Abstract
The mucosal immune system, via a dynamic immune network, serves as the first line of defense against exogenous antigens. Mucosal immune system dysregulation is closely associated with the pathogenesis of immunoglobulin A nephropathy (IgAN), as illustrated by IgAN having the clinical feature of gross hematuria, often concurrent with mucosal infections. Notably, previous studies have demonstrated the efficacy of tonsillectomy and found that a targeted-release formulation of budesonide reduced proteinuria in patients with IgAN. However, it remains unclear how exogenous antigens interact with the mucosal immune system to induce or exacerbate IgAN. Thus, in this review, we focus on the dysregulation of mucosal immune response in the pathogenesis of IgAN.
1. Introduction
Immunoglobulin A nephropathy (IgAN) is characterized by the deposition of IgA and complement (C) 3 in the mesangial region. IgAN was first reported by Jean Berger and Hinglais in 1968 and accounts for 30%–50% of patients with primary glomerulonephritis (GN) [1,2]. According to long-term follow-up studies, 20%–40% of untreated patients develop end-stage kidney disease and require renal replacement therapy within 20 years [3,4]. The multi-hit theory was proposed regarding the development of IgAN [5]. Patients with IgAN have genetically increased levels of serum IgA1 that contains galactose-deficient O-glycans in its hinge region (galactose-deficient IgA1; Gd-IgA1) (Hit 1). Anti-glycan autoantibodies that recognize this Gd-IgA1 develop (Hit 2) and form pathogenic immune complexes (ICs) (Hit 3). These pathogenic ICs deposit in the glomeruli and activate mesangial cells, resulting in glomerular injury (Hit 4). However, the main site that produces Gd-IgA1 is still unknown.
A potential association between the mucosal immune system and IgAN has been proposed because the main production site of IgA is the mucosa and a common clinical feature of IgAN is gross hematuria, often concurrent with upper respiratory tract infection [6,7]. In support of this possibility, it was reported that the nephritis in IgAN-onset ddY mice was not exacerbated when these animals were housed in germ-free conditions, whereas, after stimulation with exogenous antigens, these mice exhibited mesangial IgA deposition with renal injury [8]. Furthermore, microbial infections have been found to influence various autoimmune responses and immunity [9,10]. The mucosa is constantly exposed to various exogenous antigens, and the mucosal immune system is important for their elimination. Mucosa-associated lymphoid tissue (MALT) is mainly divided into gut-associated lymphoid tissue (GALT) and nasal-associated lymphoid tissue (NALT), which have different roles and origins [11,12].
In this article, we will first review the current leading hypothesis on the pathogenesis of IgAN, and then we will focus on the mucosal immune response disorder centered on the GALT and NALT in IgAN.
2. Multi-Hit Mechanism of IgAN
IgA1 has an O-glycan in the hinge region between heavy-chain constant region domains 1 and 2. Allen and Hiki et al. reported that the IgA extracted from the renal glomeruli of patients with IgAN was mainly IgA1, in which the O-glycan side chains in the hinge region were highly under-glycosylated [13,14]. Their finding suggests that the reduced galactosylation and sialylation on the IgA1 hinge glycopeptide plays a pivotal role in the glomerular deposition of IgA. This scenario is further supported by the results of experiments conducted using KM55, an anti-Gd-IgA1 monoclonal antibody that we recently established [15]. The human α1 heavy chain contains several O-linked glycan chains, at positions 3–6, attached to the Serine and Threonine residues in the hinge region [16,17]. This glycosylation is performed stepwise by glycosyltransferases present in the Golgi apparatus of plasma cells that secrete IgA1, and diversity is observed in the O-glycan structures of IgA1 molecules [18]. Recently, it was found that Gd-IgA1 is associated with increased α-2,6-sialyltransferase 2 (ST6GalNAc-II) activity and decreased core 1 β1,3-galactosyltransferase (C1β3GalT) activity in patients with IgAN [19]. In addition, a portion of Gd-IgA1 derived from glomerular deposits in patients with IgAN is excreted in the urine. A recent report showed that the levels of urinary Gd-IgA1 were significantly higher in IgAN patients compared with disease controls [20]. Furthermore, levels of urinary Gd-IgA1 were significantly correlated with histopathological severity in IgAN patients, indicating that Gd-IgA1 plays a central role in the pathogenesis of IgAN [21].
In 70%–80% of patients with IgAN, their serum Gd-IgA1 levels are above the 90th percentile of levels in healthy controls [22]. Moreover, first-degree relatives of patients with IgAN exhibit elevated serum Gd-IgA1 levels comparable to patient levels, indicating that this condition has significant heritability [23]; however, because most of these relatives with aberrant IgA1 glycosylation were asymptomatic, it is difficult to conclude that increased Gd-IgA1 levels alone are enough to generate clinical IgAN. Thus, additional genetic or environmental cofactors may be required for the expression and development of IgAN [23,24]. Yanagihara et al. stimulated human mesangial cells, using IgA1 extracted from IgA myeloma patients, and found that only polymeric IgA1 or ICs, but not monomeric IgA1, activated the mesangium [25]. This finding suggests that the formation of ICs triggered by Gd-IgA1 contributes to IgAN exacerbation. However, the mechanisms of how the Gd-IgA1-specific IgG/IgA antibodies are produced and how the autoantibodies against Gd-IgA1 drive the formation of pathogenic ICs remain uncertain.
Tomana et al. reported that the reactivity of serum IgG extracted from patients with IgAN to IgA1 was significantly decreased by the removal of O-glycans from the IgA1 hinge region, indicating that IgG antibodies against nephritic IgA may recognize O-glycans in the hinge region [26]. Furthermore, Suzuki et al. generated Epstein–Barr Virus-immortalized IgG-secreting cells from IgAN patients and found that the secreted IgG formed glycan-dependent ICs with Gd-IgA1 [27]. Recently, Rizk et al. demonstrated the presence of IgG that exhibited specificity for Gd-IgA1 in the deposits in glomeruli of patients with IgAN, including IgG that is not detectable by routine immunofluorescence microscopy [28]. Collectively, these findings further support the possibility that Gd-IgA1-specific IgG autoantibodies have a pivotal role in the pathogenesis of IgAN.
Previous work found that the injection of aberrantly glycosylated IgA from IgAN-onset mice into nude mice induced rapid IgA deposition along the glomerular capillary wall and in the mesangium [29]. Furthermore, we showed that the injection of in vitro-formed human Gd-IgA1–IgG ICs into nude mice resulted in the formation of glomerular immunodeposits and induced pathological changes similar to those associated with human IgAN [30]. In addition, Gd-IgA1-containing ICs induced glomerular endothelial cell activation in nude mice and caused the upregulation of chemokines, pro-inflammatory cytokines, and adhesion molecules, resulting in endothelial injury that alters the endothelial permeability, thus facilitating the entry of ICs into the mesangial region [30]. Together, these findings suggest that, in patients with IgAN, Gd-IgA1-containing ICs are continuously supplied to glomerular regions and deposited in the mesangium, resulting in the activation of mesangial cells and subsequent induction of glomerular injury. The multi-hit mechanism is summarized in Figure 1.
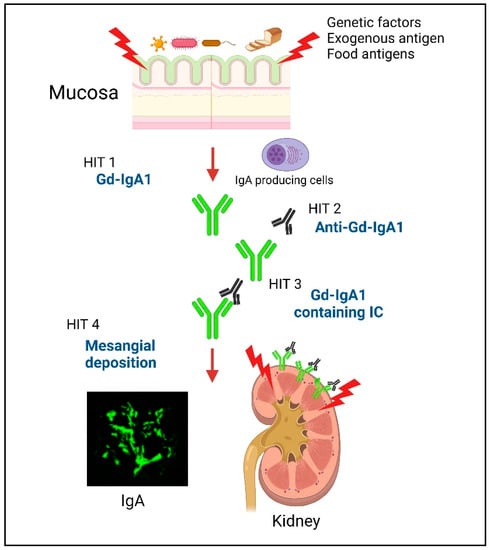
Figure 1.
Schematic depicting the multi-hit model of IgAN development. Hit 1: Patients with IgAN have genetically increased levels of serum IgA1 that contains galactose-deficient O-glycans in its hinge region (galactose-deficient IgA1; Gd-IgA1). In addition, a disorder of the mucosal immune response against exogenous antigens in the MALT, causes an increase in the levels of Gd-IgA1 through Toll-like receptors (TLRs). Hits 2 and 3: Anti-glycan autoantibodies that recognize this Gd-IgA1 develop and form pathogenic immune complexes (ICs). Hit 4: These pathogenic ICs deposit in the glomeruli and activate mesangial cells, resulting in glomerular injury. Created with BioRender.com.
3. The Main Production Site of Gd-IgA1 in IgAN
Gd-IgA1 is recognized as a key molecule in the pathogenesis of IgAN; however, it is still unclear where it is produced. A connection between the mucosa-associated immune system and IgAN is indicated by the following: (1) a main clinical feature of IgAN is gross hematuria after an upper respiratory tract infection, and (2) the renal deposits of IgA consist of polymeric IgA1 and secretory IgA, which are mostly produced by the MALT [31,32,33]. Humans have two IgA isotypes. IgA1 accounts for 80%–85% of total serum IgA, which, at mucosal sites, is matched by local plasma cells (PCs) in the tonsils and nasal mucosa, whereas IgA2 usually predominates in the normal large bowel mucosa [34]. Intestinal bacteria cleave the amino acids in the hinge region of IgA1; in contrast, IgA2 is resistant to such cleavage by intestinal bacteria owing to the lack of 13 amino acids in its hinge region [35]. Consequently, the abundance of IgA2-producing PCs on the intestinal mucosal surface is considered to help efficiently defend against infection [35].
Interestingly, Sakai et al. showed that in patients with IgAN and chronic myeloblastic leukemia, bone marrow transplantation resulted in remission of IgAN as well as leukemia [36]. Furthermore, Imazawa et al. found that the transplantation of bone marrow stem cells from IgAN-onset mice to normal mice resulted in IgA deposition in their glomeruli, and that bone marrow transplantation from normal mice to IgAN-onset mice reduced their renal lesions, proteinuria, and serum IgA levels [37]. Suzuki et al. suggested that IgA-producing bone marrow cells in patients with IgAN may drive the development of IgAN [38]. On the basis of these findings, immunoregulatory impairment in the “mucosa–bone-marrow axis” has been hypothesized to occur in patients with IgAN [39].
The mucosal immune system is mainly divided into the GALT and NALT. However, it is unclear which sites significantly contribute to the production of Gd-IgA1 and the pathogenesis of IgAN.
4. Gut-Associated Lymphoid Tissue (GALT)
Patients with celiac disease or inflammatory bowel disease often develop IgAN, especially in Europe [40,41]. The unexpectedly high co-occurrence of IgAN with celiac disease, which is developed by intolerance to gluten, led to the proposal of a possible association between IgAN and intestinal disorder. One report indicated that a gluten-free diet reduced both gliadin-specific IgA and IgA-containing ICs and also altered the intestinal microbiota [42]. Furthermore, Papista et al. showed, in a double transgenic IgAN model mouse co-expressing human IgA1 and human CD89, that a gluten-free diet prevented the deposition of IgA and renal injury associated with IgAN, whereas the introduction of dietary gluten caused IgAN to develop [43]. These results suggest that digestive antigens, especially gluten, may be involved in the pathogenesis of IgAN [44].
A genome-wide association study recently revealed the role of gut microbial exposure in individuals at genetic risk of developing IgAN and found a relationship between IgAN and genes involved in immunity to gut pathogens [45]. Dietary and climate factors were related with the genetic risk; however, it was most strongly associated with regional pathogen diversity [45]. Most loci found to pose a risk for developing IgAN, such as ITGAM-ITGAX and CARD9, were related to inflammatory bowel disease as well as to maintaining the intestinal barrier and regulating the GALT against gut pathogens [45].
A targeted-release formulation of budesonide (TRF-budesonide) is designed to deliver budesonide to the distal ileum, a major site of mucosal B-cell localization within the GALT. Because this formulation is rapidly metabolized in the liver, which clears most of the drug, less than 10% of the drug is systemically effective, and it is, therefore, expected to have relatively fewer systemic side effects. In a clinical trial (named the NEFIGAN study), the TRF-budesonide group exhibited a significant decrease in the urinary protein creatinine ratio (UPCR), whereas the placebo group exhibited an increase in the UPCR after 9 months [46,47]. On the basis of this encouraging finding, a Phase 3, multicenter, randomized, double-blind, placebo-controlled trial is currently underway to confirm the safety and efficacy of TRF-budesonide. However, steroid-related adverse events were reported more frequently in the TRF-budesonide group; thus, the systemic effects of TRF-budesonide should be considered with caution.
B-cell-activating factor (BAFF) belonging to the tumor necrosis factor (TNF) family is a type 2 transmembrane protein belonging to the TNF superfamily of ligands, along with a proliferation-inducing ligand (APRIL) [48]. BAFF is produced by multiple cell types, such as dendritic cells and monocytes, and regulates the differentiation and survival of B cells by binding to receptors on these cells [49]. BAFF-transgenic (BAFF-Tg) mice exhibit a similar pathology to that of IgAN unless they are kept under germ-free conditions. In the absence of pre-existing commensal bacteria, following the colonization of germ-free BAFF-Tg mice with a limited commensal microbiota containing Lactobacillus murinus, IgA against L. murinus was produced, and IgAN was reconstituted in these colonized BAFF-Tg mice [50]. Therefore, commensal bacteria appear to be necessary for the development of IgAN in BAFF-Tg mice. However, the clinical trial of blisibimod, an anti-BAFF antibody, was discontinued in the phase 3 trial following the results of the phase 2 trial, and the role of BAFF in the pathogenesis of IgAN needs further verification.
IgA produced by intestinal tract plasma cells is secreted as a dimer or polymer linked by J-chain binding. This dimer or polymer then binds to the polymeric immunoglobulin receptor (pIgR), which is expressed on the basement membrane of mucosal epithelial cells and is secreted to the luminal side as secretory IgA [51]. However, inflammation, especially in inflammatory bowel disease, reportedly causes pIgR dysfunction. The secretion of IgA produced by the lamina propria to the mucosal surface is consequently inhibited, resulting in the transfer of this IgA into the blood instead. Patients with inflammatory bowel disease were found to have an elevated serum level of secretory IgA owing to pIgR dysfunction [52,53].
In other words, if the intestinal mucosa is constantly exposed to inflammation, such as in inflammatory bowel disease, antigen-specific IgA or mucosal IgA may migrate into the blood. However, most patients with persistent inflammatory bowel disease or gastrointestinal symptoms do not have IgAN, suggesting that IgA produced in the intestinal tract can migrate into the blood without causing secondary IgAN. Aizawa et al. demonstrated that bone marrow transplantation from IgAN-onset mice can experimentally reconstitute mouse IgAN, not only in normal mice but also in alymphoplasia mice that lack Peyer’s patches, lamina propria and all lymph nodes, doing so in a manner independent of IgA-producing-cell homing to secondary lymphoid tissues or mucosa [54]. This finding indicates that homing to the intestinal mucosa by the cells responsible for the production of aberrantly glycosylated IgA is not necessary.
Generally, specific antibodies against dietary proteins or gut microbiota members are not normally observed in serum, except in special circumstances when members of the gut microbiota enter the bloodstream. The involvement of gluten in IgAN has been demonstrated, particularly in Europe; however, a report from the United States of America indicated that the levels of antibodies to gliadin and TG2 (serological markers specific for celiac disease) did not differ between patients with IgAN and disease controls [55]. Thus, the involvement of celiac disease and gliadin in IgAN is still controversial. However, previous reports showed that from approximately one-third to one-half of patients with IgAN have an intestinal mucosa with sensitivity to gluten without the development of celiac disease [56,57]. In addition, anti-gliadin IgA antibodies were found to be correlated with high levels of ICs, and 71% of patients with IgAN showed a decrease in proteinuria and/or hematuria after adopting a gluten-free diet [58]. These findings together suggest that subclinical inflammation of the intestinal mucosa in response to antigens such as gluten may be involved in the pathogenesis of IgAN. Thus, it is necessary to elucidate the detailed mechanism by which antigen-specific IgA produced in the intestinal tract moves into the blood.
5. Nasal-Associated Lymphoid Tissue (NALT)
NALT is an immune-inducing tissue that consists mainly of the Waldeyer pharyngeal ring, composed of adenoids and palatine tonsils in humans. Regarding IgA subclasses, NALT-derived plasma cells produce significantly more IgA1 than IgA2, with a IgA1:IgA2 ratio of 9:1, whereas GALT-derived plasma cells produce IgA with a subtype ratio of 1:1 [34]. The effects of tonsillectomy (described in detail below) and the specific involvement of IgA1, rather than IgA2, in causing nephritis suggest that the main pathophysiology for IgAN is the homing of NALT-induced B cells to the bone marrow, centering on the tonsils.
To date, many reports have shown the efficacy of tonsillectomy in treating IgAN [59,60,61,62], and one nationwide multicenter cohort study in Japan found that, among patients with IgAN, undergoing a tonsillectomy was associated with a lower risk of renal outcomes [63]. In addition to these reports from Japan, a meta-analysis also provides credible evidence to support the use of tonsillectomy for patients with IgAN, especially those under long-term treatment [64], and the efficacy of tonsillectomy was also demonstrated in 98 Caucasian patients with IgAN in a Hungarian report [65].
Notably, the tonsils of patients with IgAN contain a significantly elevated proportion of IgA-positive cells and cells producing IgA polymers with J-chains [66,67]. It was found by mass spectrometry that the O-glycan structure of IgA1 produced by tonsillar lymphocytes was galactose-deficient in the hinge region [68]. Furthermore, the levels of gene expression of β1,3-galactosyltransferase (β3GalT), the polypeptide N-acetylgalactosaminyl-transferase 2, C1β3GalT-specific molecular chaperone, and Cosmc were significantly lower in CD19-positive B cells from the tonsils of patients with IgAN than in those from the tonsils of control volunteers [69]. These lower gene expression levels are related to the similar abnormal expression of glycosylation enzymes in IgA1-producing cell lines derived from patients with IgAN [19]. It has also been reported that, in patients with IgAN, the T-cell area in the tonsils is significantly expanded and the ratio of IgA/IgG-producing cells is high compared with that in patients with chronic tonsillitis. Furthermore, tonsillectomy performed at 1 year after kidney transplantation significantly decreased the level of serum Gd-IgA1 and the recurrence rate of histological IgAN after kidney transplantation [70]. On the bases of these findings, serum Gd-IgA1 derived from the tonsillar B cells likely plays an important role in the pathogenesis of IgAN.
There have been reports of the detection of specific exogenous antigens from pathogens such as methicillin-resistant Staphylococcus aureus and Haemophilus parainfluenzae in the glomeruli of patients with IgAN [71,72]. Consequently, it has been debated for many years whether specific antigens and IgA form ICs in the mucous membranes, blood, or kidneys, and if the IgA-containing ICs induce nephritis [73]. However, the reported antigen detection had low reproducibility in many cases; thus, it is not clear whether there is an exogenous-specific antigen involved in the pathogenesis of IgAN. Recently, the involvement of Toll-like receptors (TLRs), which are a group of central innate immune system receptors that recognize the common antigen structures of bacteria and viruses and work for host defense, has been suggested for IgAN. Activation of the innate immune system may increase the production of nephritic IgA, independently of specific antigens. Our research team established a grouped ddY (gddY) mouse model that develops IgA in the early period by selectively crossing ddY mice [74,75]. A genetic analysis revealed a strong correlation supporting the involvement of the gene for a TLR-signaling molecule (MyD88) in the development of IgAN. In addition, we confirmed that the expression of TLR9, which recognizes unmethylated microbial DNA as a ligand, is enhanced in the spleen of IgAN-onset mice [75]. Furthermore, nasal stimulation of these mice with the TLR9 ligand CpG-oligodeoxynucleotide (CpG-ODN) exacerbated renal injury and increased the levels of serum and mesangial IgA [76]. In human studies, we found that two SNPs in the TLR9 gene (rs352139 and rs352140) may be associated with IgAN histologic severity [76]. Additionally, tonsillar TLR9 and TLR9 SNP expression levels correlated with the efficacy of tonsillectomy with steroid pulse therapy in patients with IgAN [77]. Furthermore, the group in which serum Gd-IgA1 levels decreased after tonsillectomy alone exhibited significantly higher levels of tonsillar TLR9 expression and increased improvements in hematuria immediately after tonsillectomy compared with the group in which Gd-IgA1 levels only decreased after the addition of steroid pulse therapy after tonsillectomy. The results of these studies indicate that the Gd-IgA1-producing cells are present in palatine tonsils [78]. Recently, a genome-wide association study revealed that a TNFSF13 (APRIL) variant is involved in IgAN susceptibility [79]. TNFSF13 encodes a member of the TNF ligand family that is important to the IgA class-switch recombination and the development and survival of B cells [80,81]. Encouragingly, the use of an anti-APRIL antibody in IgAN model mice suppressed the levels of IgA and IgG and decreased the level urinary protein, and clinical trials assessing this antibody treatment are currently underway [82,83].
Muto et al. found that tonsillar germinal centers (GCs) of patients with IgAN included abnormal APRIL-producing cells, contributing to a significant upregulation of overall APRIL expression in the tonsils, in contrast with the GCs of control patients with tonsillitis. Furthermore, the aberrant APRIL expression in tonsillar GCs was positively associated with proteinuria, and those with an overexpression of aberrant APRIL in their tonsillar GCs were more likely to respond beneficially to tonsillectomy with a decrease in their levels of serum Gd-IgA1 [84]. Notably, the stimulation of tonsillar mononuclear cells with CpG-ODN increased the levels of BAFF and interferon-γ production by these cells in patients with IgAN, and tonsil cell BAFF expression was also elevated in patients with IgAN as compared that in patients with chronic tonsillitis [85]. Thus, IgAN may also develop owing to the overexpression of BAFF molecules on tonsil cells. Increased serum APRIL and BAFF levels in patients with IgAN and their correlation with prognosis suggest that the activation of APRIL/BAFF, mainly in the tonsils, contributes to the development and progression of IgAN [50,86].
6. The Difference between the NALT and GALT
Interestingly, despite being structurally and functionally very similar, Peyer’s patches and the NALT have completely different origin and developmental processes. Peyer’s patches begin to form during the embryonic stage of development and are completed at birth, whereas NALT formation begins postnatally [11,87]. In general, Peyer’s patch-targeted immunization induces a protective immunity that is broadly effective against antigens in the GALT, whereas NALT-targeted immunization efficiently generates antigen-specific immunity, mainly in the respiratory tract [12].
Plasmablasts derived from a mucosal immune response can be detected in the blood [88]. Interestingly, a recent report showed that gut-derived IgA+ plasmablast and/or plasma cells play an important role in dampening neuroinflammation in mice with experimental autoimmune encephalomyelitis [89]. Previously, basic homing of IgA-producing B cells has been reported [90]. The homing of B cells to the mucosa and peripheral tissues is fundamentally dependent on a unique combination of adhesion molecules and chemokine receptors; these expression patterns differ between IgA-producing B cells stimulated in the NALT and those stimulated in the GALT [90]. Over the course of the IgA plasmablast differentiation in the intestinal lymphoid tissue, CC Chemokine Receptor (CCR) 9 and α4β7 upregulation directs their homing to locations in the small intestine expressing C-C motif chemokine ligand (CCL) 25 and mucosal-addressing cell adhesion molecule (MAdCAM)-1 [90]. Thus, in general, plasmablasts induced by the GALT have difficulty migrating to the systemic immune system owing to its unique combination of adhesion molecules and chemokine receptor expression.
The NALT also induces the expression of α4β1, which interacts with vascular cell adhesion molecule (VCAM)-1, and the molecules l-selectin (CD62L) and CCR7, resulting in systemic homing [91]. This probably reflects that the NALT acts as a “cross-road” between mucosal and systemic immunity [92]. The basic dichotomy of B-cells homing is summarized in Figure 2.
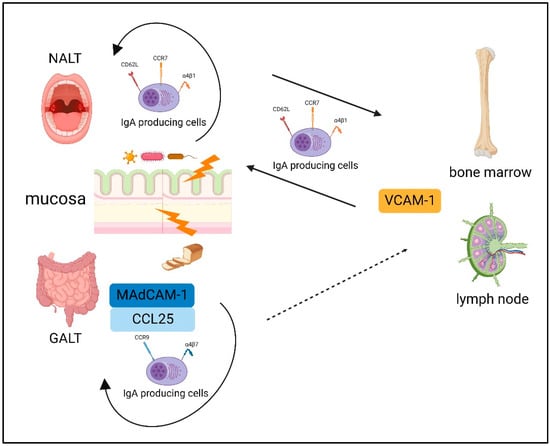
Figure 2.
The basic dichotomy of B-cells homing. The expression patterns differ between IgA-producing B cells stimulated in the NALT and those stimulated in the GALT. The GALT induced CC Chemokine Receptor (CCR) 9 and α4β7 upregulation, which directs their homing to locations in the small intestine expressing mucosal-addressing cell adhesion molecule (MAdCAM)-1 and C-C motif chemokine ligand (CCL) 25. NALT induces the expression of α4β1, which interacts with vascular cell adhesion molecule (VCAM)-1, and the molecules l-selectin (CD62L) and CCR7, resulting in systemic homing. Created with BioRender.com.
A recent report revealed that patients with IgAN showed increased levels of Gd-IgA1+ λ+ B cells expressing CCR10 and CCR9 [93]. However, it is unclear how NALT- and GALT-mediated integrin and chemokine receptors interactions in IgA-producing cells are involved in the pathogenesis of IgAN. Thus, further basic and clinical studies are required.
A recent survey demonstrated obvious differences between nations in their populations’ frequencies of intestinal complications, such as celiac disease and intestinal bowel disease, which were more frequent in European patients with IgAN [94]. In contrast, the frequencies of gross hematuria related with upper respiratory tract infections are similar according to the same survey, thus highlighting the pathogenic role of the NALT in patients with IgAN [94].
There are many studies, mainly from Japan, that have reported a beneficial effect of tonsillectomy on IgAN; however, some European studies have not found favorable results for this IgAN treatment [95,96,97]. Currently, the KDIGO guidelines suggest that tonsillectomy should not be performed as a treatment for IgAN, at least in Caucasian patients [98]. However, a favorable effect of tonsillectomy for treating IgAN was confirmed by a recent meta-analysis of studies that were mainly conducted in Asia. [99]. The studies in European populations that concluded that tonsillectomy may not be effective have some limitations. First, the number of patients who received a tonsillectomy after renal biopsy was limited to only 17 patients in the VALIGA study [95], and Piccoli et al. analyzed only 15 patients with IgAN who received tonsillectomy [96]. Although tonsillectomy is more effective in the early stages of IgAN, the German study on this topic investigated only the IgAN patients with progressed kidney disease (serum creatinine level was over 2 mg/dL) [97]. However, the results of a Hungarian study conducted on 98 Caucasian patients with IgAN supported the effectiveness of tonsillectomy [65]. Given these limitations and conflicting reports, we cannot conclude that tonsillectomy is ineffective for IgAN in Europeans, and the effectiveness of tonsillectomy should be stated with caution. To definitively determine the efficacy of tonsillectomy for IgAN, further basic and clinical studies are needed.
A recent study, conducted using biopsy samples from patients with IgAN, revealed that there are CD19-positive B cells that co-localize with IgA in the kidney [100]. Additionally, Currie et al. identified an elevated prevalence of genus Neisseria carriage in the tonsils and increased presence of serum anti-Neisseria IgA in patients with IgAN [101]. Moreover, after a nasal infection with Neisseria in BAFF-Tg mice, the serum levels of anti-Neisseria IgA were significantly increased, and anti-Neisseria IgA-secreting cells were found in the kidneys. This finding indicates that IgA-producing cells induced by exogenous antigen exposure in the respiratory tract can move to the kidney in the pathogenesis of IgAN [101]. Further studies are needed to determine the role of the specific IgA-producing cells localized in the kidney and the mechanisms that are responsible for promoting the migration of these cells from the airway to the kidneys.
7. Conclusions
According to a recent systematic review, IgAN has an incidence of at least 2.5 per 100,000 adults globally [102]. However, its prevalence varies geographically, and the prevalence of IgAN is much higher in East Asia than in North America or Europe [102,103]. There are many reports, mainly in Japan, regarding the beneficial effect of tonsillectomy on IgAN; however, some European studies have not found favorable results supporting the use of tonsillectomy for treating IgAN. Furthermore, in Europe IgAN is often associated with intestinal bowel disease and celiac disease, whereas this is rarely the case in Japan.
IgAN is the most frequent biopsy-proven primary glomerular nephritis, and is simply diagnosed by the deposition of IgA in mesangial region. Given that the epidemiologic heterogeneity of this disease was shown to depend on race and sex, there is a possibility that the molecular mechanisms responsible for its pathogenesis differ between Europe and Asia. Further basic and clinical studies are needed to clarify the mechanisms of IgAN pathogenesis.
Author Contributions
Conceptualization, T.K. and H.S.; writing—original draft preparation, T.K. and H.S.; writing—review and editing, Y.M., Y.N., Y.F., M.N., M.L., R.K., R.A., K.Y., M.M. and Y.S.; visualization, T.K. and Y.N.; funding acquisition, T.K., H.S. and Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by JSPS KAKENHI Grant Number JP22K16226.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We thank Katie Oakley, from Edanz (https://jp.edanz.com/ac) accessed on 13 September for editing a draft of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Berger, J.; Hinglais, N. Intercapillary deposits of IgA-IgG. J. Urol. Nephrol. 1968, 74, 694–695. [Google Scholar]
- Strippoli, G.F.; Maione, A.; Schena, F.P.; Tognoni, G.; Craig, J.C. IgA nephropathy: A disease in search of a large-scale clinical trial to reliably inform practice. Am. J. Kidney Dis. 2009, 53, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Feehally, J. IgA nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G.; Yan, Y.; Scolari, F.; Schena, F.P.; Frasca, G.M.; Ghiggeri, G.M.; Cooper, K.; Amoroso, A.; Viola, B.F.; Battini, G.; et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nat. Genet. 2000, 26, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef]
- Tomino, Y.; Sakai, H. Exacerbating factors in patients with IgA nephropathy. Semin. Nephrol. 1987, 7, 315–317. [Google Scholar]
- Xie, Y.; Chen, X.; Nishi, S.; Narita, I.; Gejyo, F. Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy. Kidney Int. 2004, 65, 1135–1144. [Google Scholar] [CrossRef]
- Kano, T.; Suzuki, H.; Makita, Y.; Fukao, Y.; Suzuki, Y. Nasal-associated lymphoid tissue is the major induction site for nephritogenic IgA in murine IgA nephropathy. Kidney Int. 2021, 100, 364–376. [Google Scholar] [CrossRef]
- Hricik, D.E.; Chung-Park, M.; Sedor, J.R. Glomerulonephritis. N. Engl. J. Med. 1998, 339, 888–899. [Google Scholar] [CrossRef]
- Pawar, R.D.; Patole, P.S.; Wornle, M.; Anders, H.J. Microbial nucleic acids pay a Toll in kidney disease. Am. J. Physiol. Ren. Physiol. 2006, 291, F509–F516. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, H.; Fukuyama, S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Yuki, Y.; Kiyono, H. New generation of mucosal adjuvants for the induction of protective immunity. Rev. Med. Virol. 2003, 13, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Bailey, E.M.; Brenchley, P.E.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Hiki, Y.; Odani, H.; Takahashi, M.; Yasuda, Y.; Nishimoto, A.; Iwase, H.; Shinzato, T.; Kobayashi, Y.; Maeda, K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001, 59, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, J.; Suzuki, Y.; Suzuki, H.; Hiura, N.; Yanagawa, H.; Makita, Y.; Kaneko, E.; Tomino, Y. Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol. Dial. Transplant. 2015, 30, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, J.; Kornfeld, S. Structure of the Carbohydrate Units of IgA1 Immunoglobulin. J. Biol. Chem. 1974, 249, 7270–7281. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Julian, B.A.; Tomana, M.; Mesteck, J. Progress in molecular and genetic studies of IgA nephropathy. J. Clin. Immunol. 2001, 21, 310–327. [Google Scholar] [CrossRef]
- Piller, V.; Piller, F.; Fukuda, M. Biosynthesis of truncated O-glycans in the T cell line Jurkat. Localization of O-glycan initiation. J. Biol. Chem. 1990, 265, 9264–9271. [Google Scholar] [CrossRef]
- Suzuki, H.; Moldoveanu, Z.; Hall, S.; Brown, R.; Vu, H.L.; Novak, L.; Julian, B.A.; Tomana, M.; Wyatt, R.J.; Edberg, J.C.; et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Investig. 2008, 118, 629–639. [Google Scholar] [CrossRef]
- Suzuki, H.; Allegri, L.; Suzuki, Y.; Hall, S.; Moldoveanu, Z.; Wyatt, R.J.; Novak, J.; Julian, B.A. Galactose-Deficient IgA1 as a Candidate Urinary Polypeptide Marker of IgA Nephropathy? Dis. Markers 2016, 2016, 7806438. [Google Scholar] [CrossRef]
- Fukao, Y.; Suzuki, H.; Kim, J.S.; Jeong, K.H.; Makita, Y.; Kano, T.; Nihei, Y.; Nakayama, M.; Lee, M.; Kato, R.; et al. Galactose-Deficient IgA1 as a Candidate Urinary Marker of IgA Nephropathy. J. Clin. Med. 2022, 11, 3173. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, Z.; Wyatt, R.J.; Lee, J.Y.; Tomana, M.; Julian, B.A.; Mestecky, J.; Mestecky, J.; Huang, W.-Q.; Anreddy, S.R.; Hall, S.; et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007, 71, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Matsuzaki, K.; Suzuki, H.; Okazaki, K.; Yanagawa, H.; Ieiri, N.; Sato, M.; Sato, T.; Taguma, T.; Matsuoka, J.; et al. Serum levels of galactose-deficient immunoglobulin (Ig) A1 and related immune complex are associated with disease activity of IgA nephropathy. Clin. Exp. Nephrol. 2014, 18, 770–777. [Google Scholar] [CrossRef]
- Yanagihara, T.; Brown, R.; Hall, S.; Moldoveanu, Z.; Goepfert, A.; Tomana, M.; Julian, B.A.; Mestecky, J.; Novak, J. In vitro-generated immune complexes containing galactose-deficient IgA1 stimulate proliferation of mesangial cells. Results Immunol. 2012, 2, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Tomana, M.; Novak, J.; Julian, B.A.; Matousovic, K.; Konecny, K.; Mestecky, J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81. [Google Scholar] [CrossRef]
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef]
- Rizk, D.V.; Saha, M.K.; Hall, S.; Novak, L.; Brown, R.; Huang, Z.Q.; Fatima, H.; Julian, B.A.; Novak, J. Glomerular Immunodeposits of Patients with IgA Nephropathy Are Enriched for IgG Autoantibodies Specific for Galactose-Deficient IgA1. J. Am. Soc. Nephrol. 2019, 30, 2017–2026. [Google Scholar] [CrossRef]
- Yamaji, K.; Suzuki, Y.; Suzuki, H.; Satake, K.; Horikoshi, S.; Novak, J.; Tomino, Y. The kinetics of glomerular deposition of nephritogenic IgA. PLoS ONE 2014, 9, e113005. [Google Scholar] [CrossRef]
- Makita, Y.; Suzuki, H.; Nakano, D.; Yanagawa, H.; Kano, T.; Novak, J.; Nishiyama, A.; Suzuki, Y. Glomerular deposition of galactose-deficient IgA1-containing immune complexes via glomerular endothelial cell injuries. Nephrol. Dial. Transplant. 2022, 37, 1629–1636. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Julian, B.A. IgA nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Oortwijn, B.D.; Rastaldi, M.P.; Roos, A.; Mattinzoli, D.; Daha, M.R.; van Kooten, C. Demonstration of secretory IgA in kidneys of patients with IgA nephropathy. Nephrol. Dial. Transplant. 2007, 22, 3191–3195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Xu, L.X.; Liu, G.; Zhao, M.H.; Wang, H.Y. The level of serum secretory IgA of patients with IgA nephropathy is elevated and associated with pathological phenotypes. Nephrol. Dial. Transplant. 2008, 23, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Johansen, F.E. Mucosal B cells: Phenotypic characteristics, transcriptional regulation, and homing properties. Immunol. Rev. 2005, 206, 32–63. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Baekkevold, E.S.; Farstad, I.N.; Jahnsen, F.L.; Johansen, F.E.; Nilsen, E.M.; Yamanaka, T. Regional specialization in the mucosal immune system: What happens in the microcompartments? Immunol. Today 1999, 20, 141–151. [Google Scholar] [CrossRef]
- Sakai, O. IgA nephropathy: Current concept and feature trends. Nephrology 1997, 3, 2–3. [Google Scholar]
- Imasawa, T.; Utsunomiya, Y. Stem cells in renal biology: Bone marrow transplantation for the treatment of IgA nephropathy. Exp. Nephrol. 2002, 10, 51–58. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Aizawa, M.; Yamanaka, T.; Kihara, M.; Pang, H.; Horikoshi, S.; Tomino, Y. Th1 polarization in murine IgA nephropathy directed by bone marrow-derived cells. Kidney Int. 2007, 72, 319–327. [Google Scholar] [CrossRef]
- Suzuki, Y.; Tomino, Y. The mucosa-bone-marrow axis in IgA nephropathy. Contrib. Nephrol. 2007, 157, 70–79. [Google Scholar]
- Helin, H.; Mustonen, J.; Reunala, T.; Pasternack, A. IgA nephropathy associated with celiac disease and dermatitis herpetiformis. Arch. Pathol. Lab. Med. 1983, 107, 324–327. [Google Scholar]
- Collin, P.; Syrjanen, J.; Partanen, J.; Pasternack, A.; Kaukinen, K.; Mustonen, J. Celiac disease and HLA DQ in patients with IgA nephropathy. Am. J. Gastroenterol. 2002, 97, 2572–2576. [Google Scholar] [CrossRef]
- Coppo, R.; Basolo, B.; Rollino, C.; Roccatello, D.; Martina, G.; Amore, A.; Piccoli, G. Dietary gluten and primary IgA nephropathy. N. Engl. J. Med. 1986, 315, 1167–1168. [Google Scholar] [PubMed]
- Papista, C.; Lechner, S.; Ben Mkaddem, S.; LeStang, M.B.; Abbad, L.; Bex-Coudrat, J.; Pillebout, E.; Chemouny, J.M.; Jablonski, M.; Flamant, M.; et al. Gluten exacerbates IgA nephropathy in humanized mice through gliadin-CD89 interaction. Kidney Int. 2015, 88, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Emancipator, S.N.; Gallo, G.R.; Lamm, M.E. Experimental IgA nephropathy induced by oral immunization. J. Exp. Med. 1983, 157, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef]
- Coppo, R.; Mariat, C. Systemic corticosteroids and mucosal-associated lymphoid tissue-targeted therapy in immunoglobulin A nephropathy: Insight from the NEFIGAN study. Nephrol. Dial. Transplant. 2020, 35, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Hahne, M.; Kataoka, T.; Schroter, M.; Hofmann, K.; Irmler, M.; Bodmer, J.L.; Schneider, P.; Bornand, T.; Holler, N.; French, L.R.; et al. APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J. Exp. Med. 1998, 188, 1185–1190. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef]
- McCarthy, D.D.; Kujawa, J.; Wilson, C.; Papandile, A.; Poreci, U.; Porfilio, E.A.; Ward, L.; Lawson, M.A.E.; Macpherson, A.J.; McCoy, K.D.; et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J. Clin. Investig. 2011, 121, 3991–4002. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Transport models for secretory IgA and secretory IgM. Clin. Exp. Immunol. 1981, 44, 221–232. [Google Scholar] [PubMed]
- Wang, J.; Anders, R.A.; Wu, Q.; Peng, D.; Cho, J.H.; Sun, Y.; Karaliukas, R.; Kang, H.-S.; Turner, J.R.; Fu, Y.-X. Dysregulated LIGHT expression on T cells mediates intestinal inflammation and contributes to IgA nephropathy. J. Clin. Investig. 2004, 113, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Arsenescu, R.; Bruno, M.E.; Rogier, E.W.; Stefka, A.T.; McMahan, A.E.; Wright, T.B.; Nasser, M.S.; Villiers, W.J.S.; Kaetzel, C.S. Signature biomarkers in Crohn’s disease: Toward a molecular classification. Mucosal. Immunol. 2008, 1, 399–411. [Google Scholar] [CrossRef]
- Aizawa, M.; Suzuki, Y.; Suzuki, H.; Pang, H.; Kihara, M.; Nakata, J.; Yamaji, K.; Horikoshi, S.; Tomino, Y. Uncoupling of glomerular IgA deposition and disease progression in alymphoplasia mice with IgA nephropathy. PLoS ONE 2014, 9, e95365. [Google Scholar] [CrossRef]
- Moeller, S.; Canetta, P.A.; Taylor, A.K.; Arguelles-Grande, C.; Snyder, H.; Green, P.H.; Kiryluk, K.; Alaedini, A. Lack of serologic evidence to link IgA nephropathy with celiac disease or immune reactivity to gluten. PLoS ONE 2014, 9, e94677. [Google Scholar] [CrossRef] [PubMed]
- McKnight, A.J.; O’Donoghue, D.; Peter Maxwell, A. Annotated chromosome maps for renal disease. Hum. Mutat. 2009, 30, 314–320. [Google Scholar] [CrossRef]
- Kloster Smerud, H.; Fellstrom, B.; Hallgren, R.; Osagie, S.; Venge, P.; Kristjansson, G. Gastrointestinal sensitivity to soy and milk proteins in patients with IgA nephropathy. Clin. Nephrol. 2010, 74, 364–371. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Roccatello, D. Dietary antigens and primary immunoglobulin A nephropathy. J. Am. Soc. Nephrol. 1992, 2 (Suppl. 10), S173–S180. [Google Scholar] [CrossRef]
- Komatsu, H.; Fujimoto, S.; Hara, S.; Sato, Y.; Yamada, K.; Kitamura, K. Effect of tonsillectomy plus steroid pulse therapy on clinical remission of IgA nephropathy: A controlled study. Clin. J. Am. Soc. Nephrol. 2008, 3, 1301–1307. [Google Scholar] [CrossRef]
- Hirano, K.; Amano, H.; Kawamura, T.; Watanabe, K.; Koike, K.; Shimizu, A.; Endo, S.; Tsunoi, N.; Okonogi, H.; Niyazaki, Y.; et al. Tonsillectomy reduces recurrence of IgA nephropathy in mesangial hypercellularity type categorized by the Oxford classification. Clin. Exp. Nephrol. 2016, 20, 425–432. [Google Scholar] [CrossRef][Green Version]
- Yang, D.; He, L.; Peng, X.; Liu, H.; Peng, Y.; Yuan, S.; Liu, Y.; Chen, X.; Liu, F.; Liu, C. The efficacy of tonsillectomy on clinical remission and relapse in patients with IgA nephropathy: A randomized controlled trial. Ren. Fail. 2016, 38, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, Z.; Wang, Q.; Yu, Y.; Zeng, C.; Chen, H.; Liu, Z.H.; Li, L.S. Long-term efficacy of tonsillectomy in Chinese patients with IgA nephropathy. Am. J. Nephrol. 2007, 27, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association Between Tonsillectomy and Outcomes in Patients with Immunoglobulin A Nephropathy. JAMA Netw. Open 2019, 2, e194772. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Liu, D.; Duan, G.; Liu, Z. Long-term efficacy of tonsillectomy as a treatment in patients with IgA nephropathy: A meta-analysis. Int. Urol. Nephrol. 2017, 49, 103–112. [Google Scholar] [CrossRef]
- Kovacs, T.; Vas, T.; Kovesdy, C.P.; Degrell, P.; Nagy, G.; Rekasi, Z.; Wittmann, I.; Nagy, J. Effect of tonsillectomy and its timing on renal outcomes in Caucasian IgA nephropathy patients. Int. Urol. Nephrol. 2014, 46, 2175–2182. [Google Scholar] [CrossRef]
- Nagy, J.; Brandtzaeg, P. Tonsillar distribution of IgA and IgG immunocytes and production of IgA subclasses and J chain in tonsillitis vary with the presence or absence of IgA nephropathy. Scand. J. Immunol. 1988, 27, 393–399. [Google Scholar] [CrossRef]
- Bene, M.C.; Hurault De Ligny, B.; Kessler, M.; Faure, G.C. Confirmation of tonsillar anomalies in IgA nephropathy: A multicenter study. Nephron 1991, 58, 425–428. [Google Scholar] [CrossRef]
- Horie, A.; Hiki, Y.; Odani, H.; Yasuda, Y.; Takahashi, M.; Kato, M.; Iwase, H.; Kobayashi, Y.; Nakashima, I.; Maeda, K. IgA1 molecules produced by tonsillar lymphocytes are under-O-glycosylated in IgA nephropathy. Am. J. Kidney Dis. 2003, 42, 486–496. [Google Scholar] [CrossRef]
- Inoue, T.; Sugiyama, H.; Hiki, Y.; Takiue, K.; Morinaga, H.; Kitagawa, M.; Maeshima, Y.; Fukushima, K.; Nishizaki, K.; Akagi, H.; et al. Differential expression of glycogenes in tonsillar B lymphocytes in association with proteinuria and renal dysfunction in IgA nephropathy. Clin. Immunol. 2010, 136, 447–455. [Google Scholar] [CrossRef]
- Kawabe, M.; Yamamoto, I.; Yamakawa, T.; Katsumata, H.; Isaka, N.; Katsuma, A.; Nakada, Y.; Kobayashi, A.; Koike, K.; Ueda, H.; et al. Association Between Galactose-Deficient IgA1 Derived from the Tonsils and Recurrence of IgA Nephropathy in Patients Who Underwent Kidney Transplantation. Front. Immunol. 2020, 11, 2068. [Google Scholar] [CrossRef]
- Suzuki, S.; Nakatomi, Y.; Sato, H.; Tsukada, H.; Arakawa, M. Haemophilus parainfluenzae antigen and antibody in renal biopsy samples and serum of patients with IgA nephropathy. Lancet 1994, 343, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Shimizu, Y.; Hagiwara, M.; Yoh, K.; Hirayama, K.; Yamagata, K.; Ohba, S.; Nagata, M.; Koyama, A. Post-MRSA infection glomerulonephritis with marked Staphylococcus aureus cell envelope antigen deposition in glomeruli. J. Nephrol. 2006, 19, 215–219. [Google Scholar] [PubMed]
- Coppo, R.; Amore, A.; Peruzzi, L.; Vergano, L.; Camilla, R. Innate immunity and IgA nephropathy. J. Nephrol. 2010, 23, 626–632. [Google Scholar] [PubMed]
- Okazaki, K.; Suzuki, Y.; Otsuji, M.; Suzuki, H.; Kihara, M.; Kajiyama, T.; Hashimoto, A.; Nishimura, H.; Brown, R.; Hall, S.; et al. Development of a model of early-onset IgA nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Suzuki, Y.; Yamanaka, T.; Hirose, S.; Nishimura, H.; Toei, J.; Horikoshi, S.; Tomino, Y. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J. Am. Soc. Nephrol. 2005, 16, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Suzuki, Y.; Narita, I.; Aizawa, M.; Kihara, M.; Yamanaka, T.; Kanou, T.; Tsukaguchi, H.; Novak, J.; Horikoshi, S.; et al. Toll-like receptor 9 affects severity of IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Sato, D.; Suzuki, Y.; Kano, T.; Suzuki, H.; Matsuoka, J.; Yokoi, H.; Horikoshi, S.; Ikeda, K.; Tomino, Y. Tonsillar TLR9 expression and efficacy of tonsillectomy with steroid pulse therapy in IgA nephropathy patients. Nephrol. Dial. Transplant. 2012, 27, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Nakata, J.; Suzuki, Y.; Suzuki, H.; Sato, D.; Kano, T.; Yanagawa, H.; Matsuzaki, K.; Horikoshi, S.; Novak, J.; Tomino, Y. Changes in nephritogenic serum galactose-deficient IgA1 in IgA nephropathy following tonsillectomy and steroid therapy. PLoS ONE 2014, 9, e89707. [Google Scholar] [CrossRef]
- Yu, X.Q.; Li, M.; Zhang, H.; Low, H.Q.; Wei, X.; Wang, J.Q.; Sun, L.D.; Sim, K.S.; Li, Y.; Foo, J.N.; et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 44, 178–182. [Google Scholar] [CrossRef]
- Stein, J.V.; López-Fraga, M.; Elustondo, F.A.; Carvalho-Pinto, C.E.; Rodríguez, D.; Gómez-Caro, R.; Jong, J.; Martinez-A, C.; Medema, J.P.; Hahne, M. APRIL modulates B and T cell immunity. J. Clin. Investig. 2002, 109, 1587–1598. [Google Scholar] [CrossRef]
- He, B.; Xu, W.; Santini, P.A.; Polydorides, A.D.; Chiu, A.; Estrella, J.; Shan, M.; Chadburn, A.; Villanacci, V.; Plebani, A.; et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 2007, 26, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Alvarez, M.; Suzuki, H.; Hirose, S.; Izui, S.; Tomino, Y.; Huard, B.; Suzuki, Y. Pathogenic Role of a Proliferation-Inducing Ligand (APRIL) in Murine IgA Nephropathy. PLoS ONE 2015, 10, e0137044. [Google Scholar] [CrossRef] [PubMed]
- Myette, J.R.; Kano, T.; Suzuki, H.; Sloan, S.E.; Szretter, K.J.; Ramakrishnan, B.; Adari, H.; Deotale, K.D.; Engler, F.; Shriver, Z.; et al. A Proliferation Inducing Ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney Int. 2019, 96, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Muto, M.; Manfroi, B.; Suzuki, H.; Joh, K.; Nagai, M.; Wakai, S.; Righini, C.; Maiguma, M.; Izui, S.; Tomino, Y.; et al. Toll-Like Receptor 9 Stimulation Induces Aberrant Expression of a Proliferation-Inducing Ligand by Tonsillar Germinal Center B Cells in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1227–1238. [Google Scholar] [CrossRef]
- Goto, T.; Bandoh, N.; Yoshizaki, T.; Nozawa, H.; Takahara, M.; Ueda, S.; Hayashi, T.; Harabuchi, Y. Increase in B-cell-activation factor (BAFF) and IFN-gamma productions by tonsillar mononuclear cells stimulated with deoxycytidyl-deoxyguanosine oligodeoxynucleotides (CpG-ODN) in patients with IgA nephropathy. Clin. Immunol. 2008, 126, 260–269. [Google Scholar] [CrossRef]
- Li, W.; Peng, X.; Liu, Y.; Liu, H.; Liu, F.; He, L.; Liu, Y.; Zhang, F.; Guo, C.; Chen, G.; et al. TLR9 and BAFF: Their expression in patients with IgA nephropathy. Mol. Med. Rep. 2014, 10, 1469–1474. [Google Scholar] [CrossRef]
- Yoshida, H.; Honda, K.; Shinkura, R.; Adachi, S.; Nishikawa, S.; Maki, K.; Ikuta, K.; Nishikawa, S. IL-7 receptor alpha+ CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int. Immunol. 1999, 11, 643–655. [Google Scholar] [CrossRef]
- Mei, H.E.; Yoshida, T.; Sime, W.; Hiepe, F.; Thiele, K.; Manz, R.A.; Radbruch, A.; Dorner, T. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood 2009, 113, 2461–2469. [Google Scholar] [CrossRef]
- Rojas, O.L.; Probstel, A.K.; Porfilio, E.A.; Wang, A.A.; Charabati, M.; Sun, T.; Lee, D.S.W.; Galicia, G.; Ramaglia, V.; Ward, L.A.; et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell 2019, 176, 610–624 e18. [Google Scholar] [CrossRef]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal. Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Secretory IgA: Designed for Anti-Microbial Defense. Front. Immunol. 2013, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Potential of nasopharynx-associated lymphoid tissue for vaccine responses in the airways. Am. J. Respir. Crit. Care Med. 2011, 183, 1595–1604. [Google Scholar] [CrossRef]
- Zachova, K.; Jemelkova, J.; Kosztyu, P.; Ohyama, Y.; Takahashi, K.; Zadrazil, J.; Orsag, J.; Matousovic, K.; Galuszkova, D.; Petejova, N.; et al. Galactose-Deficient IgA1 B cells in the Circulation of IgA Nephropathy Patients Carry Preferentially Lambda Light Chains and Mucosal Homing Receptors. J. Am. Soc. Nephrol. 2022, 33, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Monteiro, R.C.; Coppo, R.; Suzuki, H. The Phenotypic Difference of IgA Nephropathy and its Race/Gender-dependent Molecular Mechanisms. Kidney360 2021, 2, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J.; Coppo, R.; Troyanov, S.; Bellur, S.S.; Cattran, D.; Cook, T.; Roberts, I.S.D.; Verhave, J.C.; Camilla, R.; Vergano, L.; et al. Tonsillectomy in a European Cohort of 1,147 Patients with IgA Nephropathy. Nephron 2016, 132, 15–24. [Google Scholar] [CrossRef]
- Piccoli, A.; Codognotto, M.; Tabbi, M.G.; Favaro, E.; Rossi, B. Influence of tonsillectomy on the progression of mesangioproliferative glomerulonephritis. Nephrol. Dial. Transplant. 2010, 25, 2583–2589. [Google Scholar] [CrossRef]
- Rasche, F.M.; Schwarz, A.; Keller, F. Tonsillectomy does not prevent a progressive course in IgA nephropathy. Clin. Nephrol. 1999, 51, 147–152. [Google Scholar]
- Kidney Disease: Improving Global Outcomes Glomerular Diseases Work G. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [CrossRef]
- Liu, L.L.; Wang, L.N.; Jiang, Y.; Yao, L.; Dong, L.P.; Li, Z.L.; Li, X.L. Tonsillectomy for IgA nephropathy: A meta-analysis. Am. J. Kidney Dis. 2015, 65, 80–87. [Google Scholar] [CrossRef]
- Taylor, S.; Pieri, K.; Nanni, P.; Tica, J.; Barratt, J.; Didangelos, A. Phosphatidylethanolamine binding protein-4 (PEBP4) is increased in IgA nephropathy and is associated with IgA-positive B-cells in affected kidneys. J. Autoimmun. 2019, 105, 102309. [Google Scholar] [CrossRef]
- Currie, E.G.; Coburn, B.; Porfilio, E.A.; Lam, P.; Rojas, O.L.; Novak, J.; Yang, S.; Chowdhury, R.B.; Ward, L.A.; Wang, P.W.; et al. Immunoglobulin A nephropathy is characterized by anticommensal humoral immune responses. JCI Insight 2022, 7, e141289. [Google Scholar] [CrossRef] [PubMed]
- McGrogan, A.; Franssen, C.F.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Schena, F.P.; Nistor, I. Epidemiology of IgA Nephropathy: A Global Perspective. Semin. Nephrol. 2018, 38, 435–442. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).