
Refining the Clinical Spectrum of the 17p13.3 Microduplication Syndrome: Case-Report of a Familial Small Microduplication
 , and
, and
Abstract
1. Introduction
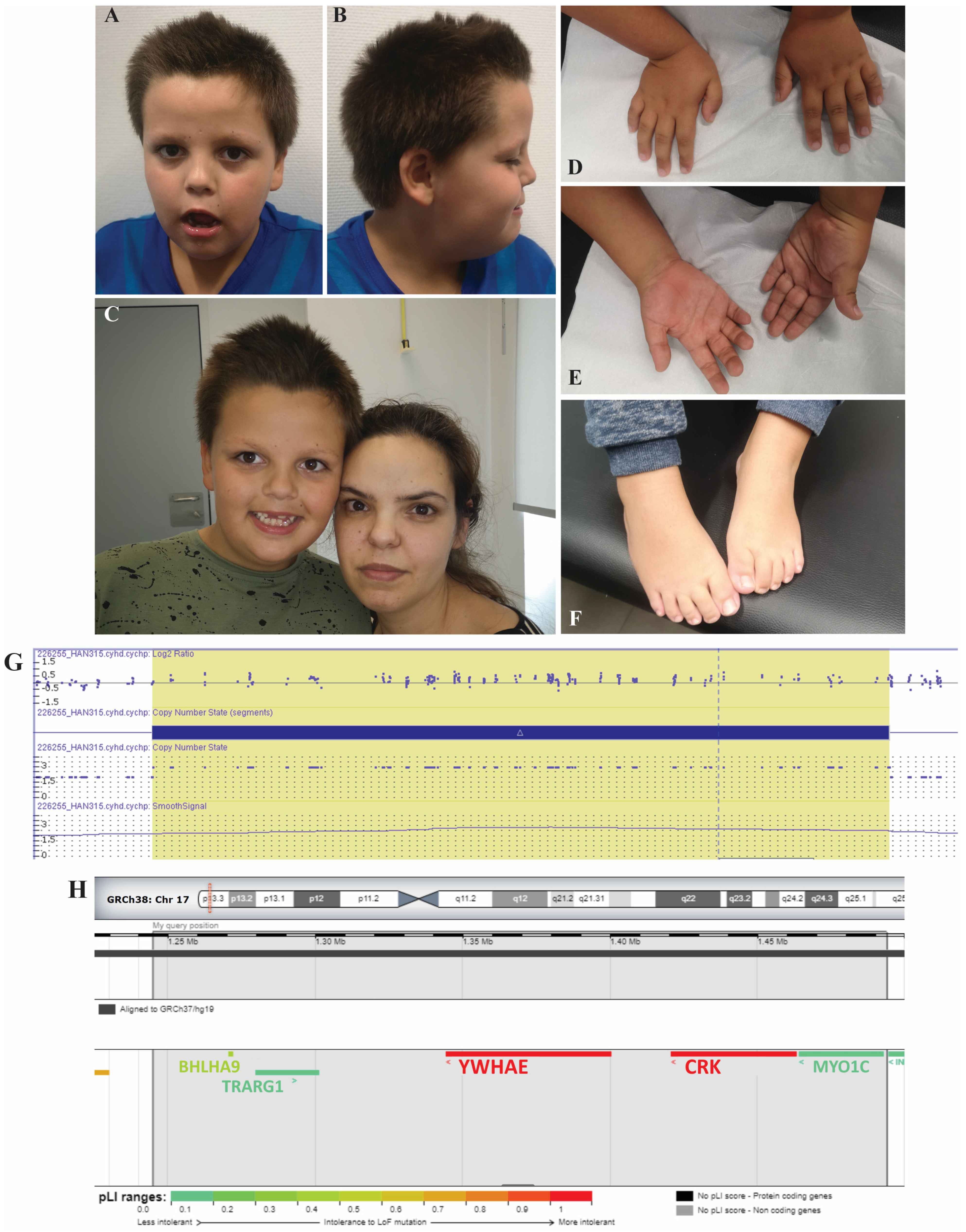
2. Case Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platt, R.N., 2nd; Vandewege, M.W.; Ray, D.A. Mammalian Transposable Elements and Their Impacts on Genome Evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, K.; Sugiura, C.; Takahashi, H.; Ikegami, M.; Takahashi, Y.; Ohno, K.; Matsuo, M.; Saito, K.; Yamamoto, T. Genomic Copy Number Variations at 17p13.3 and Epileptogenesis. Epilepsy Res. 2010, 89, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Blazejewski, S.M.; Bennison, S.A.; Smith, T.H.; Toyo-Oka, K. Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3. Front. Genet. 2018, 9, 80. [Google Scholar] [CrossRef]
- Farra, C.; Abdouni, L.; Hani, A.; Dirani, L.; Hamdar, L.; Souaid, M.; Awwad, J. 17p13.3 Microduplication Syndrome: Further Delineating the Clinical Spectrum. J. Pediatric Genet. 2021, 10, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Bruno, D.L.; Anderlid, B.-M.; Lindstrand, A.; van Ravenswaaij-Arts, C.; Ganesamoorthy, D.; Lundin, J.; Martin, C.L.; Douglas, J.; Nowak, C.; Adam, M.P.; et al. Further Molecular and Clinical Delineation of Co-Locating 17p13.3 Microdeletions and Microduplications That Show Distinctive Phenotypes. J. Med. Genet. 2010, 47, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bennison, S.A.; Robinson, L.; Toyo-Oka, K. Responsible Genes for Neuronal Migration in the Chromosome 17p13.3: Beyond Pafah1b1(Lis1), Crk and Ywhae(14-3-3ε). Brain Sci. 2021, 12, 56. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Hoppman-Chaney, N.; Wain, K.; Seger, P.R.; Superneau, D.W.; Hodge, J.C. Identification of Single Gene Deletions at 15q13.3: Further Evidence That CHRNA7 Causes the 15q13.3 Microdeletion Syndrome Phenotype. Clin. Genet. 2013, 83, 345–351. [Google Scholar] [CrossRef]
- Hannes, F.D.; Sharp, A.J.; Mefford, H.C.; de Ravel, T.; Ruivenkamp, C.A.; Breuning, M.H.; Fryns, J.-P.; Devriendt, K.; Van Buggenhout, G.; Vogels, A.; et al. Recurrent Reciprocal Deletions and Duplications of 16p13.11: The Deletion Is a Risk Factor for MR/MCA While the Duplication May Be a Rare Benign Variant. J. Med. Genet. 2009, 46, 223–232. [Google Scholar] [CrossRef]
- Cornell, B.; Wachi, T.; Zhukarev, V.; Toyo-Oka, K. Regulation of Neuronal Morphogenesis by 14-3-3epsilon (Ywhae) via the Microtubule Binding Protein, Doublecortin. Hum. Mol. Genet. 2016, 25, 4405–4418. [Google Scholar] [CrossRef]
- Kataoka, K.; Matsushima, T.; Ito, Y.; Sato, T.; Yokoyama, S.; Asahara, H. Bhlha9 Regulates Apical Ectodermal Ridge Formation during Limb Development. J. Bone Miner. Metab. 2018, 36, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.; Jourdain, A.-S.; Andrieux, J.; Baujat, G.; Baumann, C.; Beneteau, C.; David, A.; Faivre, L.; Gaillard, D.; Gilbert-Dussardier, B.; et al. Split Hand/Foot Malformation with Long-Bone Deficiency and BHLHA9 Duplication: Report of 13 New Families. Clin. Genet. 2014, 85, 464–469. [Google Scholar] [CrossRef]
- Malik, S.; Percin, F.E.; Bornholdt, D.; Albrecht, B.; Percesepe, A.; Koch, M.C.; Landi, A.; Fritz, B.; Khan, R.; Mumtaz, S.; et al. Mutations Affecting the BHLHA9 DNA-Binding Domain Cause MSSD, Mesoaxial Synostotic Syndactyly with Phalangeal Reduction, Malik-Percin Type. Am. J. Hum. Genet. 2014, 95, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Park, T.-J.; Curran, T. Essential Roles of Crk and CrkL in Fibroblast Structure and Motility. Oncogene 2014, 33, 5121–5132. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.K.; Astbury, C.; Stratakis, C.A.; Hickey, S.E. 17p13.3 Microduplication Including CRK Leads to Overgrowth and Elevated Growth Factors: A Case Report. Eur. J. Med. Genet. 2016, 59, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Deodati, A.; Inzaghi, E.; Germani, D.; Fausti, F.; Cianfarani, S. Crk Haploinsufficiency Is Associated with Intrauterine Growth Retardation and Severe Postnatal Growth Failure. Horm. Res. Paediatr. 2021, 94, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Park, T.-J.; Boyd, K.; Curran, T. Cardiovascular and Craniofacial Defects in Crk-Null Mice. Mol. Cell. Biol. 2006, 26, 6272–6282. [Google Scholar] [CrossRef][Green Version]
- Solanki, A.K.; Biswal, M.R.; Walterhouse, S.; Martin, R.; Kondkar, A.A.; Knölker, H.-J.; Rahman, B.; Arif, E.; Husain, S.; Montezuma, S.R.; et al. Loss of Motor Protein MYO1C Causes Rhodopsin Mislocalization and Results in Impaired Visual Function. Cells 2021, 10, 1322. [Google Scholar] [CrossRef]
- Zadro, C.; Alemanno, M.S.; Bellacchio, E.; Ficarella, R.; Donaudy, F.; Melchionda, S.; Zelante, L.; Rabionet, R.; Hilgert, N.; Estivill, X.; et al. Are MYO1C and MYO1F Associated with Hearing Loss? Biochim. Biophys. Acta 2009, 1792, 27–32. [Google Scholar] [CrossRef][Green Version]
- Fazakerley, D.J.; Naghiloo, S.; Chaudhuri, R.; Koumanov, F.; Burchfield, J.G.; Thomas, K.C.; Krycer, J.R.; Prior, M.J.; Parker, B.L.; Murrow, B.A.; et al. Proteomic Analysis of GLUT4 Storage Vesicles Reveals Tumor Suppressor Candidate 5 (TUSC5) as a Novel Regulator of Insulin Action in Adipocytes. J. Biol. Chem. 2015, 290, 23528–23542. [Google Scholar] [CrossRef]
- Beaton, N.; Rudigier, C.; Moest, H.; Müller, S.; Mrosek, N.; Röder, E.; Rudofsky, G.; Rülicke, T.; Ukropec, J.; Ukropcova, B.; et al. TUSC5 Regulates Insulin-Mediated Adipose Tissue Glucose Uptake by Modulation of GLUT4 Recycling. Mol. Metab. 2015, 4, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lee, W.H.; Sobott, F.; Papagrigoriou, E.; Robinson, C.V.; Grossmann, J.G.; Sundström, M.; Doyle, D.A.; Elkins, J.M. Structural Basis for Protein-Protein Interactions in the 14-3-3 Protein Family. Proc. Natl. Acad. Sci. USA 2006, 103, 17237–17242. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.J.; Rosenfeld, J.A.; Grant, E.; Gripp, K.W.; Anderson, C.; Aylsworth, A.S.; Ben Saad, T.; Chizhikov, V.V.; Dybose, G.; Fagerberg, C.; et al. The Duplication 17p13.3 Phenotype: Analysis of 21 Families Delineates Developmental, Behavioral and Brain Abnormalities, and Rare Variant Phenotypes. Am. J. Med. Genet. A 2013, 161A, 1833–1852. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Bogatan, S.; Watkins, N.; Meschino, W.S.; Stavropoulos, D.J. Disruption of YWHAE Gene at 17p13.3 Causes Learning Disabilities and Brain Abnormalities. Clin. Genet. 2018, 93, 365–367. [Google Scholar] [CrossRef]
- Toyo-oka, K.; Shionoya, A.; Gambello, M.J.; Cardoso, C.; Leventer, R.; Ward, H.L.; Ayala, R.; Tsai, L.-H.; Dobyns, W.; Ledbetter, D.; et al. 14-3-3epsilon Is Important for Neuronal Migration by Binding to NUDEL: A Molecular Explanation for Miller-Dieker Syndrome. Nat. Genet. 2003, 34, 274–285. [Google Scholar] [CrossRef]
- Paththinige, C.S.; Sirisena, N.D.; Escande, F.; Manouvrier, S.; Petit, F.; Dissanayake, V.H.W. Split Hand/Foot Malformation with Long Bone Deficiency Associated with BHLHA9 Gene Duplication: A Case Report and Review of Literature. BMC Med. Genet. 2019, 20, 108. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Protein | Gene Function | Gene Duplication | Monoallelic LoF | Biallelic LoF | LoF in Mice |
|---|---|---|---|---|---|---|
| BHLHA9 | Basic Helix-Loop-Helix Family Member A9 | Transcription factor that regulates limb development [11] | Split hand/foot malformation with long-bone deficiency [12] | No phenotype [13] (pLI = 0.30) | Mesoaxial synostotic syndactyly with phalangeal reduction [13] | Syndactyly in the forelimb bud and poliosis in the hindlimb finger [11] |
| CRK | Adapter Molecule Crk | Tyrosine kinase signaling for cell growth and migration [14] | Overgrowth due to growth factor increase [15] | Intrauterine and postnatal growth failure [16] (pLI = 0.96) | ? | Embryonically lethal [17] |
| MYO1C | Myosin 1C | Actin-binding motor protein that regulates cell trafficking [18] | ? | Sensorineural hearing loss? (unconfirmed) [19] (pLI = 0.00) | ? | Loss of retinal photoreceptors [18] |
| TRARG1 | Trafficking Regulator of GLUT4 | Regulates insulin-stimulated GLUT4 trafficking and insulin sensitivity [20] | ? | ? (pLI = 0.00) | ? | Increased body weight and insulin resistance [21] |
| YWHAE | 14-3-3 Protein Epsilon | Adaptor protein for phosphoprotein signaling [22] | Autism spectrum disorder, developmental delay [23] | Brain abnormalities, learning disabilities, and seizures [24] (pLI = 0.98) | ? | Cortical and hippocampus malformation [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Silva, J.D.; Gonzaga, D.; Barreta, A.; Correia, H.; Fortuna, A.M.; Soares, A.R.; Tkachenko, N. Refining the Clinical Spectrum of the 17p13.3 Microduplication Syndrome: Case-Report of a Familial Small Microduplication. Biomedicines 2022, 10, 3078. https://doi.org/10.3390/biomedicines10123078
Da Silva JD, Gonzaga D, Barreta A, Correia H, Fortuna AM, Soares AR, Tkachenko N. Refining the Clinical Spectrum of the 17p13.3 Microduplication Syndrome: Case-Report of a Familial Small Microduplication. Biomedicines. 2022; 10(12):3078. https://doi.org/10.3390/biomedicines10123078
Chicago/Turabian StyleDa Silva, Jorge Diogo, Diana Gonzaga, Ana Barreta, Hildeberto Correia, Ana Maria Fortuna, Ana Rita Soares, and Nataliya Tkachenko. 2022. "Refining the Clinical Spectrum of the 17p13.3 Microduplication Syndrome: Case-Report of a Familial Small Microduplication" Biomedicines 10, no. 12: 3078. https://doi.org/10.3390/biomedicines10123078
APA StyleDa Silva, J. D., Gonzaga, D., Barreta, A., Correia, H., Fortuna, A. M., Soares, A. R., & Tkachenko, N. (2022). Refining the Clinical Spectrum of the 17p13.3 Microduplication Syndrome: Case-Report of a Familial Small Microduplication. Biomedicines, 10(12), 3078. https://doi.org/10.3390/biomedicines10123078