

Synaptic Plasticity in the Pain-Related Cingulate and Insular Cortex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
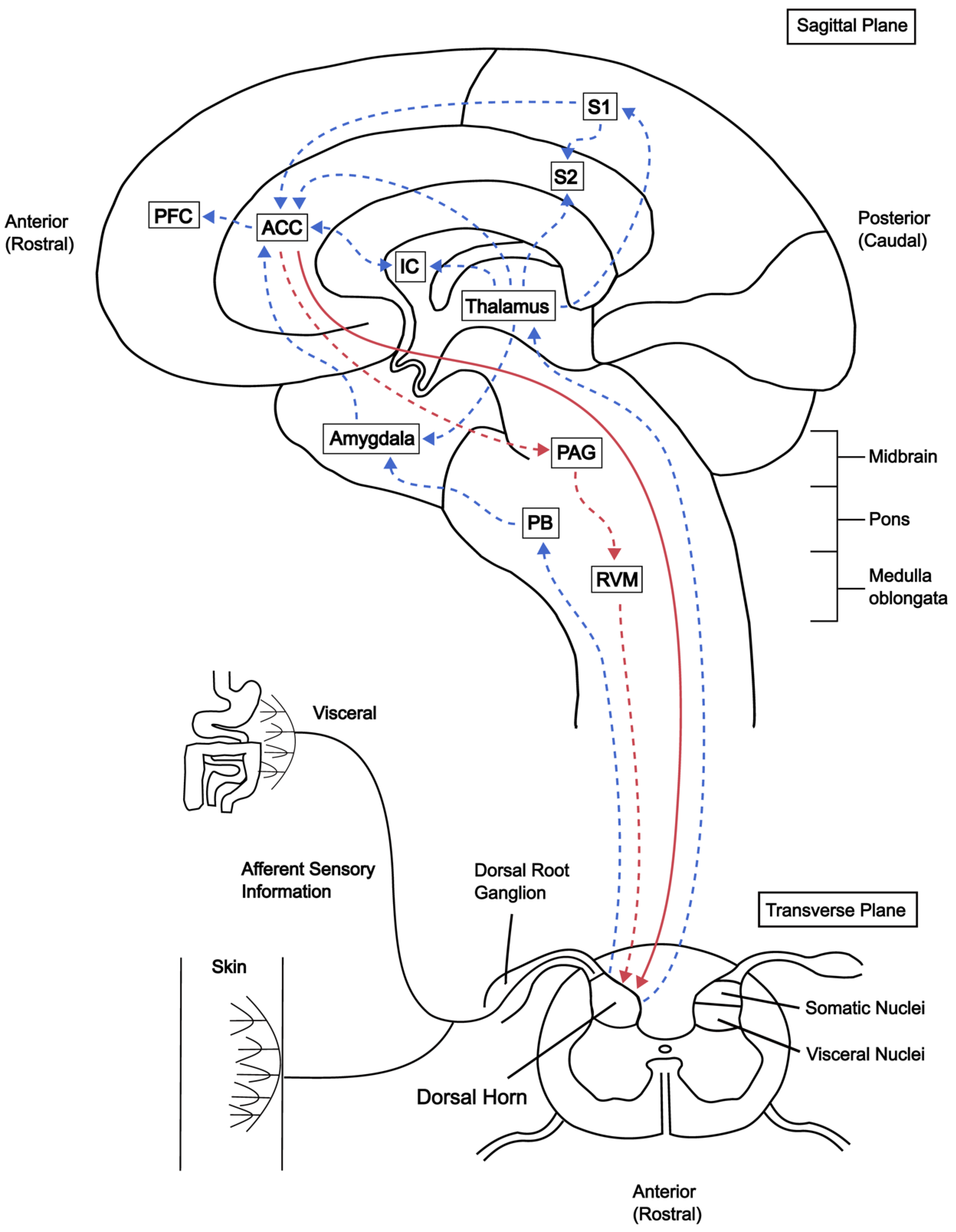
1. Introduction
2. Animal Studies of ACC
2.1. Lesions in ACC Reduce Chronic Pain
2.2. Electrical Stimulation and Glutamate Microinjection in the ACC Trigger Pain and Fear
3. Human Studies of ACC and IC
3.1. Imaging Studies
3.2. Electrophysiological Recordings from ACC
3.3. The IC
4. Synaptic Transmission in the ACC and IC
Excitatory vs. Inhibitory Transmission
5. LTPs: Pre-LTP and Post-LTP
6. LTPs: Early Phase LTP and Late-Phase LTP
7. Long-Term Depression (LTD)
8. Functional Implications of LTPs and LTDs
9. Synaptic Tagging
10. Sex-Related Studies of Cortical Plasticity
11. Neuromodulation of ACC Plasticity: Oxytocin, Norepinephrine, 5-HT, and DA
12. Synaptic Structural Changes after Injury
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LeDoux, J.E.; Moscarello, J.; Sears, R.; Campese, V. The birth, death and resurrection of avoidance: A reconceptualization of a troubled paradigm. Mol. Psychiatry 2017, 22, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Frankland, P.W.; Josselyn, S.A. Hippocampal Neurogenesis and Memory Clearance. Neuropsychopharmacology 2016, 41, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M. Cortical excitation and chronic pain. Trends Neurosci. 2008, 31, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tan, Q.; Cheng, D.; Zhang, L.; Zhang, J.; Gu, E.-W.; Fang, W.; Lu, X.; Liu, X. The changes of intrinsic excitability of pyramidal neurons in anterior cingulate cortex in neuropathic pain. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Zhuo, M. Long-term potentiation in the anterior cingulate cortex and chronic pain. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 369, 20130146. [Google Scholar] [CrossRef]
- Kang, S.J.; Kim, S.; Lee, J.; Kwak, C.; Lee, K.; Zhuo, M.; Kaang, B.-K. Inhibition of anterior cingulate cortex excitatory neuronal activity induces conditioned place preference in a mouse model of chronic inflammatory pain. Korean J. Physiol. Pharmacol. 2017, 21, 487. [Google Scholar] [CrossRef]
- Fuchs, P.N.; Peng, Y.B.; Boyette-Davis, J.A.; Uhelski, M.L. The anterior cingulate cortex and pain processing. Front Integr. Neurosci. 2014, 8, 35. [Google Scholar] [CrossRef]
- Sandkühler, J. Models and mechanisms of hyperalgesia and allodynia. Physiol. Rev. 2009, 89, 707–758. [Google Scholar] [CrossRef]
- Singh, A.; Patel, D.; Li, A.; Hu, L.; Zhang, Q.; Liu, Y.; Guo, X.; Robinson, E.; Martinez, E.; Doan, L.; et al. Mapping cortical integration of sensory and affective pain pathways. Curr. Biol. 2020, 30. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, H.; Sun, G.; Vemulapalli, B.; Jee, H.J.; Zhang, Q.; Wang, J. Frequency dependent electrical stimulation of PFC and ACC for acute pain treatment in rats. Front. Pain Res. 2021, 2, 728045. [Google Scholar] [CrossRef]
- Chen, T.; Taniguchi, W.; Chen, Q.-Y.; Tozaki-Saitoh, H.; Song, Q.; Liu, R.-H.; Koga, K.; Matsuda, T.; Kaito-Sugimura, Y.; Wang, J.; et al. Top-down descending facilitation of spinal sensory excitatory transmission from the anterior cingulate cortex. Nat. Commun. 2018, 9, 1886. [Google Scholar] [CrossRef] [PubMed]
- Calejesan, A.A.; Kim, S.J.; Zhuo, M. Descending facilitatory modulation of a behavioral nociceptive response by stimulation in the adult rat anterior cingulate cortex. Eur J. Pain 2000, 4, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Ko, S.; Ding, H.K.; Qiu, C.S.; Calejesan, A.A.; Zhuo, M. Pavlovian fear memory induced by activation in the anterior cingulate cortex. Mol. Pain 2005, 1, 6. [Google Scholar] [CrossRef]
- Zugaib, J.; Coutinho, M.R.; Ferreira, M.D.; Menescal-de-Oliveira, L. Glutamate/GABA balance in ACC modulates the nociceptive responses of vocalization: An expression of affective-motivational component of pain in guinea pigs. Physiol. Behav. 2014, 126, 8–14. [Google Scholar] [CrossRef]
- Ren, D.; Li, J.-N.; Qiu, X.-T.; Wan, F.-P.; Wu, Z.-Y.; Fan, B.-Y.; Zhang, M.-M.; Chen, T.; Li, H.; Bai, Y.; et al. Anterior cingulate cortex mediates hyperalgesia and anxiety induced by chronic pancreatitis in rats. Neurosci. Bull. 2021, 38, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, W.; Dong, Y.-L.; Zhang, M.-M.; Wang, J.; Koga, K.; Liao, Y.-H.; Li, J.-L.; Budisantoso, T.; Shigemoto, R.; et al. Postsynaptic insertion of AMPA receptor onto cortical pyramidal neurons in the anterior cingulate cortex after peripheral nerve injury. Mol. Brain 2014, 7, 76. [Google Scholar] [CrossRef]
- Zhao, R.; Zhou, H.; Huang, L.; Xie, Z.; Wang, J.; Gan, W.-B.; Yang, G. Neuropathic pain causes pyramidal neuronal hyperactivity in the anterior cingulate cortex. Front. Cell. Neurosci. 2018, 12, 107. [Google Scholar] [CrossRef]
- Brown, J.E.; Chatterjee, N.; Younger, J.; Mackey, S. Towards a physiology-based measure of pain: Patterns of human brain activity distinguish painful from non-painful thermal stimulation. PLoS ONE 2011, 6, e24124. [Google Scholar] [CrossRef]
- Ohara, S.; Crone, N.E.; Weiss, N.; Lenz, F.A. Analysis of synchrony demonstrates ‘pain networks’ defined by rapidly switching, task-specific, functional connectivity between pain-related cortical structures. Pain 2006, 123, 244–253. [Google Scholar] [CrossRef]
- Teutsch, S.; Herken, W.; Bingel, U.; Schoell, E.; May, A. Changes in brain gray matter due to repetitive painful stimulation. Neuroimage 2008, 42, 845–849. [Google Scholar] [CrossRef]
- Flor, H.; Nikolajsen, L.; Staehelin Jensen, T. Phantom limb pain: A case of maladaptive CNS plasticity? Nat. Rev. Neurosci. 2006, 7, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.X.; Yin, Y.; Xiao, H.; Lui, S.; Wen, C.B.; Dai, Y.E.; Yang, G.; Liu, J.; Gong, Q. Altered cortical reorganization and brain functional connectivity in Phantom limb pain: A functional MRI study. Pain Pract. 2020, 21, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Morgan, V.; Pickens, D.; Gautam, S.; Kessler, R.; Mertz, H. Amitriptyline reduces rectal pain related activation of the anterior cingulate cortex in patients with irritable bowel syndrome. Gut 2005, 54, 601–607. [Google Scholar] [CrossRef]
- Feitosa, A.A.; Amaro Junior, E.; Sanches, L.G.; Borba, E.F.; Jorge, L.L.; Halpern, A.S.R. Chronic low back pain and sick-leave: A functional magnetic resonance study. Adv. Rheumatol. 2020, 60, 46. [Google Scholar] [CrossRef] [PubMed]
- Ung, H.; Brown, J.E.; Johnson, K.A.; Younger, J.; Hush, J.; Mackey, S. Multivariate classification of structural MRI data detects chronic low back pain. Cereb. Cortex. 2014, 24, 1037–1044. [Google Scholar] [CrossRef]
- Hutchison, W.D.; Davis, K.D.; Lozano, A.M.; Tasker, R.R.; Dostrovsky, J.O. Pain-related neurons in the human cingulate cortex. Nat. Neurosci. 1999, 2, 403–405. [Google Scholar] [CrossRef]
- Rolls, E.T. The cingulate cortex and limbic systems for emotion, action, and memory. Brain Struct. Funct. 2019, 224, 3001–3018. [Google Scholar] [CrossRef]
- Stern, J.; Jeanmonod, D.; Sarnthein, J. Persistent EEG overactivation in the cortical pain matrix of neurogenic pain patients. Neuroimage 2006, 31, 721–731. [Google Scholar] [CrossRef]
- Mohseni, H.R.; Smith, P.P.; Parsons, C.E.; Young, K.S.; Hyam, J.A.; Stein, A.; Stein, J.F.; Green, A.L.; Aziz, T.Z.; Kringelbach, M.L. Meg can map short and long-term changes in brain activity following deep brain stimulation for chronic pain. PLoS ONE 2012, 7, e37993. [Google Scholar] [CrossRef]
- Terrasa, J.L.; Montoya, P.; Sitges, C.; van der Meulen, M.; Anton, F.; González-Roldán, A.M. Anterior Cingulate Cortex Activity During Rest Is Related to Alterations in Pain Perception in Aging. Front. Aging Neurosci. 2021, 13, 695200. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Rosenblum, K. Molecular mechanisms underlying memory consolidation of taste information in the cortex. Front. Behav. Neurosci. 2012, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Qadir, H.; Krimmel, S.R.; Mu, C.; Poulopoulos, A.; Seminowicz, D.A.; Mathur, B.N. Structural Connectivity of the Anterior Cingulate Cortex, Claustrum, and the Anterior Insula of the Mouse. Front. Neuroanat. 2018, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.D. Topographically organized projection to posterior insular cortex from the posterior portion of the ventral medial nucleus in the long-tailed macaque monkey. J. Comp. Neurol. 2014, 522, 36–63. [Google Scholar] [CrossRef]
- Peyron, R.; Fauchon, C. The posterior insular-opercular cortex: An access to the brain networks of thermosensory and nociceptive processes? Neurosci. Lett. 2019, 702, 34–39. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, Y.H.; Wang, J.Y.; Luo, F. Current Understanding of the Involvement of the Insular Cortex in Neuropathic Pain: A Narrative Review. Int J. Mol. Sci. 2021, 22, 2648. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, T.; Zhao, H.; Zhang, M.; Meng, F.; Fu, H.; Xie, Y.; Xu, H. Insular Cortex is critical for the perception, modulation, and Chronification of Pain. Neurosci. Bull. 2016, 32, 191–201. [Google Scholar] [CrossRef]
- Jonkman, L.E.; Fathy, Y.Y.; Berendse, H.W.; Schoonheim, M.M.; van de Berg, W.D.J. Structural network topology and microstructural alterations of the anterior insula associate with cognitive and affective impairment in Parkinson’s disease. Sci Rep. 2021, 11, 16021. [Google Scholar] [CrossRef]
- Fritz, H.-C.; McAuley, J.H.; Wittfeld, K.; Hegenscheid, K.; Schmidt, C.O.; Langner, S.; Lotze, M. Chronic back pain is associated with decreased prefrontal and anterior insular gray matter: Results from a population-based cohort study. J. Pain 2016, 17, 111–118. [Google Scholar] [CrossRef]
- Wei, F.; Li, P.; Zhuo, M. Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J. Neurosci. 1999, 19, 9346–9354. [Google Scholar] [CrossRef][Green Version]
- Tseng, K.Y.; O’Donnell, P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J. Neurosci. 2004, 24, 5131–5139. [Google Scholar] [CrossRef]
- Wu, L.J.; Zhao, M.G.; Toyoda, H.; Ko, S.W.; Zhuo, M. Kainate receptor-mediated synaptic transmission in the adult anterior cingulate cortex. J. Neurophysiol. 2005, 94, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Ko, S.W.; Zhuo, M. Kainate receptors and pain: From dorsal root ganglion to the anterior cingulate cortex. Curr. Pharm. Des. 2007, 13, 1597–1605. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kantrowitz, J.T.; Dong, Z.; Milak, M.S.; Rashid, R.; Kegeles, L.S.; Javitt, D.C.; Lieberman, J.A.; John Mann, J. Ventromedial prefrontal cortex/anterior cingulate cortex GLX, glutamate, and GABA levels in medication-free major depressive disorder. Transl. Psychiatry 2021, 11, 419. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Miao, Z.; Chen, Q.Y.; Li, X.H.; Zhuo, M. Multiple synaptic connections into a single cortical pyramidal cell or interneuron in the anterior cingulate cortex of adult mice. Mol. Brain. 2021, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Delevich, K.; Tucciarone, J.; Huang, Z.J.; Li, B. The mediodorsal thalamus drives feedforward inhibition in the anterior cingulate cortex via parvalbumin interneurons. J. Neurosci. 2015, 35, 5743–5753. [Google Scholar] [CrossRef] [PubMed]
- Ohara, P.T.; Granato, A.; Moallem, T.M.; Wang, B.R.; Tillet, Y.; Jasmin, L. Dopaminergic input to GABAergic neurons in the rostral agranular insular cortex of the rat. J. Neurocytol. 2003, 32, 131–141. [Google Scholar] [CrossRef]
- Wu, L.J.; Xu, H.; Ren, M.; Zhuo, M. Genetic and pharmacological studies of GluR5 modulation of inhibitory synaptic transmission in the anterior cingulate cortex of adult mice. Dev. Neurobiol. 2007, 67, 146–157. [Google Scholar] [CrossRef]
- Migita, K.; Matsuzaki, Y.; Koga, K.; Matsumoto, T.; Mishima, K.; Hara, S.; Honda, K. Involvement of GABA B receptor in the antihypersensitive effect in anterior cingulate cortex of partial sciatic nerve ligation model. J. Pharm. Sci. 2018, 137, 233–236. [Google Scholar] [CrossRef]
- Xu, H.; Wu, L.-J.; Wang, H.; Zhang, X.; Vadakkan, K.I.; Kim, S.S.; Steenland, H.W.; Zhuo, M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 2008, 28, 7445–7453. [Google Scholar] [CrossRef]
- Li, X.H.; Song, Q.; Chen, T.; Zhuo, M. Characterization of postsynaptic calcium signals in the pyramidal neurons of anterior cingulate cortex. Mol. Pain 2017, 13, 1744806917719847. [Google Scholar] [CrossRef]
- Ko, H.-G.; Ye, S.; Han, D.-H.; Park, P.; Lim, C.-S.; Lee, K.; Zhuo, M.; Kaang, B.-K. Transcription-independent expression of PKMζ in the anterior cingulate cortex contributes to chronically maintained neuropathic pain. Mol. Pain 2018, 14, 174480691878394. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Matsuura, T.; Liu, R.H.; Xue, M.; Zhuo, M. Calcitonin gene-related peptide potentiated the excitatory transmission and network propagation in the anterior cingulate cortex of adult mice. Mol. Pain 2019, 15, 1744806919832718. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Q.-Y.; Lee, J.H.; Li, X.-H.; Yu, S.; Zhuo, M. Cortical potentiation induced by calcitonin gene-related peptide (CGRP) in the insular cortex of adult mice. 2020, 36. Mol. Brain 2020, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.-H.; Miao, Z.; Pan, J.-G.; Li, X.-H.; Zhuo, M. Brain-derived neurotrophic factor produced long-term synaptic enhancement in the anterior cingulate cortex of adult mice. Mol. Brain 2021, 140. [Google Scholar] [CrossRef]
- Zhao, M.-G.; Toyoda, H.; Lee, Y.-S.; Wu, L.-J.; Ko, S.W.; Zhang, X.-H.; Jia, Y.; Shum, F.; Xu, H.; Li, B.-M.; et al. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and Contextual Fear Memory. Neuron 2005, 47, 859–872. [Google Scholar] [CrossRef]
- Grover, L.M. Evidence for postsynaptic induction and expression of NMDA receptor independent LTP. J. Neurophysiol. 1998, 79, 1167–1182. [Google Scholar] [CrossRef]
- Liauw, J.; Wu, L.J.; Zhuo, M. Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J. Neurophysiol. 2005, 94, 878–882. [Google Scholar] [CrossRef]
- Wei, F.; Qiu, C.-S.; Kim, S.J.; Muglia, L.; Maas, J.W.; Pineda, V.V.; Xu, H.-M.; Chen, Z.-F.; Storm, D.R.; Muglia, L.J.; et al. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 2002, 36, 713–726. [Google Scholar] [CrossRef]
- Wei, F.; Qiu, C.-S.; Liauw, J.; Robinson, D.A.; Ho, N.; Chatila, T.; Zhuo, M. Calcium–calmodulin-dependent protein kinase IV is required for fear memory. Nat. Neurosci. 2002, 5, 573–579. [Google Scholar] [CrossRef]
- Zhao, M.-G.; Ko, S.W.; Wu, L.-J.; Toyoda, H.; Xu, H.; Quan, J.; Li, J.; Jia, Y.; Ren, M.; Xu, Z.C.; et al. Enhanced presynaptic neurotransmitter release in the anterior cingulate cortex of mice with chronic pain. J. Neurosci. 2006, 26, 8923–8930. [Google Scholar] [CrossRef]
- Miao, H.H.; Li, X.H.; Chen, Q.Y.; Zhuo, M. Calcium-stimulated adenylyl cyclase subtype 1 is required for presynaptic long-term potentiation in the insular cortex of adult mice. Mol. Pain 2019, 15, 1744806919842961. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Zhao, M.-G.; Mercaldo, V.; Chen, T.; Descalzi, G.; Kida, S.; Zhuo, M. Calcium/calmodulin-dependent kinase IV contributes to translation-dependent early synaptic potentiation in the anterior cingulate cortex of adult mice. Mol. Brain 2010, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Wu, L.J.; Zhao, M.G.; Xu, H.; Zhuo, M. Time-dependent postsynaptic AMPA GluR1 receptor recruitment in the cingulate synaptic potentiation. Dev. Neurobiol. 2007, 67, 498–509. [Google Scholar] [CrossRef]
- Zhao, M.G.; Toyoda, H.; Ko, S.W.; Ding, H.K.; Wu, L.J.; Zhuo, M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 2005, 25, 7385–7392. [Google Scholar] [CrossRef]
- Patterson, M.; Yasuda, R. Signalling pathways underlying structural plasticity of dendritic spines. Br. J. Pharmacol. 2011, 163, 1626–1638. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 2011, 472, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Thibault, K.; Lin, W.K.; Rancillac, A.; Fan, M.; Snollaerts, T.; Sordoillet, V.; Hamon, M.; Smith, G.M.; Lenkei, Z.; Pezet, S. BDNF-dependent plasticity induced by peripheral inflammation in the primary sensory and the cingulate cortex triggers cold allodynia and reveals a major role for endogenous BDNF as a tuner of the affective aspect of pain. J. Neurosci. 2014, 34, 14739–14751. [Google Scholar] [CrossRef]
- Li, X.-Y.; Ko, H.-G.; Chen, T.; Descalzi, G.; Koga, K.; Wang, H.; Kim, S.S.; Shang, Y.; Kwak, C.; Park, S.-W.; et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMΖ in the anterior cingulate cortex. Science 2010, 330, 1400–1404. [Google Scholar] [CrossRef]
- Junho, C.V.C.; Caio-Silva, W.; Trentin-Sonoda, M.; Carneiro-Ramos, M.S. An Overview of the Role of Calcium/Calmodulin-Dependent Protein Kinase in Cardiorenal Syndrome. Front. Physiol. 2020, 11, 735. [Google Scholar] [CrossRef]
- Wayman, G.A.; Lee, Y.S.; Tokumitsu, H.; Silva, A.J.; Soderling, T.R. Calmodulin-kinases: Modulators of neuronal development and plasticity. Neuron 2008, 59, 914–931. [Google Scholar] [CrossRef]
- Baltaci, S.B.; Mogulkoc, R.; Baltaci, A.K. Molecular Mechanisms of Early and Late LTP. Neurochem Res. 2019, 44, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Bliss, T.; Collingridge, G.L. Persistent memories of long-term potentiation and the N-methyl-d-aspartate receptor. Brain Neurosci. Adv. 2019, 3, 2398212819848213. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L.; Kaang, B.K.; Zhuo, M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 2016, 17, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Koga, K.; Descalzi, G.; Qiu, S.; Wang, J.; Zhang, L.-S.; Zhang, Z.-J.; He, X.-B.; Qin, X.; Xu, F.-Q.; et al. Postsynaptic potentiation of corticospinal projecting neurons in the anterior cingulate cortex after nerve injury. Mol. Pain 2014, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; O’Den, G.; Song, Q.; Koga, K.; Zhang, M.M.; Zhuo, M. Adenylyl cyclase subtype 1 is essential for late-phase long term potentiation and spatial propagation of synaptic responses in the anterior cingulate cortex of adult mice. Mol. Pain 2014, 10, 65. [Google Scholar] [CrossRef]
- Song, Q.; Zheng, H.-W.; Li, X.-H.; Huganir, R.L.; Kuner, T.; Zhuo, M.; Chen, T. Selective phosphorylation of AMPA receptor contributes to the network of long-term potentiation in the anterior cingulate cortex. J. Neurosci. 2017, 37, 8534–8548. [Google Scholar] [CrossRef]
- Wen, J.; Xu, Y.; Yu, Z.; Zhou, Y.; Wang, W.; Yang, J.; Wang, Y.; Bai, Q.; Li, Z. The camp response element- binding protein/brain-derived neurotrophic factor pathway in anterior cingulate cortex regulates neuropathic pain and anxiodepression like behaviors in rats. Front. Mol. Neurosci. 2022, 15, 831151. [Google Scholar] [CrossRef]
- Shao, X.-M.; Sun, J.; Jiang, Y.-L.; Liu, B.-Y.; Shen, Z.; Fang, F.; Du, J.-Y.; Wu, Y.-Y.; Wang, J.-L.; Fang, J.-Q. Inhibition of the camp/PKA/CREB pathway contributes to the analgesic effects of electroacupuncture in the anterior cingulate cortex in a rat pain memory model. Neural Plast. 2016, 2016, 1–16. [Google Scholar] [CrossRef]
- Clem, R.L.; Huganir, R.L. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 2010, 330, 1108–1112. [Google Scholar] [CrossRef]
- Wang, H.X.; Gao, W.J. Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J. Physiol. 2010, 588, 2823–2838. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Zhao, M.G.; Zhuo, M. Roles of NMDA receptor NR2A and NR2B subtypes for long-term depression in the anterior cingulate cortex. Eur J. Neurosci. 2005, 22, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-J.; Toyoda, H.; Zhao, M.-G.; Lee, Y.-S.; Tang, J.; Ko, S.W.; Jia, Y.H.; Shum, F.W.F.; Zerbinatti, C.V.; Bu, G.; et al. Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J. Neurosci. 2005, 25, 11107–11116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Li, X.H.; Zhuo, M. NMDA receptors and synaptic plasticity in the anterior cingulate cortex. Neuropharmacology 2021, 197, 108749. [Google Scholar] [CrossRef] [PubMed]
- Klavir, O.; Genud-Gabai, R.; Paz, R. Low-frequency stimulation depresses the primate anterior-cingulate-cortex and prevents spontaneous recovery of aversive memories. J. Neurosci. 2012, 32, 8589–8597. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Launey, T.; Mikawa, S.; Hirai, H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000, 19, 2765–2774. [Google Scholar] [CrossRef]
- Ren, S.-Q.; Yan, J.-Z.; Zhang, X.-Y.; Bu, Y.-F.; Pan, W.-W.; Yao, W.; Tian, T.; Lu, W. PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. EMBO J. 2013, 32, 1365–1380. [Google Scholar] [CrossRef]
- Chiou, C.S.; Huang, C.C.; Liang, Y.C.; Tsai, Y.C.; Hsu, K.S. Impairment of long-term depression in the anterior cingulate cortex of mice with bone cancer pain. Pain 2012, 153, 2097–2108. [Google Scholar] [CrossRef]
- Hogrefe, N.; Blom, S.M.; Valentinova, K.; Ntamati, N.R.; Jonker, L.J.E.; Nevian, N.E.; Nevian, T. Long-lasting, pathway-specific impairment of a novel form of spike-timing-dependent long-term depression by neuropathic pain in the anterior cingulate cortex. J. Neurosci. 2022, 42, 2166–2179. [Google Scholar] [CrossRef]
- Xue, M.; Zhou, S.B.; Liu, R.H.; Chen, Q.Y.; Zhuo, M.; Li, X.H. NMDA Receptor-Dependent Synaptic Depression in Potentiated Synapses of the Anterior Cingulate Cortex of adult Mice. Mol. Pain 2021, 17, 17448069211018045. [Google Scholar] [CrossRef]
- Kang, S.J.; Liu, M.-G.; Chen, T.; Ko, H.-G.; Baek, G.-C.; Lee, H.-R.; Lee, K.; Collingridge, G.L.; Kaang, B.-K.; Zhuo, M. Plasticity of metabotropic glutamate receptor-dependent long-term depression in the anterior cingulate cortex after amputation. J. Neurosci. 2012, 32, 11318–11329. [Google Scholar] [CrossRef]
- Pick, J.E.; Khatri, L.; Sathler, M.F.; Ziff, E.B. mGluR long-term depression regulates GluA2 association with COPII vesicles and exit from the endoplasmic reticulum. EMBO J. 2017, 36, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Yao, I.; Setou, M.; Zhuo, M. SCRAPPER Selectively Contributes to Spontaneous Release and Presynaptic Long-Term Potentiation in the Anterior Cingulate Cortex. J. Neurosci. 2017, 37, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Perrone-Capano, C.; Volpicelli, F.; Penna, E.; Chun, J.T.; Crispino, M. Presynaptic protein synthesis and brain plasticity: From physiology to neuropathology. Prog. Neurobiol. 2021, 202, 102051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-T.; Shen, F.-Y.; Ma, L.-Q.; Wen, W.; Wang, B.; Peng, Y.-Z.; Wang, Z.-R.; Zhao, X. Potentiation of synaptic transmission in rat anterior cingulate cortex by chronic itch. Mol. Brain 2016, 9, 73. [Google Scholar] [CrossRef]
- Kanold, P.O.; Deng, R.; Meng, X. The Integrative Function of Silent Synapses on Subplate Neurons in Cortical Development and Dysfunction. Front. Neuroanat. 2019, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.E.; Paylor, J.W.; Suh, J.S.; Tenorio, G.; Caliaperumal, J.; Colbourne, F.; Baker, G.; Winship, I.; Kerr, B.J. Altered excitatory-inhibitory balance within somatosensory cortex is associated with enhanced plasticity and pain sensitivity in a mouse model of multiple sclerosis. J. Neuroinflammation 2016, 13, 142. [Google Scholar] [CrossRef]
- Cheriyan, J.; Sheets, P.L. Peripheral nerve injury reduces the excitation-inhibition balance of basolateral amygdala inputs to prelimbic pyramidal neurons projecting to the periaqueductal gray. Mol. Brain 2020, 13, 100. [Google Scholar] [CrossRef]
- Han, K.; Lee, M.; Lim, H.-K.; Jang, M.W.; Kwon, J.; Lee, C.J.; Kim, S.-G.; Suh, M. Excitation-inhibition imbalance leads to alteration of neuronal coherence and neurovascular coupling under acute stress. J. Neurosci. 2020, 40, 9148–9162. [Google Scholar] [CrossRef]
- Markicevic, M.; Fulcher, B.D.; Lewis, C.; Helmchen, F.; Rudin, M.; Zerbi, V.; Wenderoth, N. Cortical excitation:inhibition imbalance causes abnormal brain network dynamics as observed in neurodevelopmental disorders. Cereb. Cortex 2020, 30, 4922–4937. [Google Scholar] [CrossRef]
- Yao, I.; Takagi, H.; Ageta, H.; Kahyo, T.; Sato, S.; Hatanaka, K.; Fukuda, Y.; Chiba, T.; Morone, N.; Yuasa, S.; et al. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell 2007, 130, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Frey, U.; Morris, R.G. Synaptic tagging and long-term potentiation. Nature 1997, 385, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Redondo, R.L.; Morris, R.G. Making memories last: The synaptic tagging and capture hypothesis. Nat. Rev. Neurosci. 2011, 12, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Højgaard, K.; Privitera, L.; Bayraktar, G.; Takeuchi, T. Initial memory consolidation and the synaptic tagging and capture hypothesis. Eur J. Neurosci. 2021, 54, 6826–6849. [Google Scholar] [CrossRef]
- Nomoto, M.; Inokuchi, K. Behavioral, cellular, and synaptic tagging frameworks. Neurobiol Learn. Mem. 2018, 153, 13–20. [Google Scholar] [CrossRef]
- Gröger, N.; Mannewitz, A.; Bock, J.; Becker, S.; Guttmann, K.; Poeggel, G.; Braun, K. Infant avoidance training alters cellular activation patterns in prefronto-limbic circuits during adult avoidance learning: II. Cellular imaging of neurons expressing the activity-regulated cytoskeleton-associated protein (Arc/Arg3.1). Brain Struct. Funct. 2018, 223, 713–725. [Google Scholar] [CrossRef]
- Rogerson, T.; Cai, D.J.; Frank, A.; Sano, Y.; Shobe, J.; Lopez-Aranda, M.F.; Silva, A.J. Synaptic tagging during memory allocation. Nat. Rev. Neurosci. 2014, 15, 157–169. [Google Scholar] [CrossRef]
- Liu, M.G.; Song, Q.; Zhuo, M. Loss of Synaptic Tagging in the Anterior Cingulate Cortex after Tail Amputation in Adult Mice. J. Neurosci. 2018, 38, 8060–8070. [Google Scholar] [CrossRef]
- Zhuo, M. Neural Mechanisms Underlying Anxiety-Chronic Pain Interactions. Trends Neurosci. 2016, 39, 136–145. [Google Scholar] [CrossRef]
- Zheng, H.; Schnabel, A.; Yahiaoui-Doktor, M.; Meissner, W.; Van Aken, H.; Zahn, P.; Pogatzki-Zahn, E. Age and preoperative pain are major confounders for sex differences in postoperative pain outcome: A prospective database analysis. PLoS ONE 2017, 12, e0178659. [Google Scholar] [CrossRef]
- Bartley, E.J.; Fillingim, R.B. Sex differences in pain: A brief review of clinical and experimental findings. Br. J. Anaesth. 2013, 111, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Rea, B.J.; Wattiez, A.-S.; Waite, J.S.; Castonguay, W.C.; Schmidt, C.M.; Fairbanks, A.M.; Robertson, B.R.; Brown, C.J.; Mason, B.N.; Moldovan-Loomis, M.-C.; et al. Peripherally administered calcitonin gene–related peptide induces spontaneous pain in mice: Implications for Migraine. Pain 2018, 159, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Baratta, M.V.; Gruene, T.M.; Dolzani, S.D.; Chun, L.E.; Maier, S.F.; Shansky, R.M. Controllable stress elicits circuit-specific patterns of prefrontal plasticity in males, but not females. Brain Struct Funct. 2019, 224, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Sorge, R.E.; Mapplebeck, J.C.; Rosen, S.; Beggs, S.; Taves, S.; Alexander, J.K.; Martin, L.J.; Austin, J.-S.; Sotocinal, S.G.; Chen, D.; et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015, 18, 1081–1083. [Google Scholar] [CrossRef]
- Rosen, S.F.; Ham, B.; Drouin, S.; Boachie, N.; Chabot-Dore, A.-J.; Austin, J.-S.; Diatchenko, L.; Mogil, J.S. T-cell mediation of pregnancy analgesia affecting chronic pain in mice. J. Neurosci. 2017, 37, 9819–9827. [Google Scholar] [CrossRef]
- Dachtler, J.; Fox, K. Do cortical plasticity mechanisms differ between males and females? J. Neurosci. Res. 2017, 95, 518–526. [Google Scholar] [CrossRef]
- Hyer, M.M.; Phillips, L.L.; Neigh, G.N. Sex Differences in Synaptic Plasticity: Hormones and Beyond. Front. Mol. Neurosci. 2018, 11, 266. [Google Scholar] [CrossRef]
- Liu, R.H.; Xue, M.; Li, X.H.; Zhuo, M. Sex difference in synaptic plasticity in the anterior cingulate cortex of adult mice. Mol. Brain 2020, 13, 41. [Google Scholar] [CrossRef]
- Dachtler, J.; Hardingham, N.R.; Fox, K. The role of nitric oxide synthase in cortical plasticity is sex specific. J. Neurosci. 2012, 32, 14994–14999. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, K.; Xu, T.; Yamaki, V.N.; Wei, Z.; Huang, M.; Rose, G.M.; Cai, X. Sex differences in long-term potentiation at temporoammonic-CA1 synapses: Potential implications for memory consolidation. PLoS ONE 2016, 11, e0165891. [Google Scholar] [CrossRef]
- Zhou, Z.; Shi, W.; Fan, K.; Xue, M.; Zhou, S.; Chen, Q.-Y.; Lu, J.-S.; Li, X.-H.; Zhuo, M. Inhibition of calcium-stimulated adenylyl cyclase subtype 1 (AC1) for the treatment of neuropathic and inflammatory pain in adult female mice. Mol. Pain 2021, 17, 174480692110216. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Z.; Zhang, X.; Li, S.; Wu, W.; Li, X.; Yang, Y. A sex-dependent delayed maturation of visual plasticity induced by adverse experiences in early childhood. Neurobiol. Stress. 2020, 13, 100256. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Huang, G.Z.; Woolley, C.S. Latent Sex Differences in Molecular Signaling That Underlies Excitatory Synaptic Potentiation in the Hippocampus. J. Neurosci. 2019, 39, 1552–1565. [Google Scholar] [CrossRef]
- Bender, R.A.; Zhou, L.; Vierk, R.; Brandt, N.; Keller, A.; Gee, C.E.; Schäfer, M.K.E.; Rune, G.M. Sex-dependent regulation of aromatase-mediated synaptic plasticity in the basolateral amygdala. J. Neurosci. 2016, 37, 1532–1545. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.-J.; Miao, W.; Zhang, X.; Zheng, J.-J.; Wang, C.; Yu, X. Oxytocin regulates synaptic transmission in the sensory cortices in a developmentally dynamic manner. Front. Cell. Neurosci. 2021, 15, 673439. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Matsuura, T.; Xue, M.; Chen, Q.-Y.; Liu, R.-H.; Lu, J.-S.; Shi, W.; Fan, K.; Zhou, Z.; Miao, Z.; et al. Oxytocin in the anterior cingulate cortex attenuates neuropathic pain and emotional anxiety by inhibiting presynaptic long-term potentiation. Cell Rep. 2021, 36, 109411. [Google Scholar] [CrossRef]
- Cavalli, J.; Ruttorf, M.; Pahi, M.R.; Zidda, F.; Flor, H.; Nees, F. Oxytocin differentially modulates pavlovian cue and context fear acquisition. Soc. Cogn. Affect. Neurosci. 2017, 12, 976–983. [Google Scholar] [CrossRef]
- Esmaeilou, Y.; Tamaddonfard, E.; Erfanparast, A.; Soltanalinejad-Taghiabad, F. Behavioral and receptor expression studies on the primary somatosensory cortex and anterior cingulate cortex oxytocin involvement in modulation of sensory and affective dimensions of neuropathic pain induced by partial sciatic nerve ligation in rats. Physiol. Behav. 2022, 251, 113818. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Q.; Tian, R.; Wen, Q.; Qin, G.; Zhang, D.; Chen, L.; Zhang, Y.; Zhou, J. Repeated oxytocin prevents central sensitization by regulating synaptic plasticity via oxytocin receptor in a chronic migraine mouse model. J. Headache Pain 2021, 22, 84. [Google Scholar] [CrossRef]
- Crane, J.W.; Holmes, N.M.; Fam, J.; Westbrook, R.F.; Delaney, A.J. Oxytocin increases inhibitory synaptic transmission and blocks development of long-term potentiation in the lateral amygdala. J. Neurophysiol. 2020, 123, 587–599. [Google Scholar] [CrossRef]
- Koga, K.; Yamada, A.; Song, Q.; Li, X.-H.; Chen, Q.-Y.; Liu, R.-H.; Ge, J.; Zhan, C.; Furue, H.; Zhuo, M.; et al. Ascending noradrenergic excitation from the locus coeruleus to the anterior cingulate cortex. Mol. Brain 2020, 13, 49. [Google Scholar] [CrossRef]
- Pereira-Silva, R.; Costa-Pereira, J.T.; Alonso, R.; Serrão, P.; Martins, I.; Neto, F.L. Attenuation of the Diffuse Noxious Inhibitory Controls in Chronic Joint Inflammatory Pain Is Accompanied by Anxiodepressive-Like Behaviors and Impairment of the Descending Noradrenergic Modulation. Int. J. Mol. Sci. 2020, 21, 2973. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zuo, Z.X.; Wu, C.; Liu, L.; Feng, Z.H.; Li, X.Y. Cingulate Alpha-2A Adrenoceptors Mediate the Effects of Clonidine on Spontaneous Pain Induced by Peripheral Nerve Injury. Front. Mol. Neurosci. 2017, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Bravo, L.; Mico, J.A.; Rey-Brea, R.; Camarena-Delgado, C.; Berrocoso, E. Effect of DSP4 and desipramine in the sensorial and affective component of neuropathic pain in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 70, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Takeda, R.; Watanabe, Y.; Ikeda, T.; Abe, H.; Ebihara, K.; Matsuo, H.; Nonaka, H.; Hashiguchi, H.; Nishimori, T.; Ishida, Y. Analgesic effect of Milnacipran is associated with c-fos expression in the anterior cingulate cortex in the rat neuropathic pain model. Neurosci. Res. 2009, 64, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Yamanaka, M.; Bernabucci, M.; Zhao, M.G.; Zhuo, M. Characterization of serotonin-induced inhibition of excitatory synaptic transmission in the anterior cingulate cortex. Mol. Brain 2017, 10, 21. [Google Scholar] [CrossRef]
- Rovira, V.; Geijo-Barrientos, E. Intra- and Interhemispheric Propagation of Electrophysiological Synchronous Activity and Its Modulation by Serotonin in the Cingulate Cortex of Juvenile Mice. PLoS ONE 2016, 11, e0150092. [Google Scholar] [CrossRef]
- Santello, M.; Nevian, T. Dysfunction of cortical dendritic integration in neuropathic pain reversed by serotoninergic neuromodulation. Neuron 2015, 86, 233–246. [Google Scholar] [CrossRef]
- Santello, M.; Bisco, A.; Nevian, N.E.; Lacivita, E.; Leopoldo, M.; Nevian, T. The brain-penetrant 5-HT7 receptor agonist LP-211 reduces the sensory and affective components of neuropathic pain. Neurobiol. Dis. 2017, 106, 214–221. [Google Scholar] [CrossRef]
- Ramírez, D.; Zúñiga, R.; Concha, G.; Zúñiga, L. HCN Channels: New Therapeutic Targets for Pain Treatment. Molecules 2018, 23, 2094. [Google Scholar] [CrossRef]
- Ishikawa, T.; Yasuda, S.; Minoda, S.; Ibuki, T.; Fukuhara, K.; Iwanaga, Y.; Ariyoshi, T.; Sasaki, H. Neurotropin® ameliorates chronic pain via induction of brain-derived neurotrophic factor. Cell. Mol. Neurobiol. 2014, 35, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Lançon, K.; Qu, C.; Navratilova, E.; Porreca, F.; Séguéla, P. Decreased dopaminergic inhibition of pyramidal neurons in anterior cingulate cortex maintains chronic neuropathic pain. Cell Rep. 2021, 37, 109933. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.B. Role of central dopamine in pain and analgesia. Expert Rev. Neurother. 2008, 8, 781–797. [Google Scholar] [CrossRef]
- Darvish-Ghane, S.; Yamanaka, M.; Zhuo, M. Dopaminergic Modulation of Excitatory Transmission in the Anterior Cingulate Cortex of Adult Mice. Mol. Pain 2016, 12, 1744806916648153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Q.; Lin, W.-P.; Huang, L.-P.; Zhao, B.; Zhang, C.-C.; Yin, D.-M. Dopamine D2 receptor regulates cortical synaptic pruning in rodents. Nat. Commun. 2021, 12, 6444. [Google Scholar] [CrossRef]
- Darvish-Ghane, S.; Quintana, C.; Beaulieu, J.-M.; Martin, L.J. D1 receptors in the anterior cingulate cortex modulate basal mechanical sensitivity threshold and glutamatergic synaptic transmission. Mol. Brain 2020, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Li, M.; Zhou, Y.; Ma, L.; Qiao, Q.; Hu, W.; Li, W.; Wills, Z.P.; Gan, W.-B. Abnormal dendritic calcium activity and synaptic depotentiation occur early in a mouse model of alzheimer’s disease. Mol. Neurodegener. 2017, 12. [Google Scholar] [CrossRef]
- Short, B. How dendritic spines shape calcium dynamics. J. Gen. Physiol. 2019, 151, 970. [Google Scholar] [CrossRef]
- Huang, L.; Jin, J.; Chen, K.; You, S.; Zhang, H.; Sideris, A.; Norcini, M.; Recio-Pinto, E.; Wang, J.; Gan, W.-B.; et al. BDNF produced by Cerebral Microglia promotes cortical plasticity and pain hypersensitivity after peripheral nerve injury. PLoS Biol. 2021, 19, e3001337. [Google Scholar] [CrossRef]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2017, 18, 113. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.A.; Chen, Q.; Zhuo, M. Synaptic Plasticity in the Pain-Related Cingulate and Insular Cortex. Biomedicines 2022, 10, 2745. https://doi.org/10.3390/biomedicines10112745
Lee J-HA, Chen Q, Zhuo M. Synaptic Plasticity in the Pain-Related Cingulate and Insular Cortex. Biomedicines. 2022; 10(11):2745. https://doi.org/10.3390/biomedicines10112745
Chicago/Turabian StyleLee, Jung-Hyun Alex, Qiyu Chen, and Min Zhuo. 2022. "Synaptic Plasticity in the Pain-Related Cingulate and Insular Cortex" Biomedicines 10, no. 11: 2745. https://doi.org/10.3390/biomedicines10112745
APA StyleLee, J.-H. A., Chen, Q., & Zhuo, M. (2022). Synaptic Plasticity in the Pain-Related Cingulate and Insular Cortex. Biomedicines, 10(11), 2745. https://doi.org/10.3390/biomedicines10112745