

How Well Do Rodent Models of Parkinson’s Disease Recapitulate Early Non-Motor Phenotypes? A Systematic Review
 , , and
, , and
Abstract
1. Introduction
2. Methods
2.1. Definitions and Eligibility Criteria
2.2. Search Terms and Strategy
2.3. Data Extraction
2.4. Risk of Bias Assessment
3. Results
3.1. Study Characteristics
3.2. Quality Assessment of Studies
3.3. How Well Do Genetic Rodent Models of PD Recaptiulate MDS Criteria Phenotypes?
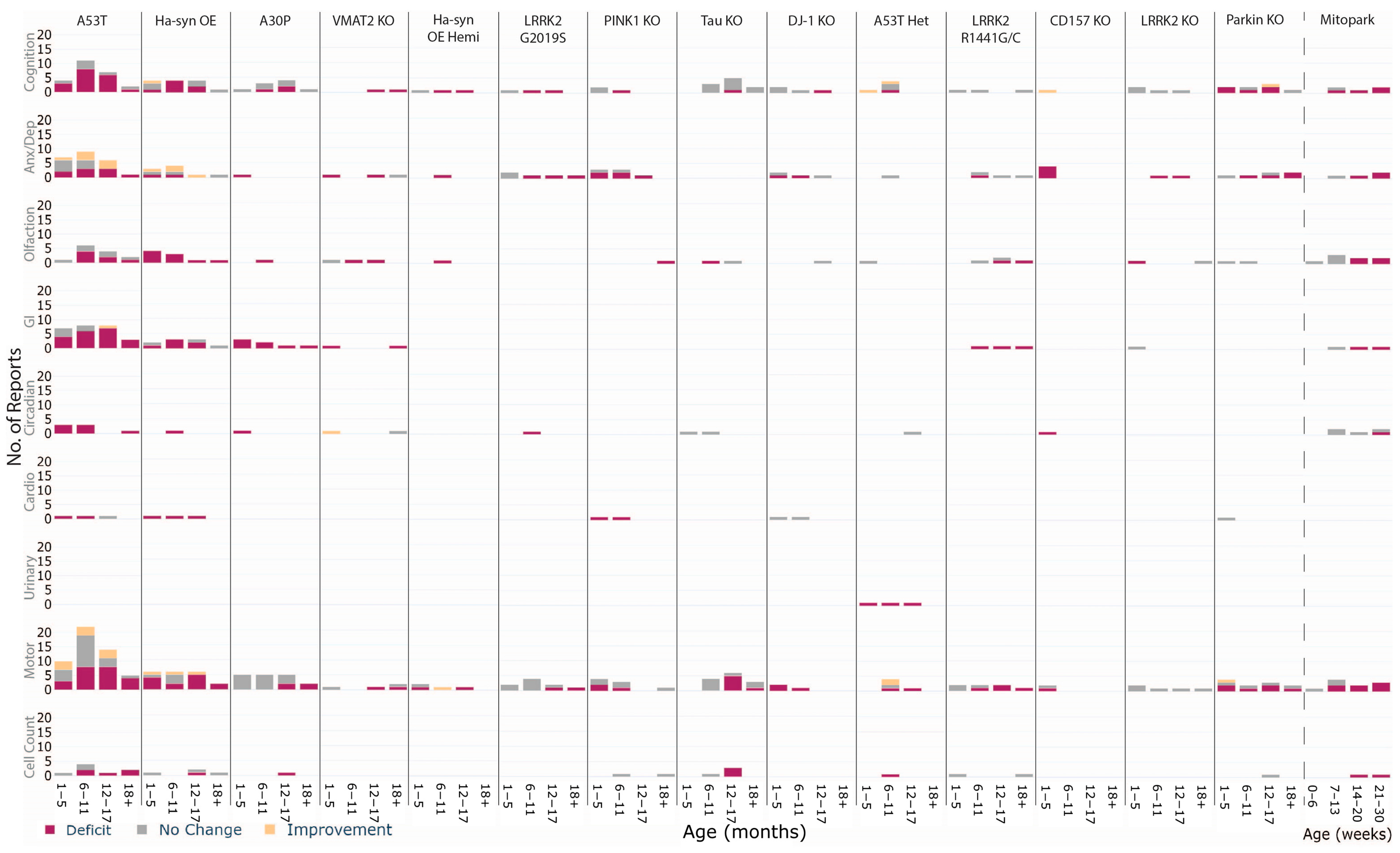
3.3.1. Homozygous A53T
3.3.2. Heterozygous A53T
3.3.3. Homozygous Hα-syn OE
3.3.4. Hemizygous Hα-syn OE (Thy1-αsyn Hemi)
3.3.5. A30P
3.3.6. Mitopark
3.3.7. VMAT2 KO
3.3.8. LRRK2 G2019S
3.3.9. PINK1 KO
3.3.10. Tau KO
3.3.11. DJ-1 KO
3.3.12. LRRK2 R1441G/C
3.3.13. CD157 KO
3.3.14. LRRK2 KO
3.3.15. Parkin KO
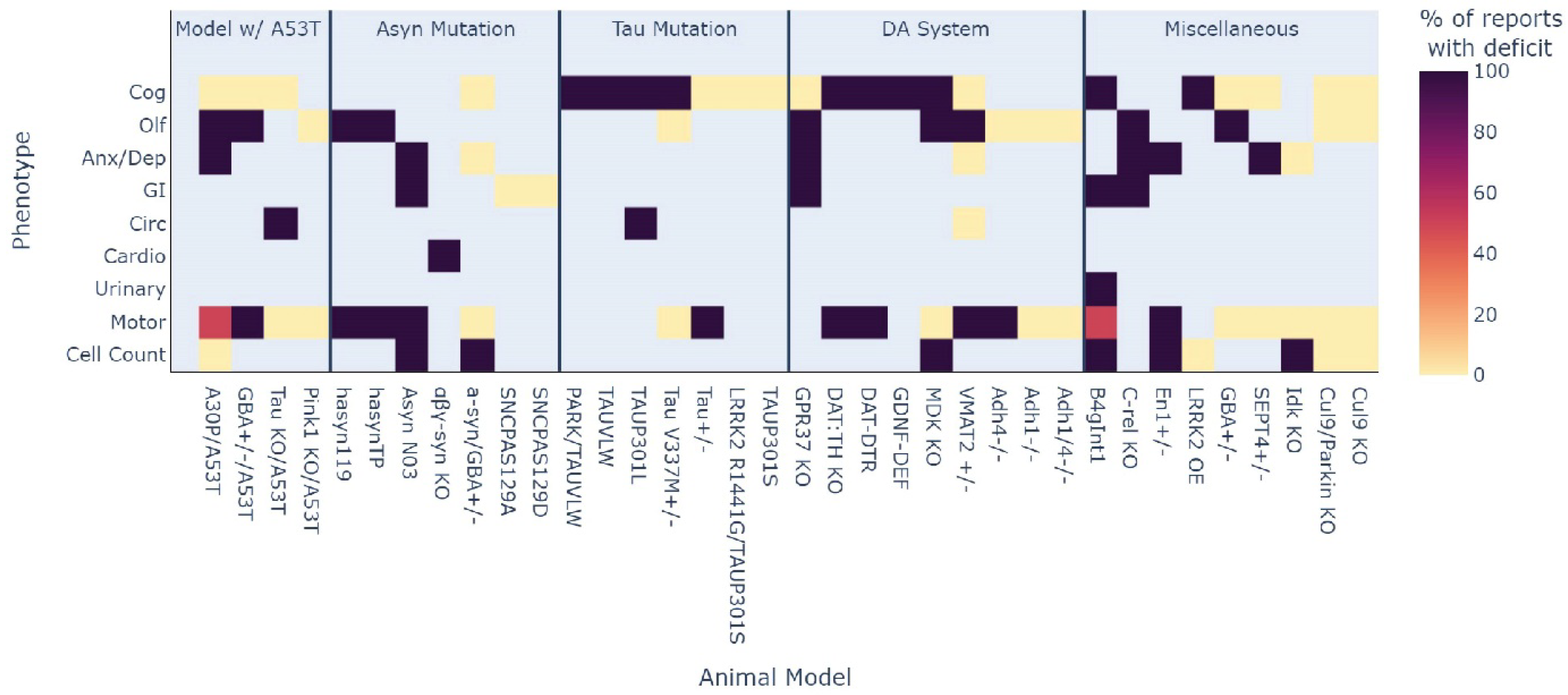
3.4. Which Phenotype Is Most Consistent across All Animal Models?
4. Discussion
4.1. The Contribution of Variability to the Reproducibility of Phenotypes
4.1.1. GI Function
4.1.2. Olfaction
4.1.3. Cognition
4.1.4. Anxiety/Depressive-Like Behaviour
4.1.5. Understudied Phenotypes
4.2. Tracking Age-Dependent Phenotypes to Understand Different Pathological Trajectories
4.3. Do PD Rodent Models Have Good Face Validity?
4.4. Limitations
4.5. Recommendations and Opportunities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Feigin, V.L.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Nguyen, G. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Murray, C.J. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.M.; Nadkarni, N.V.; Lye, W.K.; Yong, M.H.; Tan, E.K. The impact of non-motor symptoms on the quality of life of Parkinson’s disease patients: A longitudinal study. Eur. J. Neurol. 2016, 23, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Surguchov, A. Neurodegenerative Diseases Biomarkers. In Biomarkers in Parkinson’s Disease; Peplow, P.V., Martinez, B., Gennarelli, T.A., Eds.; Humana: New York, NY, USA, 2022; Volume 173, pp. 155–180. [Google Scholar]
- Scherman, D.; Desnos, C.; Darchen, F.; Pollak, P.; Javoy-Agid, F.; Agid, Y. Striatal dopamine deficiency in parkinson s disease: Role of aging. Ann. Neurol. 1989, 26, 551–557. [Google Scholar] [CrossRef]
- Lee, C.S.; Samii, A.; Sossi, V.; Ruth, T.J.; Schulzer, M.; Holden, J.E.; Wudel, J.; Pal, P.K.; Fuente-Fernandez, R.D.L.; Calne, D.B.; et al. In vivo positron emission tomographic evidence for compensatory changes in presynaptic. Ann. Neurol. 2000, 47, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef]
- Rizzo, G.; Copetti, M.; Arcuti, S.; Martino, D.; Fontana, A.; Logroscino, G. Accuracy of clinical diagnosis of Parkinson disease. Neurology 2016, 86, 566–576. [Google Scholar] [CrossRef]
- Lees, A.J. When Did Ray Kennedy’s Parkinson’s Disease Begin? Mov. Disord. 1992, 7, 110–116. [Google Scholar] [CrossRef]
- Pont-Sunyer, C.; Hotter, A.; Gaig, C.; Seppi, K.; Compta, Y.; Katzenschlager, R.; Mas, N.; Hofeneder, D.; Brucke, T.; Bayes, A.; et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov. Disord. 2015, 30, 229–237. [Google Scholar] [CrossRef]
- Scott, G.D.; Lim, M.M.; Drake, M.G.; Woltjer, R.; Quinn, J.F. Onset of Skin, Gut, and Genitourinary Prodromal Parkinson’s Disease: A Study of 1.5 Million Veterans. Mov. Disord. 2021, 36, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Gasperi, A.; Djamshidian, A.; Kiechl, S.; Stockner, H.; Willeit, P.; Willeit, J.; Rungger, G.; Poewe, W.; Seppi, K. Performance of the Movement Disorders Society criteria for prodromal Parkinson’s disease: A population-based 10-year study. Mov. Disord. 2018, 33, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Hogl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef]
- Titze-de-Almeida, R.; Titze-de-Almeida, S.S.; Ferreira, G.G.; Brito Silva, A.P.; de Paula Brandao, P.R.; Oertel, W.H.; Schenck, C.H.; Delgado Rodrigues, R.N. microRNA signatures in prodromal REM sleep behavior disorder and early Parkinson’s disease as noninvasive biomarkers. Sleep Med. 2021, 78, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Gao, X.; Yang, W.; Chang, Z.; Yang, X.; Wei, X.; Huang, Z.; Xie, H.; Yue, Z.; Zhou, F.; et al. Advances in the Research of Risk Factors and Prodromal Biomarkers of Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 973–990. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D. Advances in markers of prodromal Parkinson disease. Nat. Rev. Neurol. 2016, 12, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P. Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef]
- van der Staay, F.J.; Arndt, S.S.; Nordquist, R.E. Evaluation of animal models of neurobehavioral disorders. Behav. Brain Funct. 2009, 5, 11. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.M.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, K.J.; Rothman, S.M.; Ladenheim, B.; Wan, R.; Vranis, N.; Hutchison, E.; Okun, E.; Cadet, J.L.; Mattson, M.P. Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant alpha-synuclein. Neurobiol. Aging 2013, 34, 928–935. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Li, Z.; Jiao, Y.; Gaborit, N.; Pani, A.K.; Orrison, B.M.; Bruneau, B.G.; Giasson, B.I.; Smeyne, R.J.; Gershon, M.D.; et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 2010, 19, 1633–1650. [Google Scholar] [CrossRef] [PubMed]
- Carboni, E.; Tatenhorst, L.; Tonges, L.; Barski, E.; Dambeck, V.; Bahr, M.; Lingor, P. Deferiprone Rescues Behavioral Deficits Induced by Mild Iron Exposure in a Mouse Model of Alpha-Synuclein Aggregation. Neuromolecular Med. 2017, 19, 309–321. [Google Scholar] [CrossRef]
- Valek, L.; Tran, B.; Wilken-Schmitz, A.; Trautmann, S.; Heidler, J.; Schmid, T.; Brune, B.; Thomas, D.; Deller, T.; Geisslinger, G.; et al. Prodromal sensory neuropathy in Pink1(-/-) SNCA(A53T) double mutant Parkinson mice. Neuropathol. Appl. Neurobiol. 2021, 47, 1060–1079. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Nwankwo, E.I.; Nussbaum, R.L.; Rogers, J.; Maccecchini, M.L. Translational inhibition of α-synuclein by Posiphen normalizes distal colon motility in transgenic Parkinson mice. Am. J. Neurodegener. Dis. 2019, 8, 1–15. [Google Scholar] [PubMed]
- Seo, J.H.; Kang, S.W.; Kim, K.; Wi, S.; Lee, J.W.; Cho, S.R. Environmental Enrichment Attenuates Oxidative Stress and Alters Detoxifying Enzymes in an A53T alpha-Synuclein Transgenic Mouse Model of Parkinson’s Disease. Antioxidants 2020, 9, 928. [Google Scholar] [CrossRef]
- Farrell, K.F.; Krishnamachari, S.; Villanueva, E.; Lou, H.; Alerte, T.N.; Peet, E.; Drolet, R.E.; Perez, R.G. Non-motor parkinsonian pathology in aging A53T alpha-synuclein mice is associated with progressive synucleinopathy and altered enzymatic function. J. Neurochem. 2014, 128, 536–546. [Google Scholar] [CrossRef]
- Wi, S.; Lee, J.W.; Kim, M.; Park, C.H.; Cho, S.R. An Enriched Environment Ameliorates Oxidative Stress and Olfactory Dysfunction in Parkinson’s Disease with alpha-Synucleinopathy. Cell Transplant. 2018, 27, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiao, Q.; Le, W. Olfactory dysfunction and neurotransmitter disturbance in olfactory bulb of transgenic mice expressing human A53T mutant alpha-synuclein. PLoS ONE 2015, 10, e0119928. [Google Scholar] [CrossRef]
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Carretero Murillo, M.; Sengupta, U.; Barrett, A.; Kayed, R. Tau oligomers mediate alpha-synuclein toxicity and can be targeted by immunotherapy. Mol. Neurodegener. 2018, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Diwakarla, S.; Finkelstein, D.I.; Constable, R.; Artaiz, O.; Di Natale, M.; McQuade, R.M.; Lei, E.; Chai, X.Y.; Ringuet, M.T.; Fothergill, L.J.; et al. Chronic isolation stress is associated with increased colonic and motor symptoms in the A53T mouse model of Parkinson’s disease. Neurogastroenterol. Motil. 2020, 32, e13755. [Google Scholar] [CrossRef] [PubMed]
- Diwakarla, S.; McQuade, R.M.; Constable, R.; Artaiz, O.; Lei, E.; Barnham, K.J.; Adlard, P.A.; Cherny, R.A.; Di Natale, M.R.; Wu, H.; et al. ATH434 Reverses Colorectal Dysfunction in the A53T Mouse Model of Parkinson’s Disease. J. Park. Dis. 2021, 11, 1821–1832. [Google Scholar] [CrossRef]
- Taguchi, T.; Ikuno, M.; Hondo, M.; Parajuli, L.K.; Taguchi, K.; Ueda, J.; Sawamura, M.; Okuda, S.; Nakanishi, E.; Hara, J.; et al. alpha-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: A prodromal Parkinson’s disease model. Brain 2020, 143, 249–265. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Zhang, L.; Wang, M.; Li, Y. Dl-3-n-Butylphthalide Alleviates Behavioral and Cognitive Symptoms Via Modulating Mitochondrial Dynamics in the A53T-alpha-Synuclein Mouse Model of Parkinson’s Disease. Front. Neurosci. 2021, 15, 647266. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; Perez, G.; Berhe, B.; Tayebi, N.; Sidransky, E. Behavioral Phenotyping in a Murine Model of GBA1-Associated Parkinson Disease. Int. J. Mol. Sci. 2021, 22, 6826. [Google Scholar] [CrossRef]
- Stanojlovic, M.; Pallais, J.P.; Lee, M.K.; Kotz, C.M. Pharmacological and chemogenetic orexin/hypocretin intervention ameliorates Hipp-dependent memory impairment in the A53T mice model of Parkinson’s disease. Mol. Brain 2019, 12, 87. [Google Scholar] [CrossRef]
- Graham, D.R.; Sidhu, A. Mice expressing the A53T mutant form of human alpha-synuclein exhibit hyperactivity and reduced anxiety-like behavior. J. Neurosci. Res. 2010, 88, 1777–1783. [Google Scholar] [CrossRef]
- Costa, G.; Sisalli, M.J.; Simola, N.; Della Notte, S.; Casu, M.A.; Serra, M.; Pinna, A.; Feliciello, A.; Annunziato, L.; Scorziello, A.; et al. Gender Differences in Neurodegeneration, Neuroinflammation and Na(+)-Ca(2+) Exchangers in the Female A53T Transgenic Mouse Model of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 118. [Google Scholar] [CrossRef]
- Peters, S.T.; Fahrenkopf, A.; Choquette, J.M.; Vermilyea, S.C.; Lee, M.K.; Vossel, K. Ablating Tau Reduces Hyperexcitability and Moderates Electroencephalographic Slowing in Transgenic Mice Expressing A53T Human alpha-Synuclein. Front. Neurol. 2020, 11, 563. [Google Scholar] [CrossRef]
- Paumier, K.L.; Sukoff Rizzo, S.J.; Berger, Z.; Chen, Y.; Gonzales, C.; Kaftan, E.; Li, L.; Lotarski, S.; Monaghan, M.; Shen, W.; et al. Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PLoS ONE 2013, 8, e70274. [Google Scholar] [CrossRef] [PubMed]
- Thom, T.; Schmitz, M.; Fischer, A.L.; Correia, A.; Correia, S.; Llorens, F.; Pique, A.V.; Mobius, W.; Domingues, R.; Zafar, S.; et al. Cellular Prion Protein Mediates alpha-Synuclein Uptake, Localization, and Toxicity In Vitro and In Vivo. Mov. Disord. 2022, 37, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Covelo, A.; Martell-Martinez, H.; Nanclares, C.; Sherman, M.A.; Okematti, E.; Meints, J.; Teravskis, P.J.; Gallardo, C.; Savonenko, A.V.; et al. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of alpha-synucleinopathy. Acta Neuropathol. 2019, 138, 551–574. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Zimmermann, Z.; Gispert, S.; Auburger, G.; Korf, H.W.; von Gall, C. Impaired Photic Entrainment of Spontaneous Locomotor Activity in Mice Overexpressing Human Mutant alpha-Synuclein. Int. J. Mol. Sci. 2018, 19, 1651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.F.; Yu, X.L.; Ji, M.; Liu, S.Y.; Wu, X.L.; Wang, Y.J.; Liu, R.T. Resveratrol alleviates motor and cognitive deficits and neuropathology in the A53T alpha-synuclein mouse model of Parkinson’s disease. Food Funct. 2018, 9, 6414–6426. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Clarke, J.; Viel, C.; Panarello, N.; Treleaven, C.M.; Kim, C.; Spencer, B.; Adame, A.; Park, H.; Dodge, J.C.; et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 2645–2660. [Google Scholar] [CrossRef]
- West, C.L.; Mao, Y.K.; Delungahawatta, T.; Amin, J.Y.; Farhin, S.; McQuade, R.M.; Diwakarla, S.; Pustovit, R.; Stanisz, A.M.; Bienenstock, J.; et al. Squalamine Restores the Function of the Enteric Nervous System in Mouse Models of Parkinson’s Disease. J. Park. Dis. 2020, 10, 1477–1491. [Google Scholar] [CrossRef]
- Noorian, A.R.; Rha, J.; Annerino, D.M.; Bernhard, D.; Taylor, G.M.; Greene, J.G. Alpha-synuclein transgenic mice display age-related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol. Dis. 2012, 48, 9–19. [Google Scholar] [CrossRef]
- Tikhonova, M.A.; Shoeva, O.Y.; Tenditnik, M.V.; Ovsyukova, M.V.; Akopyan, A.A.; Dubrovina, N.I.; Amstislavskaya, T.G.; Khlestkina, E.K. Evaluating the Effects of Grain of Isogenic Wheat Lines Differing in the Content of Anthocyanins in Mouse Models of Neurodegenerative Disorders. Nutrients 2020, 12, 3877. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Song, N.; Jia, F.; Tang, T.; Bao, W.; Zuo, C.; Xie, J.; Jiang, H. Genomic DNA levels of mutant alpha-synuclein correlate with non-motor symptoms in an A53T Parkinson’s disease mouse model. NeuroChem. Int. 2018, 114, 71–79. [Google Scholar] [CrossRef]
- Li, Y.; Jiao, Q.; Du, X.; Jiang, H. Sirt1/FoxO1-Associated MAO-A Upregulation Promotes Depressive-Like Behavior in Transgenic Mice Expressing Human A53T alpha-Synuclein. ACS Chem. Neurosci. 2020, 11, 3838–3848. [Google Scholar] [CrossRef] [PubMed]
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl. Neurodegener. 2019, 8, 5. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.M.; Moon, J.; Choi, H.J. Alpha-synuclein interferes with cAMP/PKA-dependent upregulation of dopamine beta-hydroxylase and is associated with abnormal adaptive responses to immobilization stress. Exp. Neurol. 2014, 252, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Martinez, G.; Vargas-Medrano, J.; Gil-Tommee, C.; Medina, D.; Garza, N.T.; Yang, B.; Segura-Ulate, I.; Dominguez, S.J.; Perez, R.G. FTY720/Fingolimod Reduces Synucleinopathy and Improves Gut Motility in A53T Mice: CONTRIBUTIONS OF PRO-BRAIN-DERIVED NEUROTROPHIC FACTOR (PRO-BDNF) AND MATURE BDNF. J. Biol. Chem. 2016, 291, 20811–20821. [Google Scholar] [CrossRef]
- Huang, Y.R.; Xie, X.X.; Ji, M.; Yu, X.L.; Zhu, J.; Zhang, L.X.; Liu, X.G.; Wei, C.; Li, G.; Liu, R.T. Naturally occurring autoantibodies against alpha-synuclein rescues memory and motor deficits and attenuates alpha-synuclein pathology in mouse model of Parkinson’s disease. Neurobiol. Dis. 2019, 124, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.I.; Hare, D.J.; Billings, J.L.; Sedjahtera, A.; Nurjono, M.; Arthofer, E.; George, S.; Culvenor, J.G.; Bush, A.I.; Adlard, P.A. Clioquinol Improves Cognitive, Motor Function, and Microanatomy of the Alpha-Synuclein hA53T Transgenic Mice. ACS Chem. Neurosci. 2016, 7, 119–129. [Google Scholar] [CrossRef]
- Oaks, A.W.; Frankfurt, M.; Finkelstein, D.I.; Sidhu, A. Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function. PLoS ONE 2013, 8, e60378. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, X.; Zhang, Y.; Zhang, L.; Feng, L. Chronic mild stress accelerates the progression of Parkinson’s disease in A53T α-synuclein transgenic mice. Exp. Neurol. 2016, 285, 61–71. [Google Scholar] [CrossRef]
- Rothman, S.M.; Griffioen, K.J.; Vranis, N.; Ladenheim, B.; Cong, W.N.; Cadet, J.L.; Haran, J.; Martin, B.; Mattson, M.P. Neuronal expression of familial Parkinson’s disease A53T alpha-synuclein causes early motor impairment, reduced anxiety and potential sleep disturbances in mice. J. Park. Dis. 2013, 3, 215–229. [Google Scholar] [CrossRef]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Lopes da Fonseca, T.; Koch, J.C.; Becker, S.; Tonges, L.; Bahr, M.; et al. Fasudil attenuates aggregation of alpha-synuclein in models of Parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef]
- Pavia-Collado, R.; Coppola-Segovia, V.; Miquel-Rio, L.; Alarcon-Aris, D.; Rodriguez-Aller, R.; Torres-Lopez, M.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Artigas, F.; et al. Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces alpha-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*alpha-Synuclein Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 2939. [Google Scholar] [CrossRef] [PubMed]
- Lelan, F.; Boyer, C.; Thinard, R.; Remy, S.; Usal, C.; Tesson, L.; Anegon, I.; Neveu, I.; Damier, P.; Naveilhan, P.; et al. Effects of Human Alpha-Synuclein A53T-A30P Mutations on SVZ and Local Olfactory Bulb Cell Proliferation in a Transgenic Rat Model of Parkinson Disease. J. Park. Dis. 2011, 2011, 987084. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Belin, A.C.; Westerlund, M.; Anvret, A.; Lindqvist, E.; Pernold, K.; Ogren, S.O.; Duester, G.; Galter, D. Modeling Parkinson’s disease genetics: Altered function of the dopamine system in Adh4 knockout mice. Behav. Brain Res. 2011, 217, 439–445. [Google Scholar] [CrossRef]
- Hallett, P.J.; McLean, J.R.; Kartunen, A.; Langston, J.W.; Isacson, O. alpha-Synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol. Dis. 2012, 47, 258–267. [Google Scholar] [CrossRef]
- Yamakado, H.; Moriwaki, Y.; Yamasaki, N.; Miyakawa, T.; Kurisu, J.; Uemura, K.; Inoue, H.; Takahashi, M.; Takahashi, R. alpha-Synuclein BAC transgenic mice as a model for Parkinson’s disease manifested decreased anxiety-like behavior and hyperlocomotion. Neurosci. Res. 2012, 73, 173–177. [Google Scholar] [CrossRef]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef]
- Wassouf, Z.; Hentrich, T.; Casadei, N.; Jaumann, M.; Knipper, M.; Riess, O.; Schulze-Hentrich, J.M. Distinct Stress Response and Altered Striatal Transcriptome in Alpha-Synuclein Overexpressing Mice. Front. Neurosci. 2018, 12, 1033. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Magen, I.; Bove, N.; Zhu, C.; Lemesre, V.; Dutta, G.; Elias, C.J.; Lester, H.A.; Chesselet, M.F. Chronic nicotine improves cognitive and social impairment in mice overexpressing wild type alpha-synuclein. Neurobiol. Dis. 2018, 117, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Baker, L.S.; Seroogy, K.B.; Genter, M.B. Intranasal Carnosine Mitigates alpha-Synuclein Pathology and Motor Dysfunction in the Thy1-aSyn Mouse Model of Parkinson’s Disease. ACS Chem. Neurosci. 2021, 12, 2347–2359. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef]
- Schwab, K.; Frahm, S.; Horsley, D.; Rickard, J.E.; Melis, V.; Goatman, E.A.; Magbagbeolu, M.; Douglas, M.; Leith, M.G.; Baddeley, T.C.; et al. A Protein Aggregation Inhibitor, Leuco-Methylthioninium Bis(Hydromethanesulfonate), Decreases alpha-Synuclein Inclusions in a Transgenic Mouse Model of Synucleinopathy. Front. Mol. Neurosci. 2017, 10, 447. [Google Scholar] [CrossRef]
- Wang, L.; Magen, I.; Yuan, P.Q.; Subramaniam, S.R.; Richter, F.; Chesselet, M.F.; Tache, Y. Mice overexpressing wild-type human alpha-synuclein display alterations in colonic myenteric ganglia and defecation. Neurogastroenterol. Motil. 2012, 24, e425–e436. [Google Scholar] [CrossRef] [PubMed]
- McDowell, K.A.; Shin, D.; Roos, K.P.; Chesselet, M.F. Sleep dysfunction and EEG alterations in mice overexpressing alpha-synuclein. J. Park. Dis. 2014, 4, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.L.; Seroogy, K.B.; Genter, M.B. Evaluation of Carnosine Intervention in the Thy1-aSyn Mouse Model of Parkinson’s Disease. Neuroscience 2019, 411, 270–278. [Google Scholar] [CrossRef]
- Biju, K.C.; Shen, Q.; Hernandez, E.T.; Mader, M.J.; Clark, R.A. Reduced cerebral blood flow in an alpha-synuclein transgenic mouse model of Parkinson’s disease. J. Cereb. Blood Flow Metab. 2020, 40, 2441–2453. [Google Scholar] [CrossRef]
- Fleming, S.M.; Tetreault, N.A.; Mulligan, C.K.; Hutson, C.B.; Masliah, E.; Chesselet, M.F. Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur. J. Neurosci. 2008, 28, 247–256. [Google Scholar] [CrossRef]
- Fleming, S.M.; Mulligan, C.K.; Richter, F.; Mortazavi, F.; Lemesre, V.; Frias, C.; Zhu, C.; Stewart, A.; Gozes, I.; Morimoto, B.; et al. A pilot trial of the microtubule-interacting peptide (NAP) in mice overexpressing alpha-synuclein shows improvement in motor function and reduction of alpha-synuclein inclusions. Mol. Cell. Neurosci. 2011, 46, 597–606. [Google Scholar] [CrossRef]
- Fleming, S.M.; Jordan, M.C.; Mulligan, C.K.; Masliah, E.; Holden, J.G.; Millard, R.W.; Chesselet, M.F.; Roos, K.P. Impaired baroreflex function in mice overexpressing alpha-synuclein. Front. Neurol. 2013, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Magen, I.; Fleming, S.M.; Zhu, C.; Garcia, E.C.; Cardiff, K.M.; Dinh, D.; De La Rosa, K.; Sanchez, M.; Torres, E.R.; Masliah, E.; et al. Cognitive deficits in a mouse model of pre-manifest Parkinson’s disease. Eur. J. Neurosci. 2012, 35, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.R.S.; Stanojlovic, M.; Zelikowsky, M.; Bonsberger, J.; Hean, S.; Mulligan, C.; Baldauf, L.; Fleming, S.; Masliah, E.; Chesselet, M.F.; et al. Alpha-synuclein pathology, microgliosis, and parvalbumin neuron loss in the amygdala associated with enhanced fear in the Thy1-aSyn model of Parkinson’s disease. Neurobiol. Dis. 2021, 158, 105478. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fleming, S.M.; Chesselet, M.F.; Tache, Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. Neuroreport 2008, 19, 873–876. [Google Scholar] [CrossRef]
- Li, R.; Lu, Y.; Zhang, Q.; Liu, W.; Yang, R.; Jiao, J.; Liu, J.; Gao, G.; Yang, H. Piperine promotes autophagy flux by P2RX4 activation in SNCA/alpha-synuclein-induced Parkinson disease model. Autophagy 2022, 18, 559–575. [Google Scholar] [CrossRef]
- Grant, L.M.; Richter, F.; Miller, J.E.; White, S.A.; Fox, C.M.; Zhu, C.; Chesselet, M.F.; Ciucci, M.R. Vocalization deficits in mice over-expressing alpha-synuclein, a model of pre-manifest Parkinson’s disease. Behav. Neurosci. 2014, 128, 110–121. [Google Scholar] [CrossRef]
- Beccano-Kelly, D.A.; Volta, M.; Munsie, L.N.; Paschall, S.A.; Tatarnikov, I.; Co, K.; Chou, P.; Cao, L.P.; Bergeron, S.; Mitchell, E.; et al. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum. Mol. Genet. 2015, 24, 1336–1349. [Google Scholar] [CrossRef]
- Volta, M.; Cataldi, S.; Beccano-Kelly, D.; Munsie, L.; Tatarnikov, I.; Chou, P.; Bergeron, S.; Mitchell, E.; Lim, R.; Khinda, J.; et al. Chronic and acute LRRK2 silencing has no long-term behavioral effects, whereas wild-type and mutant LRRK2 overexpression induce motor and cognitive deficits and altered regulation of dopamine release. Park. Relat. Disord. 2015, 21, 1156–1163. [Google Scholar] [CrossRef]
- Keane, P.C.; Hanson, P.S.; Patterson, L.; Blain, P.G.; Hepplewhite, P.; Khundakar, A.A.; Judge, S.J.; Kahle, P.J.; LeBeau, F.E.N.; Morris, C.M. Trichloroethylene and its metabolite TaClo lead to degeneration of substantia nigra dopaminergic neurones: Effects in wild type and human A30P mutant alpha-synuclein mice. Neurosci. Lett. 2019, 711, 134437. [Google Scholar] [CrossRef]
- Gureviciene, I.; Gurevicius, K.; Tanila, H. Aging and alpha-synuclein affect synaptic plasticity in the dentate gyrus. J. Neural Transm. 2009, 116, 13–22. [Google Scholar] [CrossRef]
- Marxreiter, F.; Ettle, B.; May, V.E.; Esmer, H.; Patrick, C.; Kragh, C.L.; Klucken, J.; Winner, B.; Riess, O.; Winkler, J.; et al. Glial A30P alpha-synuclein pathology segregates neurogenesis from anxiety-related behavior in conditional transgenic mice. Neurobiol. Dis. 2013, 59, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Schell, H.; Boden, C.; Chagas, A.M.; Kahle, P.J. Impaired c-Fos and polo-like kinase 2 induction in the limbic system of fear-conditioned alpha-synuclein transgenic mice. PLoS ONE 2012, 7, e50245. [Google Scholar] [CrossRef] [PubMed]
- Neuner, J.; Filser, S.; Michalakis, S.; Biel, M.; Herms, J. A30P alpha-Synuclein interferes with the stable integration of adult-born neurons into the olfactory network. Sci. Rep. 2014, 4, 3931. [Google Scholar] [CrossRef] [PubMed]
- Freichel, C.; Neumann, M.; Ballard, T.; Muller, V.; Woolley, M.; Ozmen, L.; Borroni, E.; Kretzschmar, H.A.; Haass, C.; Spooren, W.; et al. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol. Aging 2007, 28, 1421–1435. [Google Scholar] [CrossRef]
- Stylianou, M.; Zaaimi, B.; Thomas, A.; Taylor, J.P.; LeBeau, F.E.N. Early Disruption of Cortical Sleep-Related Oscillations in a Mouse Model of Dementia with Lewy Bodies (DLB) Expressing Human Mutant (A30P) Alpha-Synuclein. Front. Neurosci. 2020, 14, 579867. [Google Scholar] [CrossRef] [PubMed]
- Gries, M.; Christmann, A.; Schulte, S.; Weyland, M.; Rommel, S.; Martin, M.; Baller, M.; Roth, R.; Schmitteckert, S.; Unger, M.; et al. Parkinson mice show functional and molecular changes in the gut long before motoric disease onset. Mol. Neurodegener. 2021, 16, 34. [Google Scholar] [CrossRef]
- Veenit, V.; Zhang, X.; Ambrosini, A.; Sousa, V.; Svenningsson, P. The Effect of Early Life Stress on Emotional Behaviors in GPR37KO Mice. Int. J. Mol. Sci. 2021, 23, 410. [Google Scholar] [CrossRef]
- Mandillo, S.; Golini, E.; Marazziti, D.; Di Pietro, C.; Matteoni, R.; Tocchini-Valentini, G.P. Mice lacking the Parkinson’s related GPR37/PAEL receptor show non-motor behavioral phenotypes: Age and gender effect. Genes Brain Behav. 2013, 12, 465–477. [Google Scholar] [CrossRef]
- Langley, M.R.; Ghaisas, S.; Palanisamy, B.N.; Ay, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Characterization of nonmotor behavioral impairments and their neurochemical mechanisms in the MitoPark mouse model of progressive neurodegeneration in Parkinson’s disease. Exp. Neurol. 2021, 341, 113716. [Google Scholar] [CrossRef]
- Li, X.; Redus, L.; Chen, C.; Martinez, P.A.; Strong, R.; Li, S.; O’Connor, J.C. Cognitive dysfunction precedes the onset of motor symptoms in the MitoPark mouse model of Parkinson’s disease. PLoS ONE 2013, 8, e71341. [Google Scholar] [CrossRef]
- Cong, L.; Muir, E.R.; Chen, C.; Qian, Y.; Liu, J.; Biju, K.C.; Clark, R.A.; Li, S.; Duong, T.Q. Multimodal MRI Evaluation of the MitoPark Mouse Model of Parkinson’s Disease. PLoS ONE 2016, 11, e0151884. [Google Scholar] [CrossRef]
- Ghaisas, S.; Langley, M.R.; Palanisamy, B.N.; Dutta, S.; Narayanaswamy, K.; Plummer, P.J.; Sarkar, S.; Ay, M.; Jin, H.; Anantharam, V.; et al. MitoPark transgenic mouse model recapitulates the gastrointestinal dysfunction and gut-microbiome changes of Parkinson’s disease. Neurotoxicology 2019, 75, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Fifel, K.; Cooper, H.M. Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 359–369. [Google Scholar] [CrossRef]
- Pass, T.; Assfalg, M.; Tolve, M.; Blaess, S.; Rothermel, M.; Wiesner, R.J.; Ricke, K.M. The Impact of Mitochondrial Dysfunction on Dopaminergic Neurons in the Olfactory Bulb and Odor Detection. Mol. Neurobiol. 2020, 57, 3646–3657. [Google Scholar] [CrossRef] [PubMed]
- Langley, M.R.; Ghaisas, S.; Ay, M.; Luo, J.; Palanisamy, B.N.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Manganese exposure exacerbates progressive motor deficits and neurodegeneration in the MitoPark mouse model of Parkinson’s disease: Relevance to gene and environment interactions in metal neurotoxicity. Neurotoxicology 2018, 64, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Gibbs, J.T.; Melief, E.J.; Postupna, N.O.; Sherfield, E.E.; Wilson, A.; Keene, C.D.; Montine, T.J.; Palmiter, R.D.; Darvas, M. Relative contributions of severe dopaminergic neuron ablation and dopamine depletion to cognitive impairment. Exp. Neurol. 2015, 271, 205–214. [Google Scholar] [CrossRef]
- Baumann, A.; Moreira, C.G.; Morawska, M.M.; Masneuf, S.; Baumann, C.R.; Noain, D. Preliminary Evidence of Apathetic-Like Behavior in Aged Vesicular Monoamine Transporter 2 Deficient Mice. Front. Hum. Neurosci. 2016, 10, 587. [Google Scholar] [CrossRef]
- Cui, K.; Yang, F.; Tufan, T.; Raza, M.U.; Zhan, Y.; Fan, Y.; Zeng, F.; Brown, R.W.; Price, J.B.; Jones, T.C.; et al. Restoration of Noradrenergic Function in Parkinson’s Disease Model Mice. ASN Neuro 2021, 13, 17590914211009730. [Google Scholar] [CrossRef]
- Taylor, T.N.; Caudle, W.M.; Shepherd, K.R.; Noorian, A.; Jackson, C.R.; Iuvone, P.M.; Weinshenker, D.; Greene, J.G.; Miller, G.W. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J. Neurosci. 2009, 29, 8103–8113. [Google Scholar] [CrossRef]
- Ikuno, M.; Yamakado, H.; Akiyama, H.; Parajuli, L.K.; Taguchi, K.; Hara, J.; Uemura, N.; Hatanaka, Y.; Higaki, K.; Ohno, K.; et al. GBA haploinsufficiency accelerates alpha-synuclein pathology with altered lipid metabolism in a prodromal model of Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1894–1904. [Google Scholar] [CrossRef]
- Buhusi, M.; Olsen, K.; Yang, B.Z.; Buhusi, C.V. Stress-Induced Executive Dysfunction in GDNF-Deficient Mice, A Mouse Model of Parkinsonism. Front. Behav. Neurosci. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Rabl, R.; Breitschaedel, C.; Flunkert, S.; Duller, S.; Amschl, D.; Neddens, J.; Niederkofler, V.; Rockenstein, E.; Masliah, E.; Roemer, H.; et al. Early start of progressive motor deficits in Line 61 alpha-synuclein transgenic mice. BMC Neurosci. 2017, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Gabrielyan, L.; Liang, H.; Minalyan, A.; Hatami, A.; John, V.; Wang, L. Behavioral Deficits and Brain alpha-Synuclein and Phosphorylated Serine-129 alpha-Synuclein in Male and Female Mice Overexpressing Human alpha-Synuclein. J. Alzheimers Dis. 2021, 79, 875–893. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, N.R.S.; Caesar, J.; Park, A.; Sedgh, S.; Finogenov, G.; Masliah, E.; Davis, J.; Blurton-Jones, M. Neural Stem Cells Rescue Cognitive and Motor Dysfunction in a Transgenic Model of Dementia with Lewy Bodies through a BDNF-Dependent Mechanism. Stem Cell Rep. 2015, 5, 791–804. [Google Scholar] [CrossRef]
- Hosford, P.S.; Ninkina, N.; Buchman, V.L.; Smith, J.C.; Marina, N.; SheikhBahaei, S. Synuclein Deficiency Results in Age-Related Respiratory and Cardiovascular Dysfunctions in Mice. Brain Sci. 2020, 10, 583. [Google Scholar] [CrossRef] [PubMed]
- Prediger, R.D.; Rojas-Mayorquin, A.E.; Aguiar, A.S., Jr.; Chevarin, C.; Mongeau, R.; Hamon, M.; Lanfumey, L.; Del Bel, E.; Muramatsu, H.; Courty, J.; et al. Mice with genetic deletion of the heparin-binding growth factor midkine exhibit early preclinical features of Parkinson’s disease. J. Neural Transm. 2011, 118, 1215–1225. [Google Scholar] [CrossRef]
- Lim, J.; Bang, Y.; Choi, J.H.; Han, A.; Kwon, M.S.; Liu, K.H.; Choi, H.J. LRRK2 G2019S Induces Anxiety/Depression-like Behavior before the Onset of Motor Dysfunction with 5-HT1A Receptor Upregulation in Mice. J. Neurosci. 2018, 38, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Matikainen-Ankney, B.A.; Kezunovic, N.; Menard, C.; Flanigan, M.E.; Zhong, Y.; Russo, S.J.; Benson, D.L.; Huntley, G.W. Parkinson’s Disease-Linked LRRK2-G2019S Mutation Alters Synaptic Plasticity and Promotes Resilience to Chronic Social Stress in Young Adulthood. J. Neurosci. 2018, 38, 9700–9711. [Google Scholar] [CrossRef]
- Crown, L.M.; Bartlett, M.J.; Wiegand, J.L.; Eby, A.J.; Monroe, E.J.; Gies, K.; Wohlford, L.; Fell, M.J.; Falk, T.; Cowen, S.L. Sleep Spindles and Fragmented Sleep as Prodromal Markers in a Preclinical Model of LRRK2-G2019S Parkinson’s Disease. Front. Neurol. 2020, 11, 324. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Hou, X.; Zheng, B.; Melrose, H.L.; Mosley, T.; Wang, J.M. Human LRRK2 G2019S mutation represses post-synaptic protein PSD95 and causes cognitive impairment in transgenic mice. Neurobiol. Learn. Mem. 2017, 142, 182–189. [Google Scholar] [CrossRef]
- Tian, Y.; He, M.; Pan, L.; Yuan, X.; Xiong, M.; Meng, L.; Yao, Z.; Yu, Z.; Ye, K.; Zhang, Z. Transgenic Mice Expressing Human alpha-Synuclein 1-103 Fragment as a Novel Model of Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 760781. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Berger, S.; Hu, Y.; Bartsch, D.; Tian, Y. Alterations of the Motor and Olfactory Functions Related to Parkinson’s Disease in Transgenic Mice with a VMAT2-Deficiency in Dopaminergic Neurons. Front. Neurosci. 2020, 14, 356. [Google Scholar] [CrossRef] [PubMed]
- Glasl, L.; Kloos, K.; Giesert, F.; Roethig, A.; Benedetto, B.D.; Kuhn, R.; Zhang, J.; Hafen, U.; Zerle, J.; Hofmann, A.; et al. Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp. Neurol. 2012, 235, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.E.; Redell, J.B.; Kobori, N.; Underwood, E.L.; Fischer, T.D.; Hood, K.N.; LaRoche, V.; Waxham, M.N.; Moore, A.N.; Dash, P.K. Loss of PTEN-induced kinase 1 (Pink1) reduces hippocampal tyrosine hydroxylase and impairs learning and memory. Exp. Neurol. 2020, 323, 113081. [Google Scholar] [CrossRef]
- Agnihotri, S.K.; Sun, L.; Yee, B.K.; Shen, R.; Akundi, R.S.; Zhi, L.; Duncan, M.J.; Cass, W.A.; Bueler, H. PINK1 deficiency is associated with increased deficits of adult hippocampal neurogenesis and lowers the threshold for stress-induced depression in mice. Behav. Brain Res. 2019, 363, 161–172. [Google Scholar] [CrossRef]
- Ferris, C.F.; Morrison, T.R.; Iriah, S.; Malmberg, S.; Kulkarni, P.; Hartner, J.C.; Trivedi, M. Evidence of Neurobiological Changes in the Presymptomatic PINK1 Knockout Rat. J. Park. Dis. 2018, 8, 281–301. [Google Scholar] [CrossRef]
- Hoffmeister, J.D.; Kelm-Nelson, C.A.; Ciucci, M.R. Quantification of brainstem norepinephrine relative to vocal impairment and anxiety in the Pink1-/- rat model of Parkinson disease. Behav. Brain Res. 2021, 414, 113514. [Google Scholar] [CrossRef]
- Marquis, J.M.; Lettenberger, S.E.; Kelm-Nelson, C.A. Early-onset Parkinsonian behaviors in female Pink1-/- rats. Behav. Brain Res. 2020, 377, 112175. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Martinez Hernandez, A.; Silbern, I.; Geffers, I.; Tatenhorst, L.; Becker, S.; Urlaub, H.; Zweckstetter, M.; Griesinger, C.; Eichele, G. Low-Expressing Synucleinopathy Mouse Models Based on Oligomer-Forming Mutations and C-Terminal Truncation of alpha-Synuclein. Front. Neurosci. 2021, 15, 643391. [Google Scholar] [CrossRef]
- Sonnier, L.; Le Pen, G.; Hartmann, A.; Bizot, J.C.; Trovero, F.; Krebs, M.O.; Prochiantz, A. Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1. J. Neurosci. 2007, 27, 1063–1071. [Google Scholar] [CrossRef]
- Beauchamp, L.C.; Chan, J.; Hung, L.W.; Padman, B.S.; Vella, L.J.; Liu, X.M.; Coleman, B.; Bush, A.I.; Lazarou, M.; Hill, A.F.; et al. Ablation of tau causes an olfactory deficit in a murine model of Parkinson’s disease. Acta Neuropathol. Commun. 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hall, A.M.; Kelinske, M.; Roberson, E.D. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol. Aging 2014, 35, 2617–2624. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Moon, S.; Zhang, Q.; Volitakis, I.; Finkelstein, D.I.; Bush, A.I. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 2014, 9, 29. [Google Scholar] [CrossRef]
- Morris, M.; Hamto, P.; Adame, A.; Devidze, N.; Masliah, E.; Mucke, L. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol. Aging 2013, 34, 1523–1529. [Google Scholar] [CrossRef]
- Escobar, V.D.; Kuo, Y.M.; Orrison, B.M.; Giasson, B.I.; Nussbaum, R.L. Transgenic mice expressing S129 phosphorylation mutations in alpha-synuclein. Neurosci. Lett. 2014, 563, 96–100. [Google Scholar] [CrossRef][Green Version]
- Gil-Tommee, C.; Vidal-Martinez, G.; Annette Reyes, C.; Vargas-Medrano, J.; Herrera, G.V.; Martin, S.M.; Chaparro, S.A.; Perez, R.G. Parkinsonian GM2 synthase knockout mice lacking mature gangliosides develop urinary dysfunction and neurogenic bladder. Exp. Neurol. 2019, 311, 265–273. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Seo, J.H.; Alselehdar, S.K.; DeFrees, S.; Ledeen, R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020, 329, 113284. [Google Scholar] [CrossRef] [PubMed]
- Dongworth, R.K.; Mukherjee, U.A.; Hall, A.R.; Astin, R.; Ong, S.B.; Yao, Z.; Dyson, A.; Szabadkai, G.; Davidson, S.M.; Yellon, D.M.; et al. DJ-1 protects against cell death following acute cardiac ischemia-reperfusion injury. Cell Death Dis. 2014, 5, e1082. [Google Scholar] [CrossRef]
- Pham, T.T.; Giesert, F.; Rothig, A.; Floss, T.; Kallnik, M.; Weindl, K.; Holter, S.M.; Ahting, U.; Prokisch, H.; Becker, L.; et al. DJ-1-deficient mice show less TH-positive neurons in the ventral tegmental area and exhibit non-motoric behavioural impairments. Genes Brain Behav. 2010, 9, 305–317. [Google Scholar] [CrossRef]
- Li, M.; Xu, H.; Chen, G.; Sun, S.; Wang, Q.; Liu, B.; Wu, X.; Zhou, L.; Chai, Z.; Sun, X.; et al. Impaired D2 receptor-dependent dopaminergic transmission in prefrontal cortex of awake mouse model of Parkinson’s disease. Brain 2019, 142, 3099–3115. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Grothe, D.; Konecny, F.; Rao, V.; Kim, R.H.; Mak, T.W. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6085–6090. [Google Scholar] [CrossRef] [PubMed]
- Kyser, T.L.; Dourson, A.J.; McGuire, J.L.; Hemmerle, A.M.; Williams, M.T.; Seroogy, K.B. Characterization of Motor and Non-Motor Behavioral Alterations in the Dj-1 (PARK7) Knockout Rat. J. Mol. Neurosci. 2019, 69, 298–311. [Google Scholar] [CrossRef]
- Navarro, P.; Guerrero, R.; Gallego, E.; Avila, J.; Luquin, R.; Ruiz, P.J.; Sanchez, M.P. Memory and exploratory impairment in mice that lack the Park-2 gene and that over-express the human FTDP-17 mutant Tau. Behav. Brain Res. 2008, 189, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Parrella, E.; Bellucci, A.; Porrini, V.; Benarese, M.; Lanzillotta, A.; Faustini, G.; Longhena, F.; Abate, G.; Uberti, D.; Pizzi, M. NF-kappaB/c-Rel deficiency causes Parkinson’s disease-like prodromal symptoms and progressive pathology in mice. Transl. Neurodegener. 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, Y.; Wan, W.; Ma, Z. An unexpected improvement in spatial learning and memory ability in alpha-synuclein A53T transgenic mice. J. Neural Transm. 2018, 125, 203–210. [Google Scholar] [CrossRef]
- Uemura, N.; Ueda, J.; Yoshihara, T.; Ikuno, M.; Uemura, M.T.; Yamakado, H.; Asano, M.; Trojanowski, J.Q.; Takahashi, R. alpha-Synuclein Spread from Olfactory Bulb Causes Hyposmia, Anxiety, and Memory Loss in BAC-SNCA Mice. Mov. Disord. 2021, 36, 2036–2047. [Google Scholar] [CrossRef] [PubMed]
- Hamill, R.W.; Tompkins, J.D.; Girard, B.M.; Kershen, R.T.; Parsons, R.L.; Vizzard, M.A. Autonomic dysfunction and plasticity in micturition reflexes in human alpha-synuclein mice. Dev. Neurobiol. 2012, 72, 918–936. [Google Scholar] [CrossRef]
- La Vitola, P.; Balducci, C.; Baroni, M.; Artioli, L.; Santamaria, G.; Castiglioni, M.; Cerovic, M.; Colombo, L.; Caldinelli, L.; Pollegioni, L.; et al. Peripheral inflammation exacerbates alpha-synuclein toxicity and neuropathology in Parkinson’s models. Neuropathol. Appl. Neurobiol. 2021, 47, 43–60. [Google Scholar] [CrossRef]
- Tikhonova, M.A.; Tikhonova, N.G.; Tenditnik, M.V.; Ovsyukova, M.V.; Akopyan, A.A.; Dubrovina, N.I.; Amstislavskaya, T.G.; Khlestkina, E.K. Effects of Grape Polyphenols on the Life Span and Neuroinflammatory Alterations Related to Neurodegenerative Parkinson Disease-Like Disturbances in Mice. Molecules 2020, 25, 5339. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, F.; Calingasan, N.; Parolari, L.; Subramanian, A.; Yang, L.; Flint Beal, M. Lack of exacerbation of neurodegeneration in a double transgenic mouse model of mutant LRRK2 and tau. Hum. Mol. Genet. 2015, 24, 3545–3556. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Joers, V.; Nakamura, A.; Swahari, V.; Tansey, M.G.; Moy, S.S.; Deshmukh, M. Characterization of a Cul9-Parkin double knockout mouse model for Parkinson’s disease. Sci. Rep. 2020, 10, 16886. [Google Scholar] [CrossRef] [PubMed]
- Giesert, F.; Glasl, L.; Zimprich, A.; Ernst, L.; Piccoli, G.; Stautner, C.; Zerle, J.; Holter, S.M.; Vogt Weisenhorn, D.M.; Wurst, W. The pathogenic LRRK2 R1441C mutation induces specific deficits modeling the prodromal phase of Parkinson’s disease in the mouse. Neurobiol. Dis. 2017, 105, 179–193. [Google Scholar] [CrossRef]
- Bichler, Z.; Lim, H.C.; Zeng, L.; Tan, E.K. Non-motor and motor features in LRRK2 transgenic mice. PLoS ONE 2013, 8, e70249. [Google Scholar] [CrossRef]
- Dranka, B.P.; Gifford, A.; McAllister, D.; Zielonka, J.; Joseph, J.; O’Hara, C.L.; Stucky, C.L.; Kanthasamy, A.G.; Kalyanaraman, B. A novel mitochondrially-targeted apocynin derivative prevents hyposmia and loss of motor function in the leucine-rich repeat kinase 2 (LRRK2(R1441G)) transgenic mouse model of Parkinson’s disease. Neurosci. Lett. 2014, 583, 159–164. [Google Scholar] [CrossRef]
- Craven, K.M.; Kochen, W.R.; Hernandez, C.M.; Flinn, J.M. Zinc Exacerbates Tau Pathology in a Tau Mouse Model. J. Alzheimers Dis. 2018, 64, 617–630. [Google Scholar] [CrossRef]
- Ageta-Ishihara, N.; Yamakado, H.; Morita, T.; Hattori, S.; Takao, K.; Miyakawa, T.; Takahashi, R.; Kinoshita, M. Chronic overload of SEPT4, a parkin substrate that aggregates in Parkinson’s disease, causes behavioral alterations but not neurodegeneration in mice. Mol. Brain 2013, 6, 35. [Google Scholar] [CrossRef]
- Lopatina, O.; Yoshihara, T.; Nishimura, T.; Zhong, J.; Akther, S.; Fakhrul, A.A.; Liang, M.; Higashida, C.; Sumi, K.; Furuhara, K.; et al. Anxiety- and depression-like behavior in mice lacking the CD157/BST1 gene, a risk factor for Parkinson’s disease. Front. Behav. Neurosci. 2014, 8, 133. [Google Scholar] [CrossRef]
- Mizuno, A.; Cherepanov, S.M.; Kikuchi, Y.; Fakhrul, A.A.; Akther, S.; Deguchi, K.; Yoshihara, T.; Ishihara, K.; Shuto, S.; Higashida, H. Lipo-oxytocin-1, a Novel Oxytocin Analog Conjugated with Two Palmitoyl Groups, Has Long-Lasting Effects on Anxiety-Related Behavior and Social Avoidance in CD157 Knockout Mice. Brain Sci. 2015, 5, 3–13. [Google Scholar] [CrossRef]
- Kasai, S.; Yoshihara, T.; Lopatina, O.; Ishihara, K.; Higashida, H. Selegiline Ameliorates Depression-Like Behavior in Mice Lacking the CD157/BST1 Gene, a Risk Factor for Parkinson’s Disease. Front. Behav. Neurosci. 2017, 11, 75. [Google Scholar] [CrossRef]
- Higashida, H.; Liang, M.; Yoshihara, T.; Akther, S.; Fakhrul, A.; Stanislav, C.; Nam, T.S.; Kim, U.H.; Kasai, S.; Nishimura, T.; et al. An immunohistochemical, enzymatic, and behavioral study of CD157/BST-1 as a neuroregulator. BMC Neurosci. 2017, 18, 35. [Google Scholar] [CrossRef]
- Havrda, M.C.; Paolella, B.R.; Ward, N.M.; Holroyd, K.B. Behavioral abnormalities and Parkinson’s-like histological changes resulting from Id2 inactivation in mice. Dis. Model Mech. 2013, 6, 819–827. [Google Scholar] [CrossRef]
- Maekawa, T.; Tsushima, H.; Kawakami, F.; Kawashima, R.; Kodo, M.; Imai, M.; Ichikawa, T. Leucine-Rich Repeat Kinase 2 Is Associated with Activation of the Paraventricular Nucleus of the Hypothalamus and Stress-Related Gastrointestinal Dysmotility. Front. Neurosci. 2019, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Maset, A.; Albanesi, M.; di Soccio, A.; Canova, M.; Dal Maschio, M.; Lodovichi, C. Aberrant Patterns of Sensory-Evoked Activity in the Olfactory Bulb of LRRK2 Knockout Mice. Cells 2021, 10, 3212. [Google Scholar] [CrossRef]
- Lambourne, S.L.; Sellers, L.A.; Bush, T.G.; Choudhury, S.K.; Emson, P.C.; Suh, Y.H.; Wilkinson, L.S. Increased tau phosphorylation on mitogen-activated protein kinase consensus sites and cognitive decline in transgenic models for Alzheimer’s disease and FTDP-17: Evidence for distinct molecular processes underlying tau abnormalities. Mol. Cell. Biol. 2005, 25, 278–293. [Google Scholar] [CrossRef]
- Lambourne, S.L.; Humby, T.; Isles, A.R.; Emson, P.C.; Spillantini, M.G.; Wilkinson, L.S. Impairments in impulse control in mice transgenic for the human FTDP-17 tauV337M mutation are exacerbated by age. Hum. Mol. Genet. 2007, 16, 1708–1719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, X.R.; Maskri, L.; Herold, C.; Bader, V.; Stichel, C.C.; Gunturkun, O.; Lubbert, H. Non-motor behavioural impairments in parkin-deficient mice. Eur. J. Neurosci. 2007, 26, 1902–1911. [Google Scholar] [CrossRef]
- Rial, D.; Castro, A.A.; Machado, N.; Garcao, P.; Goncalves, F.Q.; Silva, H.B.; Tome, A.R.; Kofalvi, A.; Corti, O.; Raisman-Vozari, R.; et al. Behavioral phenotyping of Parkin-deficient mice: Looking for early preclinical features of Parkinson’s disease. PLoS ONE 2014, 9, e114216. [Google Scholar] [CrossRef] [PubMed]
- Itier, J.M.; Ibanez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L.; et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.A.; Palmiter, R.D. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Anvret, A.; Ran, C.; Westerlund, M.; Gellhaar, S.; Lindqvist, E.; Pernold, K.; Lundstromer, K.; Duester, G.; Felder, M.R.; Galter, D.; et al. Adh1 and Adh1/4 knockout mice as possible rodent models for presymptomatic Parkinson’s disease. Behav. Brain Res. 2012, 227, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.; Duda, J.; Quinn, S.; Zhang, B.; Tojanowski, J.; Lee, V. Neuronal alpha-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human alpha-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016–E4025. [Google Scholar] [CrossRef]
- Taylor, T.N.; Potgieter, D.; Anwar, S.; Senior, S.L.; Janezic, S.; Threlfell, S.; Ryan, B.; Parkkinen, L.; Deltheil, T.; Cioroch, M.; et al. Region-specific deficits in dopamine, but not norepinephrine, signaling in a novel A30P alpha-synuclein BAC transgenic mouse. Neurobiol. Dis. 2014, 62, 193–207. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef]
- Lohr, K.M.; Masoud, S.T.; Salahpour, A.; Miller, G.W. Membrane transporters as mediators of synaptic dopamine dynamics: Implications for disease. Eur. J. Neurosci. 2017, 45, 20–33. [Google Scholar] [CrossRef]
- Mooslehner, K.A.; Chan, P.M.; Xu, W.; Liu, L.; Smadja, C.; Humby, T.; Allen, N.D.; Wilkinson, L.S.; Emson, P.C. Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: Potential mouse model for parkinsonism. Mol. Cell Biol. 2001, 21, 5321–5331. [Google Scholar] [CrossRef]
- Melrose, H.L.; Dachsel, J.C.; Behrouz, B.; Lincoln, S.J.; Yue, M.; Hinkle, K.M.; Kent, C.B.; Korvatska, E.; Taylor, J.P.; Witten, L.; et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 2010, 40, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef]
- Hinkle, K.M.; Yue, M.; Behrouz, B.; Dachsel, J.C.; Lincoln, S.J.; Bowles, E.E.; Beevers, J.E.; Dugger, B.N.; Winner, B.; Prots, I.; et al. LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol. Neurodegener. 2012, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Stichel, C.C.; Zhu, X.R.; Bader, V.; Linnartz, B.; Schmidt, S.; Lubbert, H. Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum. Mol. Genet. 2007, 16, 2377–2393. [Google Scholar] [CrossRef]
- Haehner, A.; Hummel, T.; Reichmann, H. Olfactory loss in Parkinson’s disease. J. Park. Dis. 2011, 2011, 450939. [Google Scholar] [CrossRef]
- Makaroff, L.; Gunn, A.; Gervasoni, C.; Richy, F. Gastrointestinal disorders in Parkinson’s disease: Prevalence and health outcomes in a US claims database. J. Park. Dis. 2011, 1, 65–74. [Google Scholar] [CrossRef]
- Schellinck, H.M.; Cyr, D.P.; Brown, R.E. How Many Ways Can Mouse Behavioral Experiments Go Wrong? Confounding Variables in Mouse Models of Neurodegenerative Diseases and How to Control Them. In Advances in the Study of Behavior; Academic Press: Cambridge, MA, USA, 2010; pp. 255–366. [Google Scholar] [CrossRef]
- Sare, R.M.; Lemons, A.; Smith, C.B. Behavior Testing in Rodents: Highlighting Potential Confounds Affecting Variability and Reproducibility. Brain Sci. 2021, 11, 522. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, D.P.; Lipp, H.P. Dissecting the behaviour of transgenic mice: Is it the mutation, the genetic background, or the environment? Exp. Physiol. 2000, 86, 627–634. [Google Scholar] [CrossRef]
- Bouwknecht, J.A.; Paylor, R. Pitfalls in the interpretation of genetic and pharmacological effects on anxiety-like behaviour in rodents. Behav. Pharmacol. 2008, 19, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.M.; Keshavarzian, A.; Dodiya, H.B.; Jakate, S.; Kordower, J.H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov. Disord. 2012, 27, 716–719. [Google Scholar] [CrossRef] [PubMed]
- West, C.; Wu, R.Y.; Wong, A.; Stanisz, A.M.; Yan, R.; Min, K.K.; Pasyk, M.; McVey Neufeld, K.A.; Karamat, M.I.; Foster, J.A.; et al. Lactobacillus rhamnosus strain JB-1 reverses restraint stress-induced gut dysmotility. Neurogastroenterol. Motil. 2017, 29, e12903. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, M.; Mine, K.; Kubo, C. A study of intestinal dysfunction induced by restraint stress in rats. Scand. J. Gastroenterol. 1998, 33, 806–810. [Google Scholar] [CrossRef]
- Dall’Antonia, I.; Sonka, K.; Dusek, P. Olfaction and Colour Vision: What Can They Tell Us about Parkinson’s Disease? Prague Med. Rep. 2018, 119, 85–96. [Google Scholar] [CrossRef]
- Martinez-Marcos, A. On the organization of olfactory and vomeronasal cortices. Prog. Neurobiol. 2009, 87, 21–30. [Google Scholar] [CrossRef]
- Hubbard, P.S.; Esiri, M.M.; Reading, M.; McShane, R.; Nagy, Z. Alpha-synuclein pathology in the olfactory pathways of dementia patients. J. Anat. 2007, 211, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.D.; Rub, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.K.B.; Hawkes, C.H.; Daniel, S.E. The anterior olfactory nucleus in Parkinson s disease. Mov. Disord. 1995, 10, 283–287. [Google Scholar] [CrossRef]
- Pro-Sistiaga, P.; Mohedano-Moriano, A.; Ubeda-Banon, I.; Del Mar Arroyo-Jimenez, M.; Marcos, P.; Artacho-Perula, E.; Crespo, C.; Insausti, R.; Martinez-Marcos, A. Convergence of olfactory and vomeronasal projections in the rat basal telencephalon. J. Comp. Neurol. 2007, 504, 346–362. [Google Scholar] [CrossRef]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.J.; Duyckaerts, C.; Mizuno, Y.; Broe, G.A.; Cummings, J.; Dickson, D.W.; Gauthier, S.; et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 2007, 22, 1689–1707. [Google Scholar] [CrossRef]
- Chaudhary, S.; Kumaran, S.S.; Kaloiya, G.S.; Goyal, V.; Sagar, R.; Kalaivani, M.; Jaganathan, N.R.; Mehta, N.; Srivastava, A. Domain specific cognitive impairment in Parkinson’s patients with mild cognitive impairment. J. Clin. Neurosci. 2020, 75, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Janvin, C.C.; Larsen, J.P.; Salmon, D.P.; Galasko, D.; Hugdahl, K.; Aarsland, D. Cognitive profiles of individual patients with Parkinson’s disease and dementia: Comparison with dementia with lewy bodies and Alzheimer’s disease. Mov. Disord. 2006, 21, 337–342. [Google Scholar] [CrossRef]
- Lambert, C.T.; Guillette, L.M. The impact of environmental and social factors on learning abilities: A meta-analysis. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2871–2889. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, T.P.; Brown, R.E. Optimization of apparatus design and behavioral measures for the assessment of visuo-spatial learning and memory of mice on the Barnes maze. Learn. Mem. 2013, 20, 85–96. [Google Scholar] [CrossRef]
- Richter, S.H.; Garner, J.P.; Zipser, B.; Lewejohann, L.; Sachser, N.; Touma, C.; Schindler, B.; Chourbaji, S.; Brandwein, C.; Gass, P.; et al. Effect of population heterogenization on the reproducibility of mouse behavior: A multi-laboratory study. PLoS ONE 2011, 6, e16461. [Google Scholar] [CrossRef]
- Robinson, L.; Spruijt, B.; Riedel, G. Between and within laboratory reliability of mouse behaviour recorded in home-cage and open-field. J. Neurosci. Methods 2018, 300, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, D.S.; Engler-Chiurazzi, E.B.; Kotilinek, L.A.; Ashe, K.H.; Reed, M.N. Morris Water Maze Test: Optimization for Mouse Strain and Testing Environment. J. Vis. Exp. 2015, 100, e52706. [Google Scholar] [CrossRef]
- Sullivan, J.A.; Dumont, J.R.; Memar, S.; Skirzewski, M.; Wan, J.; Mofrad, M.H.; Ansari, H.Z.; Li, Y.; Muller, L.; Prado, V.F.; et al. New frontiers in translational research: Touchscreens, open science, and the mouse translational research accelerator platform. Genes Brain Behav. 2021, 20, e12705. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Anxiety disorders and depressive disorders preceding Parkinson s disease. Mov. Disord. 2000, 15, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, K.; Hurst, J.L. Improving the practicality of using non-aversive handling methods to reduce background stress and anxiety in laboratory mice. Sci. Rep. 2019, 9, 20305. [Google Scholar] [CrossRef] [PubMed]
- Sorge, R.E.; Martin, L.J.; Isbester, K.A.; Sotocinal, S.G.; Rosen, S.; Tuttle, A.H.; Wieskopf, J.S.; Acland, E.L.; Dokova, A.; Kadoura, B.; et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat. Methods 2014, 11, 629–632. [Google Scholar] [CrossRef]
- Bogdanova, O.V.; Kanekar, S.; D’Anci, K.E.; Renshaw, P.F. Factors influencing behavior in the forced swim test. Physiol. Behav. 2013, 118, 227–239. [Google Scholar] [CrossRef]
- Obernier, J.A.; Baldwin, R.L. Establishing an Appropriate Period of Acclimatization Following Transportation of Laboratory Animals. ILAR J. 2006, 47, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Adamec, R.; Head, D.; Blundell, J.; Burton, P.; Berton, O. Lasting anxiogenic effects of feline predator stress in mice: Sex differences in vulnerability to stress and predicting severity of anxiogenic response from the stress experience. Physiol. Behav. 2006, 88, 12–29. [Google Scholar] [CrossRef]
- Strekalova, T.; Spanagel, R.; Dolgov, O.; Bartsch, D. Stress-induced hyperlocomotion as a confounding factor in anxiety and depression models in mice. Behav. Pharmacol. 2005, 16, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Suvrathan, A.; Tomar, A.; Chattarji, S. Effects of chronic and acute stress on rat behaviour in the forced-swim test. Stress 2010, 13, 533–540. [Google Scholar] [CrossRef]
- Ennaceur, A. Tests of unconditioned anxiety—Pitfalls and disappointments. Physiol. Behav. 2014, 135, 55–71. [Google Scholar] [CrossRef]
- Molendijk, M.L.; de Kloet, E.R. Immobility in the forced swim test is adaptive and does not reflect depression. Psychoneuroendocrinology 2015, 62, 389–391. [Google Scholar] [CrossRef]
- Ennaceur, A.; Chazot, P.L. Preclinical animal anxiety research—Flaws and prejudices. Pharmacol. Res. Perspect. 2016, 4, e00223. [Google Scholar] [CrossRef]
- Bailey, K.R.; Rustay, N.R.; Crawley, J.N. Behavioral Phenotyping of Transgenic and Knockout Mice: Practical Concerns and Potential Pitfalls. ILAR J. 2006, 47, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Sousa, N.; Almeida, O.F.; Wotjak, C.T. A hitchhiker’s guide to behavioral analysis in laboratory rodents. Genes Brain Behav. 2006, 5 (Suppl. S2), 5–24. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Wheaton, M.; Murphy, D.L. What’s wrong with my mouse model? Advances and strategies in animal modeling of anxiety and depression. Behav. Brain Res. 2007, 179, 1–18. [Google Scholar] [CrossRef]
- Yu, Z.; Li, Y.; the Parkinson’s Progression Markers Initiative. Association of autonomic symptoms with cerebrospinal fluid biomarkers in Parkinson disease and scans without evidence of dopaminergic deficit. Medicine 2021, 100, e24837. [Google Scholar] [CrossRef] [PubMed]
- Bencsik, A.; Muselli, L.; Leboidre, M.; Lakhdar, L.; Baron, T. Early and Persistent Expression of Phosphorylated a-Synuclein in the Enteric Nervous System of A53T Mutant Human a-Synuclein Transgenic Mice. J. Neuropathol. Exp. Neurol. 2014, 73, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Borghammer, P.; Fereshtehnejad, S.M.; Heinzel, S.; Horsager, J.; Schaeffer, E.; Postuma, R.B. Prodromal Parkinson disease subtypes—Key to understanding heterogeneity. Nat. Rev. Neurol. 2021, 17, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Mariqueo, L.; Gimenez-Llort, L. Impact of Behavioral Assessment and Re-Test as Functional Trainings That Modify Survival, Anxiety and Functional Profile (Physical Endurance and Motor Learning) of Old Male and Female 3xTg-AD Mice and NTg Mice with Normal Aging. Biomedicines 2022, 10, 973. [Google Scholar] [CrossRef]
- Prasuhn, J.; Piskol, L.; Vollstedt, E.J.; Graf, J.; Schmidt, A.; Tadic, V.; Tunc, S.; Hampf, J.; Warrlich, E.; Bibergeil, C.; et al. Non-motor symptoms and quality of life in subjects with mild parkinsonian signs. Acta Neurol. Scand. 2017, 136, 495–500. [Google Scholar] [CrossRef]
- Marras, C.; Chaudhuri, K.R. Nonmotor features of Parkinson’s disease subtypes. Mov. Disord. 2016, 31, 1095–1102. [Google Scholar] [CrossRef]
- Sauerbier, A.; Jenner, P.; Todorova, A.; Chaudhuri, K.R. Non motor subtypes and Parkinson’s disease. Park. Relat. Disord. 2016, 22 (Suppl. S1), S41–S46. [Google Scholar] [CrossRef]
- Schneider, S.A.; Hizli, B.; Alcalay, R.N. Emerging Targeted Therapeutics for Genetic Subtypes of Parkinsonism. Neurotherapeutics 2020, 17, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Foltynie, T.; Blackwell, A.D.; Robbins, T.W.; Owen, A.M.; Barker, R.A. Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J. Neurol. Neurosurg. Psychiatry 2005, 76, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, J.S.; Ehrt, U.; Lousberg, R.; Aarsland, D.; Leentjens, A.F. The association between motor subtypes and psychopathology in Parkinson’s disease. Park. Relat. Disord. 2009, 15, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Selikhova, M.; Williams, D.R.; Kempster, P.A.; Holton, J.L.; Revesz, T.; Lees, A.J. A clinico-pathological study of subtypes in Parkinson’s disease. Brain 2009, 132, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Ross, O.A.; Vilarino-Guell, C.; Lincoln, S.J.; Kachergus, J.M.; Cobb, S.A.; Lindquist, S.G.; Nielsen, J.E.; Wszolek, Z.K.; Farrer, M.; et al. A Swedish family with de novo alpha-synuclein A53T mutation: Evidence for early cortical dysfunction. Park. Relat. Disord. 2009, 15, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.M. Mechanisms of Gene-Environment Interactions in Parkinson’s Disease. Curr. Environ. Health Rep. 2017, 4, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Albin, R.; Alcalay, R.; Babcock, D.; Bajaj, V.; Bowman, D.; Buko, A.; Cedarbaum, J.; Chelsky, D.; Cookson, M.R.; et al. Finding useful biomarkers for Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaam6003. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models in Main Results | Ref. No. | Lesser-Known Models | Ref. No. | Lesser-Known Models Cont. | Ref. No. |
|---|---|---|---|---|---|
| Homozygous A53T | [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64] | A30P/A53T | [65,66] | Adh4 KO | [67] |
| Homozygous human alpha-synuclein (hα-syn OE) | [45,49,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87] | PINK KO/A53T | [28] | LRRK2 OE | [88,89] |
| A30P | [26,29,46,90,91,92,93,94,95,96,97] | GBA+/−/A53T and GBA+/− | [39] | GPR37 KO | [98,99] |
| Mitopark | [100,101,102,103,104,105,106] | Tau KO/A53T | [43,46] | DAT:TH KO and DAT-DTR | [107] |
| VMAT2 KO | [108,109,110] | α-syn/GBA+/− | [111] | GDNF-deficient | [112] |
| hα-syn OE Hemi | [113,114,115] | αβγ-syn KO | [116] | MDK KO | [117] |
| LRRK2 G2019S | [89,118,119,120,121] | α-syn n103 | [122] | VMAT2 Het | [123] |
| PINK1 KO | [28,124,125,126,127,128,129,130] | hα-syn TP and hα-syn 119 | [131] | En1+/− | [132] |
| Tau KO | [43,46,133,134,135,136,137] | SNCAS129A and SNCAS129D | [138] | B4gInt1 KO | [139,140] |
| DJ-1 KO | [141,142,143,144,145] | Park KO/TauVLW and TauVLW | [146] | c-rel KO | [147] |
| A53T Het | [148,149,150,151,152] | LRRK2 R1441G/TauP301S and TauP301S | [153] | Cul9/Parkin KO and Cul9 KO | [154] |
| LRRK2 R1441G/C | [153,155,156,157] | TauP301L | [158] | SEPT4+/− | [159] |
| CD157 KO | [160,161,162,163] | Tau+/− | [137] | Id2 KO | [164] |
| LRRK2 KD/O | [88,89,155,165,166] | TauV337M hemi | [167,168] | ||
| Parkin KO | [146,154,169,170,171,172,173] | Adh1 KO and Adh1/4 KO | [174] | ||
| Opportunity to fill knowledge gaps in the areas of circadian, cardiovascular, and urinary phenotypes |
| These were highlighted as the least assessed MDS criteria phenotypes across all models. |
| Characterising the age-dependent appearance of phenotypes within animal models enables understanding of how and when genetic risk factors affect the whole system |
| The A53T and Mitopark models suggest differential trajectories of pathology. Further research into other models over age may unveil different subtypes that may align with clinical subtypes. |
| Investigating common mechanisms underlying gastrointestinal dysfunction |
| Highly consistent GI dysfunction across multiple models represents an exciting target to investigate and is also highly prevalent in clinical PD. |
| Consider the construct validity of phenotypic tasks |
| For example, using either non-social cues or both social and non-social cues in olfactory tests is better suited to targeting the main olfactory system which is clinically relevant to human PD. |
| Consider the methodological translatability of assessments to clinical PD |
| Clinical literature suggests heterogenous cognitive profiles in people with PD which represents an opportunity to extend cognitive assessments in PD mouse models to executive function, attention and language, and link underlying neuropathology to the specific cognitive domains. |
| Improving rigor in experimental design to reduce the effect of environmental variabilities |
| Variability across laboratories is significant and reduces the reproducibility of phenotypes, especially in behavioural tests susceptible to the environment. Within study and between study variations have been shown to have little effect on the phenotypic reproducibility, therefore, reducing stressor confounds, optimising protocols within individual cohorts, and performing thorough characterisation of multiple phenotypes may represent some solutions to improving inconsistent results. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.D.; Kolbe, S.C.; Beauchamp, L.C.; Woodbridge, E.K.; Finkelstein, D.I.; Burrows, E.L. How Well Do Rodent Models of Parkinson’s Disease Recapitulate Early Non-Motor Phenotypes? A Systematic Review. Biomedicines 2022, 10, 3026. https://doi.org/10.3390/biomedicines10123026
Zhang TD, Kolbe SC, Beauchamp LC, Woodbridge EK, Finkelstein DI, Burrows EL. How Well Do Rodent Models of Parkinson’s Disease Recapitulate Early Non-Motor Phenotypes? A Systematic Review. Biomedicines. 2022; 10(12):3026. https://doi.org/10.3390/biomedicines10123026
Chicago/Turabian StyleZhang, Tracy D., Scott C. Kolbe, Leah C. Beauchamp, Ella K. Woodbridge, David I. Finkelstein, and Emma L. Burrows. 2022. "How Well Do Rodent Models of Parkinson’s Disease Recapitulate Early Non-Motor Phenotypes? A Systematic Review" Biomedicines 10, no. 12: 3026. https://doi.org/10.3390/biomedicines10123026
APA StyleZhang, T. D., Kolbe, S. C., Beauchamp, L. C., Woodbridge, E. K., Finkelstein, D. I., & Burrows, E. L. (2022). How Well Do Rodent Models of Parkinson’s Disease Recapitulate Early Non-Motor Phenotypes? A Systematic Review. Biomedicines, 10(12), 3026. https://doi.org/10.3390/biomedicines10123026