
Molecular Mechanisms Linking Empagliflozin to Renal Protection in the LLC-PK1 Model of Diabetic Nephropathy

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment Protocol
2.2. Assessment of Cell Viability
2.3. Assessment of Oxidative Stress
2.4. Measurement of TGF-β1 Levels
2.5. Measurement of ECM Expression
2.6. Protein Extraction and Western Blot Method
3. Results
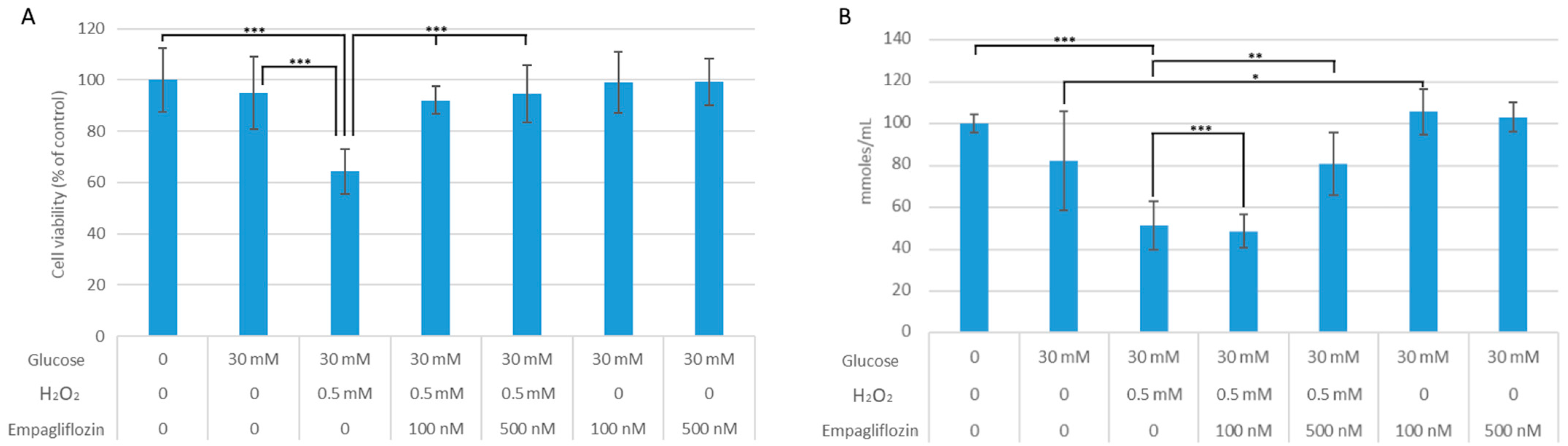
3.1. Empagliflozin Effects on Cell Viability and Oxidative Stress
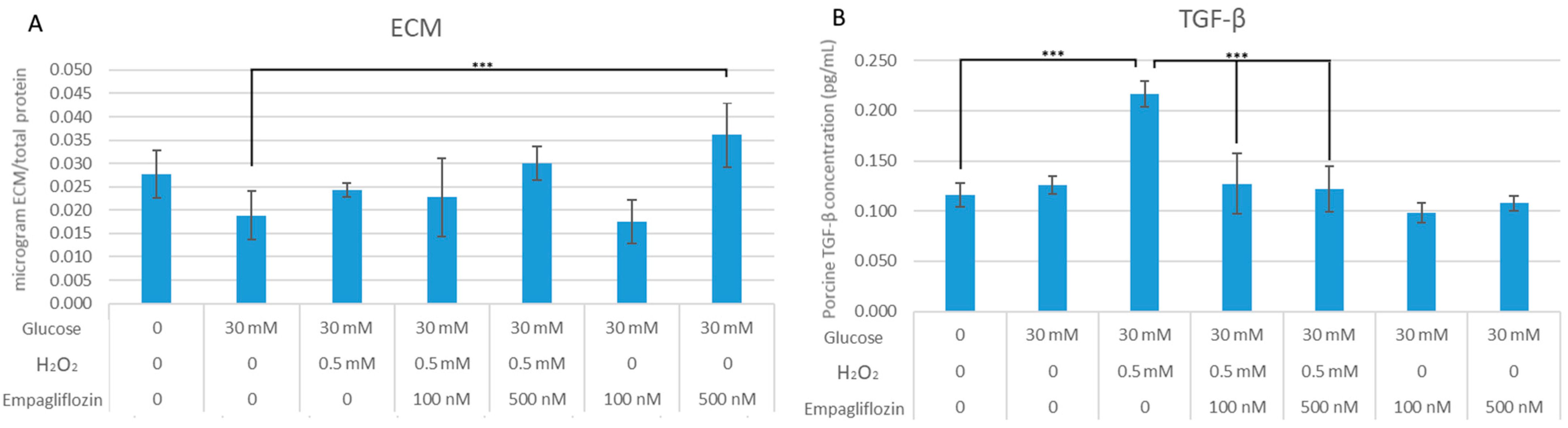
3.2. Empagliflozin Effects on Collagen Synthesis and TGF-β1 Expression
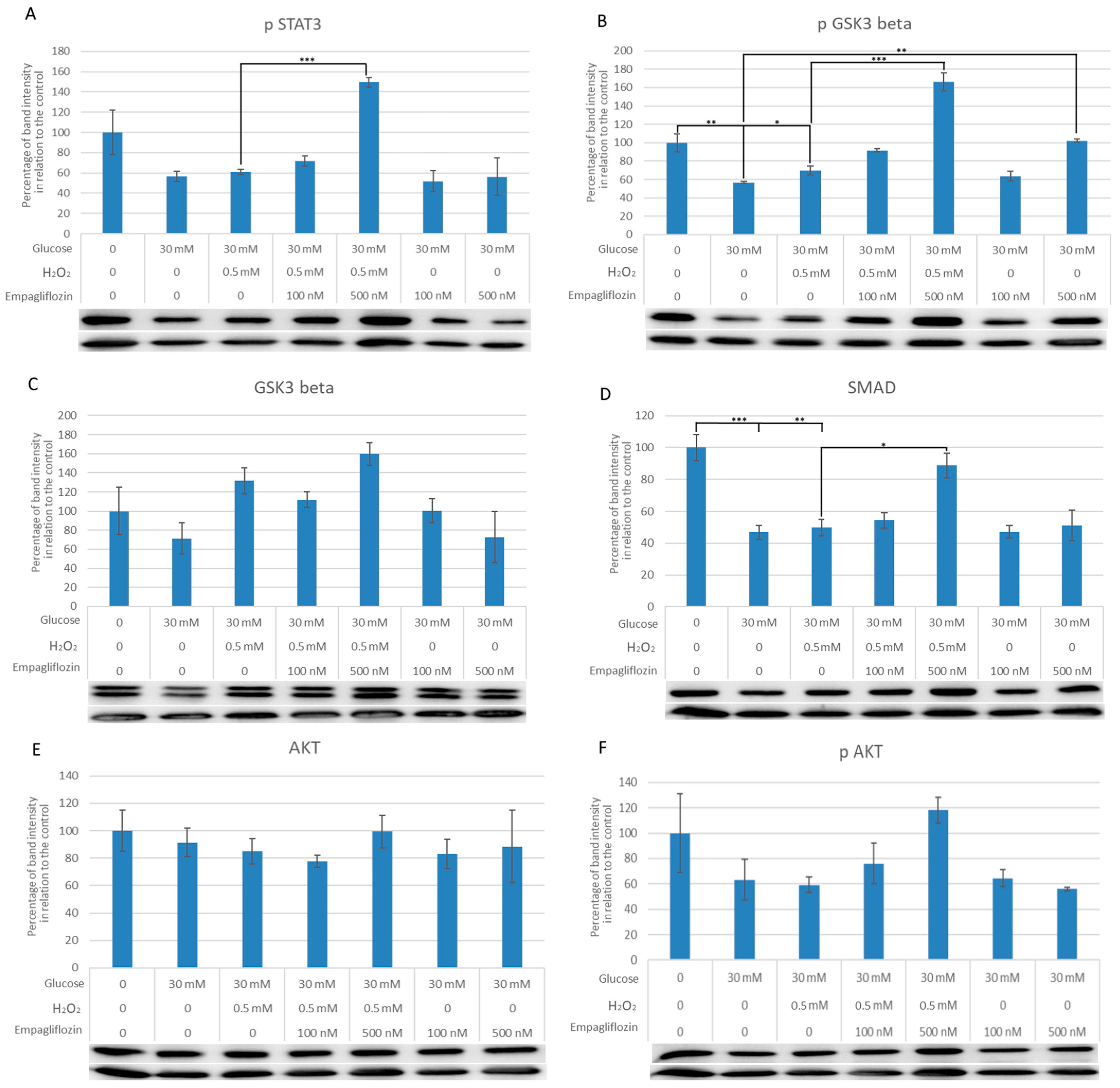
3.3. Empagliflozin Effects on the Expression of Akt, pAkt, GSK3, pGSK3, pSTAT3, and SMAD7 in LLC-PK1 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, H.; Wu, T.; Zhou, Q.; Li, H.; Liu, T.; Ma, X.; Yue, R. Protective Effect and Possible Mechanisms of Artemisinin and Its Derivatives for Diabetic Nephropathy: A Systematic Review and Meta-Analysis in Animal Models. Oxid. Med. Cell. Longev. 2022, 2022, 5401760. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M. Diabetes: Advances in Diagnosis and Treatment. JAMA 2015, 314, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Association, A.D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Guariguata, L.; Whiting, D.R.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J.E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Gembillo, G.; Cernaro, V.; Salvo, A.; Siligato, R.; Laudani, A.; Buemi, M.; Santoro, D. Role of Vitamin D Status in Diabetic Patients with Renal Disease. Medicina (Kaunas) 2019, 55, 273. [Google Scholar] [CrossRef]
- Gross, J.L.; de Azevedo, M.J.; Silveiro, S.P.; Canani, L.H.; Caramori, M.L.; Zelmanovitz, T. Diabetic nephropathy: Diagnosis, prevention, and treatment. Diabetes Care 2005, 28, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Tang, L.; Wei, W. Research progress in signalling pathway in diabetic nephropathy. Diabetes/Metab. Res. Rev. 2015, 31, 221–233. [Google Scholar] [CrossRef]
- Tanaka, S.; Sugiura, Y.; Saito, H.; Sugahara, M.; Higashijima, Y.; Yamaguchi, J.; Inagi, R.; Suematsu, M.; Nangaku, M.; Tanaka, T. Sodium-glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int. 2018, 94, 912–925. [Google Scholar] [CrossRef]
- Hu, T.; Yue, J.; Tang, Q.; Cheng, K.-W.; Chen, F.; Peng, M.; Zhou, Q.; Wang, M. The effect of quercetin on diabetic nephropathy (DN): A systematic review and meta-analysis of animal studies. Food Funct. 2022, 13, 4789–4803. [Google Scholar] [CrossRef]
- Luo, Q.; Cai, Y.; Zhao, Q.; Jiang, Y.; Tian, L.; Liu, Y.; Liu, W.J. Renal Protective Effects of Melatonin in Animal Models of Diabetes Mellitus-Related Kidney Damage: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2022, 2022, 3770417. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, X.R.; Li, A.G.; Liu, F.; Li, J.H.; Truong, L.D.; Wang, X.J.; Lan, H.Y. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: Role of Smad7. J. Am. Soc. Nephrol. 2005, 16, 1371–1383. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Ka, S.-M.; Huang, X.-R.; Lan, H.Y.; Tsai, P.-Y.; Yang, S.-M.; Shui, H.-A.; Chen, A. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J. Am. Soc. Nephrol. 2007, 18, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.R.; Chung, A.C.; Zhou, L.; Wang, X.J.; Lan, H.Y. Latent TGF-beta1 protects against crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 233–242. [Google Scholar] [CrossRef]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Landström, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Daniels, M.C.; McClain, D.A.; Crook, E.D. Transcriptional regulation of transforming growth factor beta1 by glucose: Investigation into the role of the hexosamine biosynthesis pathway. Am. J. Med. Sci. 2000, 319, 138–142. [Google Scholar]
- Orphanides, C.; Fine, L.G.; Norman, J.T. Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int. 1997, 52, 637–647. [Google Scholar] [CrossRef]
- Jones, S.C.; Saunders, H.J.; Pollock, C.A. High glucose increases growth and collagen synthesis in cultured human tubulointerstitial cells. Diabet. Med. 1999, 16, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Z.X.; Lu, Z.L.Z.; Abdul, K.S.M.; Ma, C.M.C.; Tan, K.S.T.K.S.; Jovanovic, A.; Tan, W.T.W. Isosteviol sodium protects heart embryonic H9c2 cells against oxidative stress by activating Akt/GSK-3β signaling pathway. Pharmazie 2020, 75, 36–40. [Google Scholar] [PubMed]
- Kawai, M.; Namba, N.; Mushiake, S.; Etani, Y.; Nishimura, R.; Makishima, M.; Ozono, K. Growth hormone stimulates adipogenesis of 3T3-L1 cells through activation of the Stat5A/5B-PPARgamma pathway. J. Mol. Endocrinol. 2007, 38, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Kosiborod, M.; Inzucchi, S.E.; Cherney, D.Z. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018, 94, 26–39. [Google Scholar] [CrossRef]
- Shenoy, S.V.; Nagaraju, S.P.; Bhojaraja, M.V.; Prabhu, R.A.; Rangaswamy, D.; Rao, I.R. Sodium-glucose cotransporter-2 inhibitors and non-steroidal mineralocorticoid receptor antagonists: Ushering in a new era of nephroprotection beyond renin-angiotensin system blockade. Nephrology (Carlton) 2021, 26, 858–871. [Google Scholar] [CrossRef]
- Panchapakesan, U.; Pegg, K.; Gross, S.; Komala, M.G.; Mudaliar, H.; Forbes, J.; Pollock, C.; Mather, A. Effects of SGLT2 inhibition in human kidney proximal tubular cells--renoprotection in diabetic nephropathy? PLoS ONE. 2013, 8, e54442. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Takeda, Y.; Nagai, Y.; Akamine, T.; Yokota, T.; Sango, K.; Utsunomiya, K. SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy. Int. J. Mol. Sci. 2017, 18, 1083. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Yamagishi, S. Tofogliflozin, A Highly Selective Inhibitor of SGLT2 Blocks Proinflammatory and Proapoptotic Effects of Glucose Overload on Proximal Tubular Cells Partly by Suppressing Oxidative Stress Generation. Horm. Metab. Res. 2016, 48, 191–195. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef]
- Komala, M.G.; Panchapakesan, U.; Pollock, C.; Mather, A. Sodium glucose cotransporter 2 and the diabetic kidney. Curr. Opin. Nephrol. Hypertens. 2013, 22, 113–119. [Google Scholar] [CrossRef]
- Chonchol, M.; Shlipak, M.G.; Katz, R.; Sarnak, M.J.; Newman, A.B.; Siscovick, D.S.; Kestenbaum, B.; Carney, J.K.; Fried, L.F. Relationship of uric acid with progression of kidney disease. Am. J. Kidney Dis. 2007, 50, 239–247. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1801–1802. [Google Scholar] [CrossRef]
- Yao, D.; Wang, S.; Wang, M.; Lu, W. Renoprotection of dapagliflozin in human renal proximal tubular cells via the inhibition of the high mobility group box 1-receptor for advanced glycation end products-nuclear factor-κB signaling pathway. Mol. Med. Rep. 2018, 18, 3625–3630. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Chau, Y.-Y.; Ng, H.-Y.; Chen, C.-H.; Wang, P.-W.; Liou, C.-W.; Lin, T.-K.; Chen, J.-B. Empagliflozin Protects HK-2 Cells from High Glucose-Mediated Injuries via a Mitochondrial Mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef]
- Smith, J.D.; Huang, Z.; Escobar, P.A.; Foppiano, P.; Maw, H.; Loging, W.; Yu, H.; Phillips, J.A.; Taub, M.; Ku, W.W. A Predominant Oxidative Renal Metabolite of Empagliflozin in Male Mice Is Cytotoxic in Mouse Renal Tubular Cells but not Genotoxic. Int. J. Toxicol. 2017, 36, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; Van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [PubMed]
- Baer, P.C.; Koch, B.; Freitag, J.; Schubert, R.; Geiger, H. No Cytotoxic and Inflammatory Effects of Empagliflozin and Dapagliflozin on Primary Renal Proximal Tubular Epithelial Cells under Diabetic Conditions In Vitro. Int. J. Mol. Sci. 2020, 21, 391. [Google Scholar] [CrossRef] [PubMed]
- Ndibalema, A.R.; Kabuye, D.; Wen, S.; Li, L.; Li, X.; Fan, Q. Empagliflozin Protects Against Proximal Renal Tubular Cell Injury Induced by High Glucose via Regulation of Hypoxia-Inducible Factor 1-Alpha. Diabetes Metab. Syndr. Obes. 2020, 13, 1953–1967. [Google Scholar] [CrossRef]
- Li, X.; Kimura, H.; Hirota, K.; Sugimoto, H.; Yoshida, H. Hypoxia reduces constitutive and TNF-alpha-induced expression of monocyte chemoattractant protein-1 in human proximal renal tubular cells. Biochem. Biophys. Res. Commun. 2005, 335, 1026–1034. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef]
- Kizivat, T.; Smolić, M.; Marić, I.; Tolušić Levak, M.; Smolić, R.; Bilić Čurčić, I.; Kuna, L.; Mihaljević, I.; Včev, A.; Tucak-Zorić, A. Antioxidant Pre-Treatment Reduces the Toxic Effects of Oxalate on Renal Epithelial Cells in a Cell Culture Model of Urolithiasis. Int. J. Environ. Res. Public Health 2017, 14, 109. [Google Scholar] [CrossRef]
- Turton, M.D.; O’Shea, D.; Gunn, I.; Beak, S.A.; Edwards, C.M.B.; Meeran, K.; Choi, S.J.; Taylor, G.M.; Heath, M.M.; Lambert, P.D.; et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 1996, 379, 69–72. [Google Scholar] [CrossRef]
- Tikoo, K.; Singh, K.; Kabra, D.; Sharma, V.; Gaikwad, A. Change in histone H3 phosphorylation, MAP kinase p38, SIR 2 and p53 expression by resveratrol in preventing streptozotocin induced type I diabetic nephropathy. Free Radic. Res. 2008, 42, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lee, K.; He, J.C. SIRT1 Is a Potential Drug Target for Treatment of Diabetic Kidney Disease. Front. Endocrinol. (Lausanne) 2018, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Rane, M.J.; Song, Y.; Jin, S.; Barati, M.T.; Wu, R.; Kausar, H.; Tan, Y.; Wang, Y.; Zhou, G.; Klein, J.B.; et al. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy. Am. J. Physiol. Physiol. 2010, 298, F49–F61. [Google Scholar] [CrossRef] [PubMed]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Huang, X.R.; Wang, W.; Li, J.H.; Heuchel, R.L.; Chung, A.C.; Lan, H.Y. The protective role of Smad7 in diabetic kidney disease: Mechanism and therapeutic potential. Diabetes 2011, 60, 590–601. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef]
- Zapata-Morales, J.R.; Galicia-Cruz, O.G.; Franco, M.; Martinez, Y.; Morales, F. Hypoxia-inducible factor-1α (HIF-1α) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J. Biol. Chem. 2014, 289, 346–357. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Antibody Label | Full Name of the Antibody | Antibody Classification | Host Species | Manufacturer and Catalog Number | Dilution Used for Western Blot |
|---|---|---|---|---|---|
| Akt | anti-protein kinase B | IgG monoclonal | Mouse | Cell signaling, Danvers, MA, USA Cat. No. 2920S | 1:1000 |
| pAkt-Ser 473 | anti-protein kinase B-phosphorylated on serine 473 | IgG monoclonal | Rabbit | Cell signaling, Danvers, MA, USA Cat. No. 9271S | 1:1000 |
| GSK3β | Anti-glycogen synthase kinase 3 alpha + beta | IgG monoclonal | Rabbit | Cell signaling, Danvers, MA, USA Cat. No. 5676S | 1:1000 |
| pGSK3β | Anti-glycogen synthase kinase 3-tyrosine 279 phosphorylated GSK3alpha and tyrosine 216 phosphorylated GSK3β | IgG polyclonal | Rabbit | Thermo Fisher Waltham, Rockford, IL, USA Cat. No. OPA1-03083 | 1:1000 |
| pSTAT3 | Anti-signal transducer and activator of transcription 3-phosphorylated on tyrosine 705 | IgG monoclonal | Rabbit | Cell signaling, Danvers, MA, USA Cat. No. 9145S | 1:1000 |
| SMAD7 | Anti-sterile alpha motif domain containing 7 | IgG polyclonal | Rabbit | Sigma-Aldrich, St. Louis, MO, USA Cat. No. HPA028897 | 1:1000 |
| Antibody Label | Full Name of the Antibody | Antibody Classification | Host Species | Manufacturer and Catalog Number | Dilution Used for Western Blot |
|---|---|---|---|---|---|
| αMO-biotin | Anti-mouse antibody labelled with biotin | IgG | Goat | Jackson immune research, West Grove, PA, USA Cat. No. 115-065-071 | 1:20,000 |
| αRB-HRP | Anti-rabbit antibody labelled with HRP | IgG | Goat | Jackson immune research, West Grove, PA, USA Cat. No. 111-035-144 | 1:20,000 |
| SA-HRP | Streptavidin peroxidase polymer, Ultrasensitive | - | - | Sigma-Aldrich, St. Louis, MO, USA Cat. No. S2438 | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihaljević, V.; Zjalić, M.; Kizivat, T.; Omanović Kolarić, T.; Smolić, M.; Rođak, E.; Čović, M.; Kuna, L.; Smolić, R.; Včev, A.; et al. Molecular Mechanisms Linking Empagliflozin to Renal Protection in the LLC-PK1 Model of Diabetic Nephropathy. Biomedicines 2022, 10, 2983. https://doi.org/10.3390/biomedicines10112983
Mihaljević V, Zjalić M, Kizivat T, Omanović Kolarić T, Smolić M, Rođak E, Čović M, Kuna L, Smolić R, Včev A, et al. Molecular Mechanisms Linking Empagliflozin to Renal Protection in the LLC-PK1 Model of Diabetic Nephropathy. Biomedicines. 2022; 10(11):2983. https://doi.org/10.3390/biomedicines10112983
Chicago/Turabian StyleMihaljević, Vjera, Milorad Zjalić, Tomislav Kizivat, Tea Omanović Kolarić, Martina Smolić, Edi Rođak, Marina Čović, Lucija Kuna, Robert Smolić, Aleksandar Včev, and et al. 2022. "Molecular Mechanisms Linking Empagliflozin to Renal Protection in the LLC-PK1 Model of Diabetic Nephropathy" Biomedicines 10, no. 11: 2983. https://doi.org/10.3390/biomedicines10112983
APA StyleMihaljević, V., Zjalić, M., Kizivat, T., Omanović Kolarić, T., Smolić, M., Rođak, E., Čović, M., Kuna, L., Smolić, R., Včev, A., & Bilić Ćurčić, I. (2022). Molecular Mechanisms Linking Empagliflozin to Renal Protection in the LLC-PK1 Model of Diabetic Nephropathy. Biomedicines, 10(11), 2983. https://doi.org/10.3390/biomedicines10112983