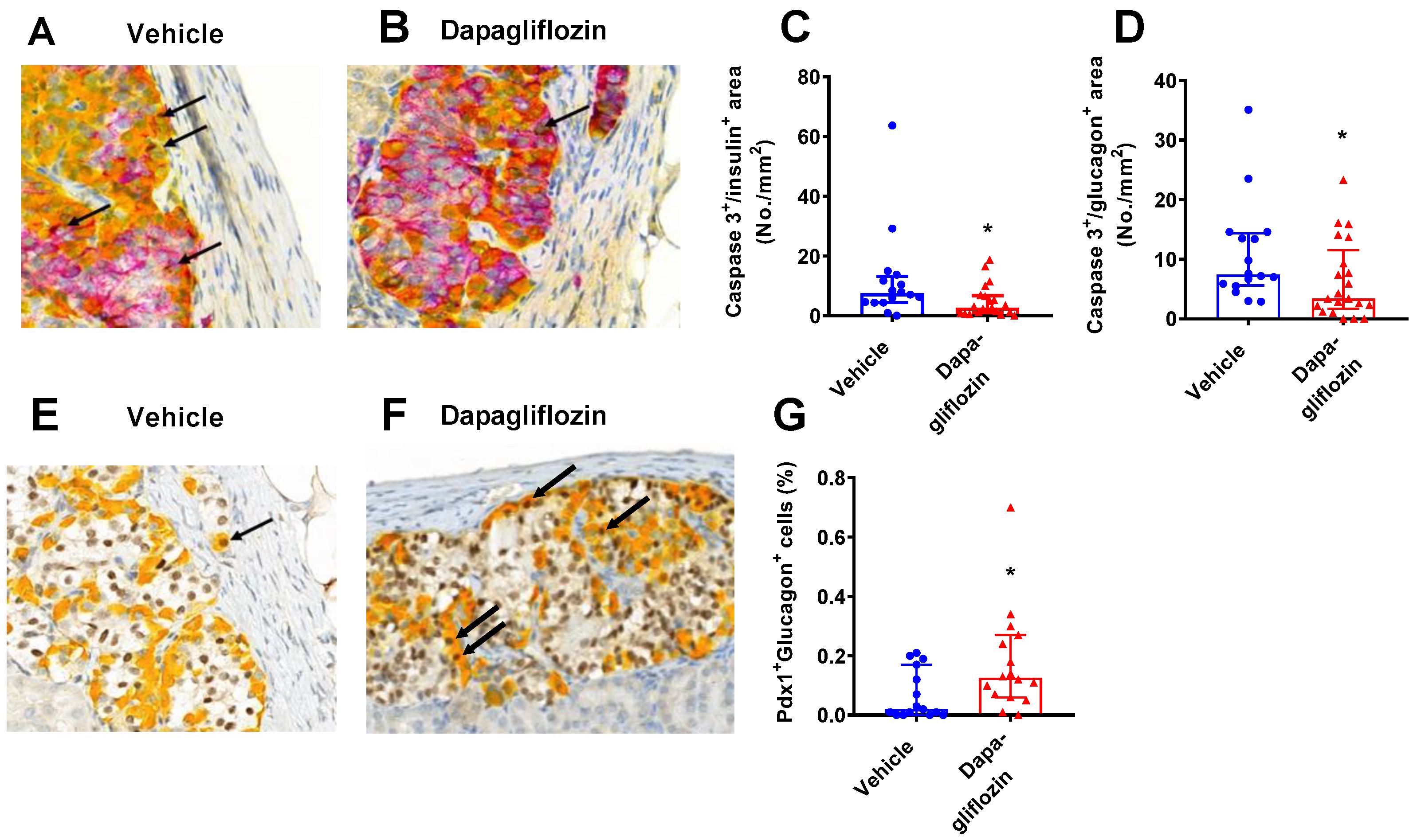
3.4. SGLT2 Inhibition Increased Human Alpha and Beta Cell Proliferation and Reduced Apoptosis
3.5. In Vitro Treatment of Human Islet with Dapagliflozin Had No Effects on Islet Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weir, G.C.; Bonner-Weir, S. Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef]
- Kaneto, H.; Matsuoka, T.A.; Kimura, T.; Obata, A.; Shimoda, M.; Kamei, S.; Mune, T.; Kaku, K. Appropriate therapy for type 2 diabetes mellitus in view of pancreatic beta-cell glucose toxicity: “the earlier, the better”. J. Diabetes 2016, 8, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Merovci, A.; Mari, A.; Solis-Herrera, C.; Xiong, J.; Daniele, G.; Chavez-Velazquez, A.; Tripathy, D.; Urban McCarthy, S.; Abdul-Ghani, M.; DeFronzo, R.A. Dapagliflozin lowers plasma glucose concentration and improves beta-cell function. J. Clin. Endocrinol. Metab. 2015, 100, 1927–1932. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Xu, W.; Shi, L.; Zhang, Q.; Zhu, D.; Hu, Y.; Zhou, Z.; Yan, X.; Tian, H.; et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel-group trial. Lancet 2008, 371, 1753–1760. [Google Scholar] [CrossRef]
- Fonolleda, M.; Murillo, M.; Vazquez, F.; Bel, J.; Vives-Pi, M. Remission Phase in Paediatric Type 1 Diabetes: New Understanding and Emerging Biomarkers. Horm. Res. Paediatr. 2017, 88, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Bonner-Weir, S. Pancreatic beta Cell Regeneration as a Possible Therapy for Diabetes. Cell Metab. 2018, 27, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Ye, L.; Robertson, M.A.; Hesselson, D.; Stainier, D.Y.; Anderson, R.M. Glucagon is essential for alpha cell transdifferentiation and beta cell neogenesis. Development 2015, 142, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, K.C.; Schulz, N.; Karampelias, C.; Charbord, J.; Hilding, A.; Rautio, L.; Bertolino, P.; Ostenson, C.G.; Brismar, K.; et al. IGFBP1 increases beta-cell regeneration by promoting alpha- to beta-cell transdifferentiation. EMBO J. 2016, 35, 2026–2044. [Google Scholar] [CrossRef]
- Chung, C.H.; Hao, E.; Piran, R.; Keinan, E.; Levine, F. Pancreatic beta-cell neogenesis by direct conversion from mature alpha-cells. Stem Cells 2010, 28, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Thorel, F.; Damond, N.; Chera, S.; Wiederkehr, A.; Thorens, B.; Meda, P.; Wollheim, C.B.; Herrera, P.L. Normal glucagon signaling and beta-cell function after near-total alpha-cell ablation in adult mice. Diabetes 2011, 60, 2872–2882. [Google Scholar] [CrossRef] [PubMed]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieira, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013, 9, e1003934. [Google Scholar] [CrossRef] [PubMed]
- Druelle, N.; Vieira, A.; Shabro, A.; Courtney, M.; Mondin, M.; Rekima, S.; Napolitano, T.; Silvano, S.; Navarro-Sanz, S.; Hadzic, B.; et al. Ectopic expression of Pax4 in pancreatic delta cells results in beta-like cell neogenesis. J. Cell Biol. 2017, 216, 4299–4311. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, K.; Chera, S.; van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef]
- Tat, V.; Forest, C.P. The role of SGLT2 inhibitors in managing type 2 diabetes. JAAPA 2018, 31, 35–40. [Google Scholar] [CrossRef]
- Dandona, P.; Mathieu, C.; Phillip, M.; Hansen, L.; Tschope, D.; Thoren, F.; Xu, J.; Langkilde, A.M.; Investigators, D. Efficacy and Safety of Dapagliflozin in Patients with Inadequately Controlled Type 1 Diabetes: The DEPICT-1 52-Week Study. Diabetes Care 2018, 41, 2552–2559. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Dandona, P.; Gillard, P.; Senior, P.; Hasslacher, C.; Araki, E.; Lind, M.; Bain, S.C.; Jabbour, S.; Arya, N.; et al. Efficacy and Safety of Dapagliflozin in Patients with Inadequately Controlled Type 1 Diabetes (the DEPICT-2 Study): 24-Week Results from a Randomized Controlled Trial. Diabetes Care 2018, 41, 1938–1946. [Google Scholar] [CrossRef]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef]
- Kuhre, R.E.; Ghiasi, S.M.; Adriaenssens, A.E.; Albrechtsen, N.J.W.; Andersen, D.B.; Aivazidis, A.; Chen, L.; Mandrup-Poulsen, T.; Orskov, C.; Gribble, F.M.; et al. No direct effect of SGLT2 activity on glucagon secretion. Diabetologia 2019, 62, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Shiraiwa, T.; Matsuoka, T.A.; Katakami, N.; Shimomura, I. Ameliorated pancreatic beta cell dysfunction in type 2 diabetic patients treated with a sodium-glucose cotransporter 2 inhibitor ipragliflozin. Endocr. J. 2015, 62, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, F.R.; Peel, J.E.; Jones, H.B.; Mayers, R.M.; Westgate, L.; Whaley, J.M.; Poucher, S.M. The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes. Metab. 2010, 12, 1004–1012. [Google Scholar] [CrossRef]
- Jurczak, M.J.; Lee, H.Y.; Birkenfeld, A.L.; Jornayvaz, F.R.; Frederick, D.W.; Pongratz, R.L.; Zhao, X.; Moeckel, G.W.; Samuel, V.T.; Whaley, J.M.; et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 2011, 60, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Jelsing, J.; Hansen, C.F.; Hansen, G.; Vrang, N.; Mark, M.; Klein, T.; Mayoux, E. The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves beta-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J. Pharmacol. Exp. Ther. 2014, 350, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.T.; Chen, L.; Li, S.Y.; Mayoux, E.; Leung, P.S. The Effects of Empagliflozin, an SGLT2 Inhibitor, on Pancreatic beta-Cell Mass and Glucose Homeostasis in Type 1 Diabetes. PLoS ONE 2016, 11, e0147391. [Google Scholar]
- Okauchi, S.; Shimoda, M.; Obata, A.; Kimura, T.; Hirukawa, H.; Kohara, K.; Mune, T.; Kaku, K.; Kaneto, H. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic beta-cells in obese type 2 diabetic db/db mice. Biochem. Biophys. Res. Commun. 2016, 470, 772–782. [Google Scholar] [CrossRef]
- Kimura, T.; Obata, A.; Shimoda, M.; Okauchi, S.; Kanda-Kimura, Y.; Nogami, Y.; Moriuchi, S.; Hirukawa, H.; Kohara, K.; Nakanishi, S.; et al. Protective effects of the SGLT2 inhibitor luseogliflozin on pancreatic beta-cells in db/db mice: The earlier and longer, the better. Diabetes Obes. Metab. 2018, 20, 2442–2457. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Diaz, R.; Dando, R.; Jacques-Silva, M.C.; Fachado, A.; Molina, J.; Abdulreda, M.H.; Ricordi, C.; Roper, S.D.; Berggren, P.O.; Caicedo, A. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat. Med. 2011, 17, 888–892. [Google Scholar] [CrossRef]
- Abdulreda, M.H.; Rodriguez-Diaz, R.; Caicedo, A.; Berggren, P.O. Liraglutide Compromises Pancreatic beta Cell Function in a Humanized Mouse Model. Cell Metab. 2016, 23, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Sahraoui, A.; Winzell, M.S.; Gorman, T.; Smith, D.M.; Skrtic, S.; Hoeyem, M.; Abadpour, S.; Johansson, L.; Korsgren, O.; Foss, A.; et al. The effects of exendin-4 treatment on graft failure: An animal study using a novel re-vascularized minimal human islet transplant model. PLoS ONE 2015, 10, e0121204. [Google Scholar] [CrossRef][Green Version]
- Kasichayanula, S.; Liu, X.; Lacreta, F.; Griffen, S.C.; Boulton, D.W. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin. Pharm. 2014, 53, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, M.; Yao, M.; Khanna, A.; Koplowitz, B.; Zhu, M.; Li, W.; Komoroski, B.; Kasichayanula, S.; Discenza, L.; Washburn, W.; et al. In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter type II inhibitor, in animals and humans. Drug Metab. Dispos. 2010, 38, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, S.J.; Vrang, N.; Larsen, L.K.; Larsen, P.J.; Jelsing, J. Stereological assessment of pancreatic beta-cell mass development in male Zucker Diabetic Fatty (ZDF) rats: Correlation with pancreatic beta-cell function. J. Anat. 2010, 217, 624–630. [Google Scholar] [CrossRef]
- Steil, G.M.; Trivedi, N.; Jonas, J.C.; Hasenkamp, W.M.; Sharma, A.; Bonner-Weir, S.; Weir, G.C. Adaptation of beta-cell mass to substrate oversupply: Enhanced function with normal gene expression. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E788–E796. [Google Scholar] [CrossRef] [PubMed]
- Moulle, V.S.; Vivot, K.; Tremblay, C.; Zarrouki, B.; Ghislain, J.; Poitout, V. Glucose and fatty acids synergistically and reversibly promote beta cell proliferation in rats. Diabetologia 2017, 60, 879–888. [Google Scholar] [CrossRef]
- Swisa, A.; Glaser, B.; Dor, Y. Metabolic Stress and Compromised Identity of Pancreatic Beta Cells. Front. Genet. 2017, 8, 21. [Google Scholar] [CrossRef]
- Dai, C.; Hang, Y.; Shostak, A.; Poffenberger, G.; Hart, N.; Prasad, N.; Phillips, N.; Levy, S.E.; Greiner, D.L.; Shultz, L.D.; et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J. Clin. Investig. 2017, 127, 3835–3844. [Google Scholar] [CrossRef] [PubMed]
- Bramswig, N.C.; Everett, L.J.; Schug, J.; Dorrell, C.; Liu, C.; Luo, Y.; Streeter, P.R.; Naji, A.; Grompe, M.; Kaestner, K.H. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J. Clin. Investig. 2013, 123, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Mezza, T.; Sorice, G.P.; Conte, C.; Sun, V.A.; Cefalo, C.M.; Moffa, S.; Pontecorvi, A.; Mari, A.; Kulkarni, R.N.; Giaccari, A. beta-Cell Glucose Sensitivity Is Linked to Insulin/Glucagon Bihormonal Cells in Nondiabetic Humans. J. Clin. Endocrinol. Metab. 2016, 101, 470–475. [Google Scholar] [CrossRef]
- Stewart, A.F.; Hussain, M.A.; Garcia-Ocana, A.; Vasavada, R.C.; Bhushan, A.; Bernal-Mizrachi, E.; Kulkarni, R.N. Human beta-cell proliferation and intracellular signaling: Part 3. Diabetes 2015, 64, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Diabetic (n = 6) | Diabetic Vehicle (n = 21) | Diabetic Dapagliflozin (n = 17) | |
|---|---|---|---|
| Fructosamine (µM) | 220 ± 2 | 278 ± 13 * | 236 ± 8 # |
| β-hydroxybutyrate (µM) | 143 ± 15 | 306 ± 29 * | 300 ± 38 * |
| Liver triglycerides (g/100 g tissue) | 0.97 ± 0.15 | 0.33 ± 0.029 *** | 0.44 ± 0.068 *** |
| Cholesterol (mM) | 2.09 ± 0.04 | 2.28 ± 0.064 | 2.45 ± 0.058 ** |
| Triglycerides (mM) | 1.39 ± 0.18 | 1.69 ± 0.23 | 1.54 ± 0.16 |
| ALT (µkat/L) | 0.44 ± 0.03 | 0.60 ± 0.031 * | 0.54 ± 0.034 |
| Haptoglobin (g/L) | 1.32 ± 0.29 | 0.44 ± 0.07 | 1.01 ± 0.30 |
| Human insulin (ng/mL) | ND | 0.055 ± 0.034 | 0.25 ± 0.072 # |
| Mouse C-peptide (ng/mL) | 0.67 ± 0.1 | 0.05 ± 0.01 *** | 0.06 ± 0.01 *** |
| Glucagon (pM) | 7.5 ± 0.8 | 40.6 ± 3.2 *** | 39.8 ± 2.9 *** |
| Pancreatic insulin (ng/µg protein) | 6.3 ± 0.4 | 0.11 ± 0.02 *** | 0.24 ± 0.0.07 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karlsson, D.; Ahnmark, A.; Sabirsh, A.; Andréasson, A.-C.; Gennemark, P.; Sandinge, A.-S.; Chen, L.; Tyrberg, B.; Lindén, D.; Sörhede Winzell, M. Inhibition of SGLT2 Preserves Function and Promotes Proliferation of Human Islets Cells In Vivo in Diabetic Mice. Biomedicines 2022, 10, 203. https://doi.org/10.3390/biomedicines10020203
Karlsson D, Ahnmark A, Sabirsh A, Andréasson A-C, Gennemark P, Sandinge A-S, Chen L, Tyrberg B, Lindén D, Sörhede Winzell M. Inhibition of SGLT2 Preserves Function and Promotes Proliferation of Human Islets Cells In Vivo in Diabetic Mice. Biomedicines. 2022; 10(2):203. https://doi.org/10.3390/biomedicines10020203
Chicago/Turabian StyleKarlsson, Daniel, Andrea Ahnmark, Alan Sabirsh, Anne-Christine Andréasson, Peter Gennemark, Ann-Sofie Sandinge, Lihua Chen, Björn Tyrberg, Daniel Lindén, and Maria Sörhede Winzell. 2022. "Inhibition of SGLT2 Preserves Function and Promotes Proliferation of Human Islets Cells In Vivo in Diabetic Mice" Biomedicines 10, no. 2: 203. https://doi.org/10.3390/biomedicines10020203
APA StyleKarlsson, D., Ahnmark, A., Sabirsh, A., Andréasson, A.-C., Gennemark, P., Sandinge, A.-S., Chen, L., Tyrberg, B., Lindén, D., & Sörhede Winzell, M. (2022). Inhibition of SGLT2 Preserves Function and Promotes Proliferation of Human Islets Cells In Vivo in Diabetic Mice. Biomedicines, 10(2), 203. https://doi.org/10.3390/biomedicines10020203