
Cellular Models of Alpha-Synuclein Aggregation: What Have We Learned and Implications for Future Study
 , , and
, , and
Abstract
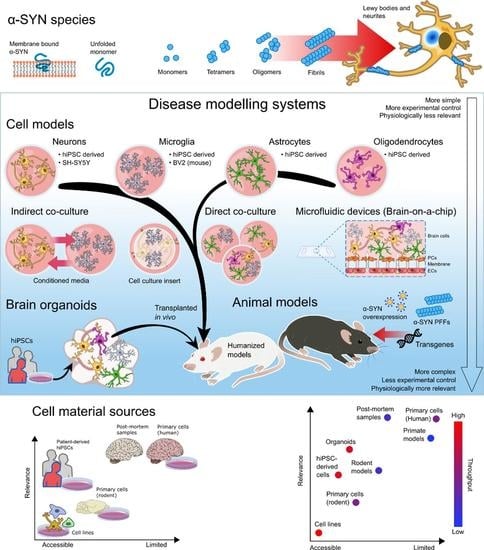
1. Introduction
2. Modeling Alpha-Synuclein Aggregation in Non-hiPSC Lines: What We Can Learn and Caution with Outcome Measures
3. Modeling Alpha-Synuclein Aggregation in hiPSC-Derived Neurons: Opportunities and Challenges
4. Effects of Alpha-Synuclein on Microglia
5. Modeling Alpha-Synuclein Pathology Using Astrocytes
6. Role of the Blood–Brain Barrier in Alpha-Synuclein Pathology
6.1. In Vivo Models
6.2. Human in Vitro Models
7. Oligodendrocytes and Synucleinopathy
8. HiPSC-Derived Brain Organoids as an Emerging Modeling Platform for Synucleinopathies
9. Conclusions and Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Lee, V.M.-Y.; Trojanowski, J.Q. Synucleinopathies: Clinical and Pathological Implications. Arch. Neurol. 2001, 58, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Lippa, C.F.; Duda, J.E.; Grossman, M.; Hurtig, H.I.; Aarsland, D.; Boeve, B.F.; Brooks, D.J.; Dickson, D.W.; Dubois, B.; Emre, M.; et al. DLB and PDD Boundary Issues: Diagnosis, Treatment, Molecular Pathology, and Biomarkers. Neurology 2007, 68, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous Alpha-Synuclein Inclusions Link Multiple System Atrophy with Parkinson’s Disease and Dementia with Lewy Bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Kasen, A.; Houck, C.; Burmeister, A.R.; Sha, Q.; Brundin, L.; Brundin, P. Upregulation of α-Synuclein Following Immune Activation: Possible Trigger of Parkinson’s Disease. Neurobiol. Dis. 2022, 166, 105654. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of α-Synuclein Binding and Conformational Switching Probed by Single-Molecule Fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef]
- Frimpong, A.K.; Abzalimov, R.R.; Uversky, V.N.; Kaltashov, I.A. Characterization of Intrinsically Disordered Proteins with Electrospray Ionization Mass Spectrometry: Conformational Heterogeneity of α-Synuclein. Proteins 2010, 78, 714–722. [Google Scholar] [CrossRef]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural Disorder of Monomeric α-Synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-Synuclein Oligomers and Fibrils: A Spectrum of Species, a Spectrum of Toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of Alpha-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Jakes, R.; Curran, M.D.; Wallace, A. Effects of the Mutations Ala30 to Pro and Ala53 to Thr on the Physical and Morphological Properties of Alpha-Synuclein Protein Implicated in Parkinson’s Disease. FEBS Lett. 1998, 440, 67–70. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The Many Faces of α-Synuclein: From Structure and Toxicity to Therapeutic Target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically Disordered Proteins: Regulation and Disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein Aggregation Nucleates through Liquid–Liquid Phase Separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein Misfolding and Parkinson’s Disease. Biochim. Et Biophys. Acta (BBA)–Mol. Basis Dis. 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.T.; Jagannath, S.; Francois, C.; Vanderstichele, H.; Stoops, E.; Lashuel, H.A. How Specific Are the Conformation-Specific α-Synuclein Antibodies? Characterization and Validation of 16 α-Synuclein Conformation-Specific Antibodies Using Well-Characterized Preparations of α-Synuclein Monomers, Fibrils and Oligomers with Distinct Structures and Morphology. Neurobiol. Dis. 2020, 146, 105086. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro Mutation in the Gene Encoding Alpha-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-Synuclein Mutation A53E Associated with Atypical Multiple System Atrophy and Parkinson’s Disease-Type Pathology. Neurobiol. Aging 2014, 35, 2180.e1-5. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841–841. [Google Scholar] [CrossRef]
- Sahay, S.; Ghosh, D.; Singh, P.K.; Maji, S.K. Alteration of Structure and Aggregation of α-Synuclein by Familial Parkinson’s Disease Associated Mutations. Curr. Protein Pept. Sci. 2017, 18, 656–676. [Google Scholar] [CrossRef]
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic Comparison of the Effects of Alpha-Synuclein Mutations on Its Oligomerization and Aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef]
- Ranjan, P.; Kumar, A. Perturbation in Long-Range Contacts Modulates the Kinetics of Amyloid Formation in α-Synuclein Familial Mutants. ACS Chem. Neurosci. 2017, 8, 2235–2246. [Google Scholar] [CrossRef]
- Zhou, T.; Lin, D.; Chen, Y.; Peng, S.; Jing, X.; Lei, M.; Tao, E.; Liang, Y. α-Synuclein Accumulation in SH-SY5Y Cell Impairs Autophagy in Microglia by Exosomes Overloading MiR-19a-3p. Epigenomics 2019, 11, 1661–1677. [Google Scholar] [CrossRef]
- Oliveira, S.R.; Dionísio, P.A.; Gaspar, M.M.; Correia Guedes, L.; Coelho, M.; Rosa, M.M.; Ferreira, J.J.; Amaral, J.D.; Rodrigues, C.M.P. MiR-335 Targets LRRK2 and Mitigates Inflammation in Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 661461. [Google Scholar] [CrossRef]
- Risiglione, P.; Cubisino, S.A.M.; Lipari, C.L.R.; De Pinto, V.; Messina, A.; Magrì, A. α-Synuclein A53T Promotes Mitochondrial Proton Gradient Dissipation and Depletion of the Organelle Respiratory Reserve in a Neuroblastoma Cell Line. Life 2022, 12, 894. [Google Scholar] [CrossRef]
- Potdar, C.; Kaushal, A.; Raj, A.; Mallick, R.; Datta, I. Reduction of Phosphorylated α-Synuclein through Downregulation of Casein Kinase 2α Alleviates Dopaminergic-Neuronal Function. Biochem. Biophys. Res. Commun. 2022, 615, 43–48. [Google Scholar] [CrossRef]
- Hivare, P.; Gadhavi, J.; Bhatia, D.; Gupta, S. α-Synuclein Fibrils Explore Actin-Mediated Macropinocytosis for Cellular Entry into Model Neuroblastoma Neurons. Traffic 2022, 23, 391–410. [Google Scholar] [CrossRef]
- Sang, J.C.; Hidari, E.; Meisl, G.; Ranasinghe, R.T.; Spillantini, M.G.; Klenerman, D. Super-Resolution Imaging Reveals α-Synuclein Seeded Aggregation in SH-SY5Y Cells. Commun. Biol. 2021, 4, 613. [Google Scholar] [CrossRef]
- Shearer, L.J.; Petersen, N.O.; Woodside, M.T. Internalization of α-Synuclein Oligomers into SH-SY5Y Cells. Biophys. J. 2021, 120, 877–885. [Google Scholar] [CrossRef]
- Li, S.; Raja, A.; Noroozifar, M.; Kerman, K. Understanding the Inhibitory and Antioxidant Effects of Pyrroloquinoline Quinone (PQQ) on Copper(II)-Induced α-Synuclein-119 Aggregation. ACS Chem. Neurosci. 2022, 13, 1178–1186. [Google Scholar] [CrossRef]
- Wang, H.; Tang, C.; Jiang, Z.; Zhou, X.; Chen, J.; Na, M.; Shen, H.; Lin, Z. Glutamine Promotes Hsp70 and Inhibits α-Synuclein Accumulation in Pheochromocytoma PC12 Cells. Exp. Med. 2017, 14, 1253–1259. [Google Scholar] [CrossRef][Green Version]
- Ortega, R.; Carmona, A.; Roudeau, S.; Perrin, L.; Dučić, T.; Carboni, E.; Bohic, S.; Cloetens, P.; Lingor, P. α-Synuclein Over-Expression Induces Increased Iron Accumulation and Redistribution in Iron-Exposed Neurons. Mol. Neurobiol. 2016, 53, 1925–1934. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Cooney, A.; Sullivan, P.; Sharabi, Y.; Goldstein, D.S. The Serotonin Aldehyde, 5-HIAL, Oligomerizes Alpha-Synuclein. Neurosci. Lett. 2015, 590, 134–137. [Google Scholar] [CrossRef][Green Version]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome Impairment by α-Synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.; Dargahi, L.; Parsafar, S.; Tayaranian Marvian, A.; Aliakbari, F.; Morshedi, D. The Primary Neuronal Cells Are More Resistant than PC12 Cells to α-Synuclein Toxic Aggregates. Neurosci. Lett. 2019, 701, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hornedo-Ortega, R.; Cerezo, A.B.; Troncoso, A.M.; Garcia-Parrilla, M.C. Protective Effects of Hydroxytyrosol against α-Synuclein Toxicity on PC12 cells and Fibril Formation. Food Chem. Toxicol. 2018, 120, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Karmacharya, M.B.; Hada, B.; Park, S.R.; Choi, B.H. Low-Intensity Ultrasound Decreases α-Synuclein Aggregation via Attenuation of Mitochondrial Reactive Oxygen Species in MPP(+)-Treated PC12 Cells. Mol. Neurobiol. 2017, 54, 6235–6244. [Google Scholar] [CrossRef] [PubMed]
- Höllerhage, M.; Stepath, M.; Kohl, M.; Pfeiffer, K.; Chua, O.W.H.; Duan, L.; Hopfner, F.; Eisenacher, M.; Marcus, K.; Höglinger, G.U. Transcriptome and Proteome Analysis in LUHMES Cells Overexpressing Alpha-Synuclein. Front. Neurol. 2022, 13, 787059. [Google Scholar] [CrossRef]
- Prahl, J.; Pierce, S.E.; Coetzee, G.A.; Tyson, T. Alpha-Synuclein Negatively Controls Cell Proliferation in Dopaminergic Neurons. Mol. Cell. Neurosci. 2022, 119, 103702. [Google Scholar] [CrossRef]
- Höllerhage, M.; Moebius, C.; Melms, J.; Chiu, W.-H.; Goebel, J.N.; Chakroun, T.; Koeglsperger, T.; Oertel, W.H.; Rösler, T.W.; Bickle, M.; et al. Protective Efficacy of Phosphodiesterase-1 Inhibition against Alpha-Synuclein Toxicity Revealed by Compound Screening in LUHMES Cells. Sci. Rep. 2017, 7, 11469. [Google Scholar] [CrossRef]
- Shaffner, K. Alpha-Synuclein Overexpression Induces Epigenomic Dysregulation of Glutamate Signaling and Locomotor Pathways|Human Molecular Genetics|Oxford Academic. Available online: https://academic.oup.com/hmg/advance-article/doi/10.1093/hmg/ddac104/6585888?login=true (accessed on 6 July 2022).
- Lv, Y.-C.; Gao, A.-B.; Yang, J.; Zhong, L.-Y.; Jia, B.; Ouyang, S.-H.; Gui, L.; Peng, T.-H.; Sun, S.; Cayabyab, F.S. Long-Term Adenosine A1 Receptor Activation-Induced Sortilin Expression Promotes α-Synuclein Upregulation in Dopaminergic Neurons. Neural. Regen. Res. 2019, 15, 712–723. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, N.; Hou, X.; Zhu, L.; Xie, Y.; Ba, Z.; Luo, Y. Icaritin Attenuates 6-OHDA-Induced MN9D Cell Damage by Inhibiting Oxidative Stress. PeerJ. 2022, 10, e13256. [Google Scholar] [CrossRef]
- Qiu, W.-Q.; Ai, W.; Zhu, F.-D.; Zhang, Y.; Guo, M.-S.; Law, B.Y.-K.; Wu, J.-M.; Wong, V.K.-W.; Tang, Y.; Yu, L.; et al. Polygala Saponins Inhibit NLRP3 Inflammasome-Mediated Neuroinflammation via SHP-2-Mediated Mitophagy. Free Radic. Biol. Med. 2022, 179, 76–94. [Google Scholar] [CrossRef]
- Yang, X.; Ma, H.; Yv, Q.; Ye, F.; He, Z.; Chen, S.; Keram, A.; Li, W.; Zhu, M. Alpha-Synuclein/MPP+ Mediated Activation of NLRP3 Inflammasome through Microtubule-Driven Mitochondrial Perinuclear Transport. Biochem. Biophys. Res. Commun. 2022, 594, 161–167. [Google Scholar] [CrossRef]
- Li, N.; Stewart, T.; Sheng, L.; Shi, M.; Cilento, E.M.; Wu, Y.; Hong, J.-S.; Zhang, J. Immunoregulation of Microglial Polarization: An Unrecognized Physiological Function of α-Synuclein. J. Neuroinflammation 2020, 17, 272. [Google Scholar] [CrossRef]
- Tu, H.; Yuan, B.; Hou, X.; Zhang, X.; Pei, C.; Ma, Y.; Yang, Y.; Fan, Y.; Qin, Z.; Liu, C.; et al. A-synuclein Suppresses Microglial Autophagy and Promotes Neurodegeneration in a Mouse Model of Parkinson’s Disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.-C.; von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C.M. Alpha-Synuclein Activates BV2 Microglia Dependent on Its Aggregation State. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef]
- Jovanovic, P.; Wang, Y.; Vit, J.-P.; Novinbakht, E.; Morones, N.; Hogg, E.; Tagliati, M.; Riera, C.E. Sustained Chemogenetic Activation of Locus Coeruleus Norepinephrine Neurons Promotes Dopaminergic Neuron Survival in Synucleinopathy. PLoS ONE 2022, 17, e0263074. [Google Scholar] [CrossRef]
- Szegö, E.M.; Van den Haute, C.; Höfs, L.; Baekelandt, V.; Van der Perren, A.; Falkenburger, B.H. Rab7 Reduces α-Synuclein Toxicity in Rats and Primary Neurons. Exp. Neurol. 2022, 347, 113900. [Google Scholar] [CrossRef]
- Nicholatos, J.W.; Tran, D.; Liu, Y.; Hirst, W.D.; Weihofen, A. Lysophosphatidylcholine Acyltransferase 1 Promotes Pathology and Toxicity in Two Distinct Cell-Based Alpha-Synuclein Models. Neurosci. Lett. 2022, 772, 136491. [Google Scholar] [CrossRef]
- Brzozowski, C.F.; Hijaz, B.A.; Singh, V.; Gcwensa, N.Z.; Kelly, K.; Boyden, E.S.; West, A.B.; Sarkar, D.; Volpicelli-Daley, L.A. Inhibition of LRRK2 Kinase Activity Promotes Anterograde Axonal Transport and Presynaptic Targeting of α-Synuclein. Acta Neuropathol. Commun. 2021, 9, 180. [Google Scholar] [CrossRef]
- Hlushchuk, I.; Barut, J.; Airavaara, M.; Luk, K.; Domanskyi, A.; Chmielarz, P. Cell Culture Media, Unlike the Presence of Insulin, Affect α-Synuclein Aggregation in Dopaminergic Neurons. Biomolecules 2022, 12, 563. [Google Scholar] [CrossRef]
- Hideshima, M.; Kimura, Y.; Aguirre, C.; Kakuda, K.; Takeuchi, T.; Choong, C.-J.; Doi, J.; Nabekura, K.; Yamaguchi, K.; Nakajima, K.; et al. Two-Step Screening Method to Identify α-Synuclein Aggregation Inhibitors for Parkinson’s Disease. Sci. Rep. 2022, 12, 351. [Google Scholar] [CrossRef]
- Szegő, É.M.; Boß, F.; Komnig, D.; Gärtner, C.; Höfs, L.; Shaykhalishahi, H.; Wördehoff, M.M.; Saridaki, T.; Schulz, J.B.; Hoyer, W.; et al. A β-Wrapin Targeting the N-Terminus of α-Synuclein Monomers Reduces Fibril-Induced Aggregation in Neurons. Front. Neurosci. 2021, 15, 696440. [Google Scholar] [CrossRef]
- Li, Y.; Glotfelty, E.J.; Karlsson, T.; Fortuno, L.V.; Harvey, B.K.; Greig, N.H. The Metabolite GLP-1 (9-36) Is Neuroprotective and Anti-Inflammatory in Cellular Models of Neurodegeneration. J. Neurochem. 2021, 159, 867–886. [Google Scholar] [CrossRef]
- Choi, D.C.; Yoo, M.; Kabaria, S.; Junn, E. MicroRNA-7 Facilitates the Degradation of Alpha-Synuclein and Its Aggregates by Promoting Autophagy. Neurosci. Lett. 2018, 678, 118–123. [Google Scholar] [CrossRef]
- Wiatrak, B.; Kubis-Kubiak, A.; Piwowar, A.; Barg, E. PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions. Cells 2020, 9, E958. [Google Scholar] [CrossRef]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T Mutant but Not Wild-Type Alpha-Synuclein in PC12 Cells Induces Alterations of the Ubiquitin-Dependent Degradation System, Loss of Dopamine Release, and Autophagic Cell Death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. α-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Barg, S.; Wiekop, P.; Lundberg, C.; Raymon, H.K.; Brundin, P. Effect of Mutant Alpha-Synuclein on Dopamine Homeostasis in a New Human Mesencephalic Cell Line. J. Biol. Chem. 2002, 277, 38884–38894. [Google Scholar] [CrossRef] [PubMed]
- Tönges, L.; Szegö, É.M.; Hause, P.; Saal, K.-A.; Tatenhorst, L.; Koch, J.C.; d’Hedouville, Z.; Dambeck, V.; Kügler, S.; Dohm, C.P.; et al. Alpha-Synuclein Mutations Impair Axonal Regeneration in Models of Parkinson’s Disease. Front. Aging Neurosci. 2014, 6, 239. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nadanaciva, S.; Berger, Z.; Shen, W.; Paumier, K.; Schwartz, J.; Mou, K.; Loos, P.; Milici, A.J.; Dunlop, J.; et al. Human A53T α-Synuclein Causes Reversible Deficits in Mitochondrial Function and Dynamics in Primary Mouse Cortical Neurons. PLoS ONE 2013, 8, e85815. [Google Scholar] [CrossRef]
- Engelender, S.; Kaminsky, Z.; Guo, X.; Sharp, A.H.; Amaravi, R.K.; Kleiderlein, J.J.; Margolis, R.L.; Troncoso, J.C.; Lanahan, A.A.; Worley, P.F.; et al. Synphilin-1 Associates with α-Synuclein and Promotes the Formation of Cytosolic Inclusions. Nat. Genet. 1999, 22, 110–114. [Google Scholar] [CrossRef]
- Tanaka, M.; Kim, Y.M.; Lee, G.; Junn, E.; Iwatsubo, T.; Mouradian, M.M. Aggresomes Formed by α-Synuclein and Synphilin-1 Are Cytoprotective. J. Biol. Chem. 2004, 279, 4625–4631. [Google Scholar] [CrossRef]
- Zhang, S.; Eitan, E.; Wu, T.-Y.; Mattson, M.P. Intercellular Transfer of Pathogenic α-Synuclein by Extracellular Vesicles Is Induced by the Lipid Peroxidation Product 4-Hydroxynonenal. Neurobiol. Aging 2018, 61, 52–65. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Valente, E.M. Phenotypic Spectrum of Alpha-Synuclein Mutations: New Insights from Patients and Cellular Models. Park. Relat. Disord. 2016, 22, S16–S20. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef]
- Liu, H.-S.; Jan, M.-S.; Chou, C.-K.; Chen, P.-H.; Ke, N.-J. Is Green Fluorescent Protein Toxic to the Living Cells? Biochem. Biophys. Res. Commun. 1999, 260, 712–717. [Google Scholar] [CrossRef]
- Detrait, E.R.; Bowers, W.J.; Halterman, M.W.; Giuliano, R.E.; Bennice, L.; Federoff, H.J.; Richfield, E.K. Reporter Gene Transfer Induces Apoptosis in Primary Cortical Neurons. Mol. Ther. 2002, 5, 723–730. [Google Scholar] [CrossRef]
- Howard, D.B.; Powers, K.; Wang, Y.; Harvey, B.K. Tropism and Toxicity of Adeno-Associated Viral Vector Serotypes 1, 2, 5, 6, 7, 8, and 9 in Rat Neurons and Glia in Vitro. Virology 2008, 372, 24–34. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-Synuclein Fibrils Seed the Formation of Lewy Body-like Intracellular Inclusions in Cultured Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M.-Y. α-Synuclein (ASyn) Preformed Fibrils Induce Endogenous ASyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M.-Y. Addition of Exogenous α-Synuclein Preformed Fibrils to Primary Neuronal Cultures to Seed Recruitment of Endogenous α-Synuclein to Lewy Body and Lewy Neurite–like Aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Intracerebral Inoculation of Pathological α-Synuclein Initiates a Rapidly Progressive Neurodegenerative α-Synucleinopathy in Mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.A.; Hasegawa, M. Prion-like Spreading of Pathological α-Synuclein in Brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The Release of Toxic Oligomers from α-Synuclein Fibrils Induces Dysfunction in Neuronal Cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef] [PubMed]
- Polinski, N.K.; Volpicelli-Daley, L.A.; Sortwell, C.E.; Luk, K.C.; Cremades, N.; Gottler, L.M.; Froula, J.; Duffy, M.F.; Lee, V.M.Y.; Martinez, T.N.; et al. Best Practices for Generating and Using Alpha-Synuclein Pre-Formed Fibrils to Model Parkinson’s Disease in Rodents. J. Parkinsons Dis. 2018, 8, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Abdelmotilib, H.; Maltbie, T.; Delic, V.; Liu, Z.; Hu, X.; Fraser, K.B.; Moehle, M.S.; Stoyka, L.; Anabtawi, N.; Krendelchtchikova, V.; et al. α-Synuclein Fibril-Induced Inclusion Spread in Rats and Mice Correlates with Dopaminergic Neurodegeneration. Neurobiol. Dis. 2017, 105, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Er, S.; Hlushchuk, I.; Airavaara, M.; Chmielarz, P.; Domanskyi, A. Studying Pre-Formed Fibril Induced α-Synuclein Accumulation in Primary Embryonic Mouse Midbrain Dopamine Neurons. J. Vis. Exp. 2020, 162, e61118. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- McCann, H.; Cartwright, H.; Halliday, G.M. Neuropathology of α-Synuclein Propagation and Braak Hypothesis. Mov. Disord. 2016, 31, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular Internalization of Alpha-Synuclein Aggregates by Cell Surface Heparan Sulfate Depends on Aggregate Conformation and Cell Type. Sci. Rep. 2017, 7, 9008. [Google Scholar] [CrossRef]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.-C.; Melki, R.; Zurzolo, C. Tunneling Nanotubes Spread Fibrillar α-Synuclein by Intercellular Trafficking of Lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Grudina, C.; Kouroupi, G.; Nonaka, T.; Hasegawa, M.; Matsas, R.; Zurzolo, C. Human NPCs Can Degrade α-Syn Fibrils and Transfer Them Preferentially in a Cell Contact-Dependent Manner Possibly through TNT-like Structures. Neurobiol. Dis. 2019, 132, 104609. [Google Scholar] [CrossRef]
- Senol, A.D.; Samarani, M.; Syan, S.; Guardia, C.M.; Nonaka, T.; Liv, N.; Latour-Lambert, P.; Hasegawa, M.; Klumperman, J.; Bonifacino, J.S.; et al. α-Synuclein Fibrils Subvert Lysosome Structure and Function for the Propagation of Protein Misfolding between Cells through Tunneling Nanotubes. PLOS Biol. 2021, 19, e3001287. [Google Scholar] [CrossRef]
- Danzer, K.M.; Ruf, W.P.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.T.; McLean, P.J. Heat-Shock Protein 70 Modulates Toxic Extracellular α-Synuclein Oligomers and Rescues Trans-Synaptic Toxicity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 326–336. [Google Scholar] [CrossRef]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-Neuron Transmission of α-Synuclein Fibrils through Axonal Transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef]
- Brahic, M.; Bousset, L.; Bieri, G.; Melki, R.; Gitler, A.D. Axonal Transport and Secretion of Fibrillar Forms of α-Synuclein, Aβ42 Peptide and HTTExon 1. Acta Neuropathol. 2016, 131, 539–548. [Google Scholar] [CrossRef]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology Underlying Clinical Variability in Patients with Synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef]
- Karpowicz, R.J.; Haney, C.M.; Mihaila, T.S.; Sandler, R.M.; Petersson, E.J.; Lee, V.M.-Y. Selective Imaging of Internalized Proteopathic α-Synuclein Seeds in Primary Neurons Reveals Mechanistic Insight into Transmission of Synucleinopathies. J. Biol. Chem. 2017, 292, 13482–13497. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The Process of Lewy Body Formation, Rather than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. PNAS 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Trinkaus, V.A.; Riera-Tur, I.; Martínez-Sánchez, A.; Bäuerlein, F.J.B.; Guo, Q.; Arzberger, T.; Baumeister, W.; Dudanova, I.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Architecture of Neuronal α-Synuclein Inclusions. Nat. Commun. 2021, 12, 2110. [Google Scholar] [CrossRef]
- Fujioka, S.; Ogaki, K.; Tacik, P.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Update on Novel Familial Forms of Parkinson’s Disease and Multiple System Atrophy. Park. Relat. Disord. 2014, 20, S29–S34. [Google Scholar] [CrossRef]
- Oliveira, L.M.A.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.-H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated α-Synuclein Caused by SNCA Gene Triplication Impairs Neuronal Differentiation and Maturation in Parkinson’s Patient-Derived Induced Pluripotent Stem Cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef]
- Lin, L.; Göke, J.; Cukuroglu, E.; Dranias, M.R.; VanDongen, A.M.J.; Stanton, L.W. Molecular Features Underlying Neurodegeneration Identified through In Vitro Modeling of Genetically Diverse Parkinson’s Disease Patients. Cell Rep. 2016, 15, 2411–2426. [Google Scholar] [CrossRef]
- Tagliafierro, L.; Zamora, M.E.; Chiba-Falek, O. Multiplication of the SNCA Locus Exacerbates Neuronal Nuclear Aging. Hum. Mol. Genet. 2019, 28, 407–421. [Google Scholar] [CrossRef]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marín-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. A Synuclein Control of Mitochondrial Homeostasis in Human-Derived Neurons Is Disrupted by Mutations Associated with Parkinson’s Disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.-M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein Oligomers Induce Early Axonal Dysfunction in Human IPSC-Based Models of Synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial Dysfunction and Mitophagy in Parkinson’s: From Familial to Sporadic Disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-Synuclein: Pathology, Mitochondrial Dysfunction and Neuroinflammation in Parkinson’s Disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Heger, L.M.; Wise, R.M.; Hees, J.T.; Harbauer, A.B.; Burbulla, L.F. Mitochondrial Phenotypes in Parkinson’s Diseases-A Focus on Human IPSC-Derived Dopaminergic Neurons. Cells 2021, 10, 3436. [Google Scholar] [CrossRef]
- Grassi, D.; Diaz-Perez, N.; Volpicelli-Daley, L.A.; Lasmézas, C.I. Pα-Syn* Mitotoxicity Is Linked to MAPK Activation and Involves Tau Phosphorylation and Aggregation at the Mitochondria. Neurobiol. Dis. 2019, 124, 248–262. [Google Scholar] [CrossRef]
- Seebauer, L.; Schneider, Y.; Drobny, A.; Plötz, S.; Koudelka, T.; Tholey, A.; Prots, I.; Winner, B.; Zunke, F.; Winkler, J.; et al. Interaction of Alpha Synuclein and Microtubule Organization Is Linked to Impaired Neuritic Integrity in Parkinson’s Patient-Derived Neuronal Cells. Int. J. Mol. Sci. 2022, 23, 1812. [Google Scholar] [CrossRef]
- Akrioti, E.; Karamitros, T.; Gkaravelas, P.; Kouroupi, G.; Matsas, R.; Taoufik, E. Early Signs of Molecular Defects in IPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease. Biomolecules 2022, 12, 876. [Google Scholar] [CrossRef] [PubMed]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s Disease Protein Alpha-Synuclein Is a Modulator of Processing Bodies and MRNA Stability. Cell 2022, 185, 2035–2056.e33. [Google Scholar] [CrossRef] [PubMed]
- Prieto Huarcaya, S.; Drobny, A.; Marques, A.R.A.; Di Spiezio, A.; Dobert, J.P.; Balta, D.; Werner, C.; Rizo, T.; Gallwitz, L.; Bub, S.; et al. Recombinant Pro-CTSD (Cathepsin D) Enhances SNCA/α-Synuclein Degradation in α-Synucleinopathy Models. Autophagy 2022, 18, 1127–1151. [Google Scholar] [CrossRef] [PubMed]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-Synuclein Aggregation in Parkinson’s Patient Neurons by Synergistic Enhancement of ER Proteostasis and Protein Trafficking. Neuron 2022, 110, 436–451.e11. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Lorente, R.; Lozano-Cruz, T.; Fernández-Carasa, I.; Miłowska, K.; de la Mata, F.J.; Bryszewska, M.; Consiglio, A.; Ortega, P.; Gómez, R.; Raya, A. Cationic Carbosilane Dendrimers Prevent Abnormal α-Synuclein Accumulation in Parkinson’s Disease Patient-Specific Dopamine Neurons. Biomacromolecules 2021, 22, 4582–4591. [Google Scholar] [CrossRef] [PubMed]
- Vajhøj, C.; Schmid, B.; Alik, A.; Melki, R.; Fog, K.; Holst, B.; Stummann, T.C. Establishment of a Human Induced Pluripotent Stem Cell Neuronal Model for Identification of Modulators of A53T α-Synuclein Levels and Aggregation. PLoS ONE 2021, 16, e0261536. [Google Scholar] [CrossRef]
- Gribaudo, S.; Tixador, P.; Bousset, L.; Fenyi, A.; Lino, P.; Melki, R.; Peyrin, J.-M.; Perrier, A.L. Propagation of α-Synuclein Strains within Human Reconstructed Neuronal Network. Stem Cell Rep. 2019, 12, 230–244. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic Alterations in Parkinson’s Disease Astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from α-Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific IPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between Astrocytes and Microglia Results in Increased Degradation of α-Synuclein and Amyloid-β Aggregates. J. Neuroinflammation 2021, 18, 124. [Google Scholar] [CrossRef]
- Russ, K.; Teku, G.; Bousset, L.; Redeker, V.; Piel, S.; Savchenko, E.; Pomeshchik, Y.; Savistchenko, J.; Stummann, T.C.; Azevedo, C.; et al. TNF-α and α-Synuclein Fibrils Differently Regulate Human Astrocyte Immune Reactivity and Impair Mitochondrial Respiration. Cell Rep. 2021, 34, 108895. [Google Scholar] [CrossRef]
- Filippini, A.; Mutti, V.; Faustini, G.; Longhena, F.; Ramazzina, I.; Rizzi, F.; Kaganovich, A.; Roosen, D.A.; Landeck, N.; Duffy, M.; et al. Extracellular Clusterin Limits the Uptake of A-synuclein Fibrils by Murine and Human Astrocytes. Glia 2021, 69, 681–696. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Kodella, K.R.; Manatakis, D.V.; Le, C.Y.; Hinojosa, C.D.; Tien-Street, W.; Manolakos, E.S.; Vekrellis, K.; Hamilton, G.A.; Ewart, L.; et al. Modeling Alpha-Synuclein Pathology in a Human Brain-Chip to Assess Blood-Brain Barrier Disruption. Nat. Commun. 2021, 12, 5907. [Google Scholar] [CrossRef]
- Azevedo, C.; Teku, G.; Pomeshchik, Y.; Reyes, J.F.; Chumarina, M.; Russ, K.; Savchenko, E.; Hammarberg, A.; Lamas, N.J.; Collin, A.; et al. Parkinson’s Disease and Multiple System Atrophy Patient IPSC-Derived Oligodendrocytes Exhibit Alpha-Synuclein–Induced Changes in Maturation and Immune Reactive Properties. Proc. Natl. Acad. Sci. USA 2022, 119, e2111405119. [Google Scholar] [CrossRef]
- Mohamed, N.-V.; Sirois, J.; Ramamurthy, J.; Mathur, M.; Lépine, P.; Deneault, E.; Maussion, G.; Nicouleau, M.; Chen, C.X.-Q.; Abdian, N.; et al. Midbrain Organoids with an SNCA Gene Triplication Model Key Features of Synucleinopathy. Brain Commun. 2021, 3, fcab223. [Google Scholar] [CrossRef]
- Brown, S.J.; Boussaad, I.; Jarazo, J.; Fitzgerald, J.C.; Antony, P.; Keatinge, M.; Blechman, J.; Schwamborn, J.C.; Krüger, R.; Placzek, M.; et al. PINK1 Deficiency Impairs Adult Neurogenesis of Dopaminergic Neurons. Sci Rep. 2021, 11, 6617. [Google Scholar] [CrossRef]
- Kano, M.; Takanashi, M.; Oyama, G.; Yoritaka, A.; Hatano, T.; Shiba-Fukushima, K.; Nagai, M.; Nishiyama, K.; Hasegawa, K.; Inoshita, T.; et al. Reduced Astrocytic Reactivity in Human Brains and Midbrain Organoids with PRKN Mutations. NPJ Park. Dis. 2020, 6, 33. [Google Scholar] [CrossRef]
- Zhu, W.; Tao, M.; Hong, Y.; Wu, S.; Chu, C.; Zheng, Z.; Han, X.; Zhu, Q.; Xu, M.; Ewing, A.G.; et al. Dysfunction of Vesicular Storage in Young-Onset Parkinson’s Patient-Derived Dopaminergic Neurons and Organoids Revealed by Single Cell Electrochemical Cytometry. Chem. Sci. 2022, 13, 6217–6223. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, W.; Ren, Y.; Fu, Y.; Martens, Y.A.; Shue, F.; Davis, M.D.; Wang, X.; Chen, K.; Li, F.; et al. Apolipoprotein E Regulates Lipid Metabolism and α-Synuclein Pathology in Human IPSC-Derived Cerebral Organoids. Acta Neuropathol. 2021, 142, 807–825. [Google Scholar] [CrossRef]
- Rodrigues, P.V.; de Godoy, J.V.P.; Bosque, B.P.; Amorim Neto, D.P.; Tostes, K.; Palameta, S.; Garcia-Rosa, S.; Tonoli, C.C.C.; de Carvalho, H.F.; de Castro Fonseca, M. Transcellular Propagation of Fibrillar α-Synuclein from Enteroendocrine to Neuronal Cells Requires Cell-to-Cell Contact and Is Rab35-Dependent. Sci. Rep. 2022, 12, 4168. [Google Scholar] [CrossRef]
- Jo, J.; Yang, L.; Tran, H.-D.; Yu, W.; Sun, A.X.; Chang, Y.Y.; Jung, B.C.; Lee, S.-J.; Saw, T.Y.; Xiao, B.; et al. Lewy Body–like Inclusions in Human Midbrain Organoids Carrying Glucocerebrosidase and α-Synuclein Mutations. Ann. Neurol. 2021, 90, 490–505. [Google Scholar] [CrossRef]
- Wulansari, N.; Darsono, W.H.W.; Woo, H.-J.; Chang, M.-Y.; Kim, J.; Bae, E.-J.; Sun, W.; Lee, J.-H.; Cho, I.-J.; Shin, H.; et al. Neurodevelopmental Defects and Neurodegenerative Phenotypes in Human Brain Organoids Carrying Parkinson’s Disease-Linked DNAJC6 Mutations. Sci. Adv. 2021, 7, eabb1540. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef]
- Kuznetsov, I.A.; Kuznetsov, A.V. Can the Lack of Fibrillar Form of Alpha-Synuclein in Lewy Bodies Be Explained by Its Catalytic Activity? Math. Biosci. 2022, 344, 108754. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ Is a Key Factor in α-Synuclein-Induced Neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef][Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Tanudjojo, B.; Shaikh, S.S.; Fenyi, A.; Bousset, L.; Agarwal, D.; Marsh, J.; Zois, C.; Heman-Ackah, S.; Fischer, R.; Sims, D.; et al. Phenotypic Manifestation of α-Synuclein Strains Derived from Parkinson’s Disease and Multiple System Atrophy in Human Dopaminergic Neurons. Nat. Commun. 2021, 12, 3817. [Google Scholar] [CrossRef]
- Yehezkel, S.; Rebibo-Sabbah, A.; Segev, Y.; Tzukerman, M.; Shaked, R.; Huber, I.; Gepstein, L.; Skorecki, K.; Selig, S. Reprogramming of Telomeric Regions during the Generation of Human Induced Pluripotent Stem Cells and Subsequent Differentiation into Fibroblast-like Derivatives. Epigenetics 2011, 6, 63–75. [Google Scholar] [CrossRef]
- Rohani, L.; Johnson, A.A.; Arnold, A.; Stolzing, A. The Aging Signature: A Hallmark of Induced Pluripotent Stem Cells? Aging Cell 2014, 13, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of Murine Microglial Cells by a V-Raf/v-Myc Carrying Retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Grozdanov, V.; Bousset, L.; Hoffmeister, M.; Bliederhaeuser, C.; Meier, C.; Madiona, K.; Pieri, L.; Kiechle, M.; McLean, P.J.; Kassubek, J.; et al. Increased Immune Activation by Pathologic α-Synuclein in Parkinson’s Disease. Ann. Neurol. 2019, 86, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-Dependent Defects of Alpha-Synuclein Oligomer Uptake in Microglia and Monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-Synuclein–Antibody Complexes Activate the NLRP3 Inflammasome in HiPSC-Derived Microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease. Cold Spring Harb. Perspect Biol. 2015, 7, a020628. [Google Scholar] [CrossRef]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-Synuclein and Its Disease-Causing Mutants Induce ICAM-1 and IL-6 in Human Astrocytes and Astrocytoma Cells. FASEB J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef]
- Rostami, J.; Holmqvist, S.; Lindström, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergström, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Sui, Y.-T.; Bullock, K.M.; Erickson, M.A.; Zhang, J.; Banks, W.A. Alpha Synuclein Is Transported into and out of the Brain by the Blood–Brain Barrier. Peptides 2014, 62, 197–202. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of α-Synuclein-Containing Erythrocyte-Derived Extracellular Vesicles across the Blood-Brain Barrier via Adsorptive Mediated Transcytosis: Another Mechanism for Initiation and Progression of Parkinson’s Disease? Acta Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.-Y.; Paul, G. Human α-Synuclein Overexpression in a Mouse Model of Parkinson’s Disease Leads to Vascular Pathology, Blood Brain Barrier Leakage and Pericyte Activation. Sci. Rep. 2021, 11, 1120. [Google Scholar] [CrossRef]
- Lan, G.; Wang, P.; Chan, R.B.; Liu, Z.; Yu, Z.; Liu, X.; Yang, Y.; Zhang, J. Astrocytic VEGFA: An Essential Mediator in Blood–Brain-Barrier Disruption in Parkinson’s Disease. Glia 2022, 70, 337–353. [Google Scholar] [CrossRef]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-Synuclein Induces Blood–Brain Barrier Dysfunction through Activated Brain Pericytes Releasing Inflammatory Mediators in Vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef]
- Kuan, W.-L.; Bennett, N.; He, X.; Skepper, J.N.; Martynyuk, N.; Wijeyekoon, R.; Moghe, P.V.; Williams-Gray, C.H.; Barker, R.A. α-Synuclein Pre-Formed Fibrils Impair Tight Junction Protein Expression without Affecting Cerebral Endothelial Cell Function. Exp. Neurol. 2016, 285, 72–81. [Google Scholar] [CrossRef]
- Richter-Landsberg, C.; Gorath, M.; Trojanowski, J.Q.; Lee, V.M. Alpha-Synuclein Is Developmentally Expressed in Cultured Rat Brain Oligodendrocytes. J. Neurosci. Res. 2000, 62, 9–14. [Google Scholar] [CrossRef]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.S.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: An In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef]
- Ahmed, Z.; Asi, Y.T.; Lees, A.J.; Revesz, T.; Holton, J.L. Identification and Quantification of Oligodendrocyte Precursor Cells in Multiple System Atrophy, Progressive Supranuclear Palsy and Parkinson’s Disease. Brain Pathol. 2012, 23, 263–273. [Google Scholar] [CrossRef]
- May, V.E.L.; Ettle, B.; Poehler, A.-M.; Nuber, S.; Ubhi, K.; Rockenstein, E.; Winner, B.; Wegner, M.; Masliah, E.; Winkler, J. α-Synuclein Impairs Oligodendrocyte Progenitor Maturation in Multiple System Atrophy. Neurobiol. Aging 2014, 35, 2357–2368. [Google Scholar] [CrossRef]
- Ettle, B.; Reiprich, S.; Deusser, J.; Schlachetzki, J.C.M.; Xiang, W.; Prots, I.; Masliah, E.; Winner, B.; Wegner, M.; Winkler, J. Intracellular Alpha-Synuclein Affects Early Maturation of Primary Oligodendrocyte Progenitor Cells. Mol. Cell Neurosci. 2014, 0, 68–78. [Google Scholar] [CrossRef]
- Sekiya, H.; Kowa, H.; Koga, H.; Takata, M.; Satake, W.; Futamura, N.; Funakawa, I.; Jinnai, K.; Takahashi, M.; Kondo, T.; et al. Wide Distribution of Alpha-Synuclein Oligomers in Multiple System Atrophy Brain Detected by Proximity Ligation. Acta Neuropathol. 2019, 137, 455–466. [Google Scholar] [CrossRef]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human Cerebral Organoids Recapitulate Gene Expression Programs of Fetal Neocortex Development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Luo, C.; Lancaster, M.A.; Castanon, R.; Nery, J.R.; Knoblich, J.A.; Ecker, J.R. Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain. Cell Rep. 2016, 17, 3369–3384. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.-D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.-P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Renner, H.; Grabos, M.; Becker, K.J.; Kagermeier, T.E.; Wu, J.; Otto, M.; Peischard, S.; Zeuschner, D.; TsyTsyura, Y.; Disse, P.; et al. A Fully Automated High-Throughput Workflow for 3D-Based Chemical Screening in Human Midbrain Organoids. eLife 2020, 9, e52904. [Google Scholar] [CrossRef] [PubMed]
- Renner, H.; Grabos, M.; Schöler, H.R.; Bruder, J.M. Generation and Maintenance of Homogeneous Human Midbrain Organoids. Bio Protoc. 2021, 11, e4049. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia Emerge from Erythromyeloid Precursors via Pu.1- and Irf8-Dependent Pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-Resident Macrophages Originate from Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly Efficient Neural Conversion of Human ES and IPS Cells by Dual Inhibition of SMAD Signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia Innately Develop within Cerebral Organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. IPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef]
- Fagerlund, I.; Dougalis, A.; Shakirzyanova, A.; Gómez-Budia, M.; Pelkonen, A.; Konttinen, H.; Ohtonen, S.; Fazaludeen, M.F.; Koskuvi, M.; Kuusisto, J.; et al. Microglia-like Cells Promote Neuronal Functions in Cerebral Organoids. Cells 2021, 11, 124. [Google Scholar] [CrossRef]
- Xu, R.; Boreland, A.J.; Li, X.; Erickson, C.; Jin, M.; Atkins, C.; Pang, Z.P.; Daniels, B.P.; Jiang, P. Developing Human Pluripotent Stem Cell-Based Cerebral Organoids with a Controllable Microglia Ratio for Modeling Brain Development and Pathology. Stem Cell Rep. 2021, 16, 1923–1937. [Google Scholar] [CrossRef]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia Integration into Human Midbrain Organoids Leads to Increased Neuronal Maturation and Functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in Vivo Model of Functional and Vascularized Human Brain Organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of Human Vascularized Brain Organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Development of Human Brain Organoids with Functional Vascular-like System. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, L.; Wang, M.; Liu, J.; Zhong, S.; Li, R.; Li, P.; Guo, L.; Fang, A.; Chen, R.; et al. Vascularized Human Cortical Organoids (VOrganoids) Model Cortical Development in Vivo. PLoS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; An, J.-H.; Yang, H.-J.; Lee, D.G.; Kim, J.; Koh, H.; Park, Y.-H.; Song, B.-S.; Sim, B.-W.; Lee, H.J.; et al. Human Blood Vessel Organoids Penetrate Human Cerebral Organoids and Form a Vessel-Like System. Cells 2021, 10, 2036. [Google Scholar] [CrossRef] [PubMed]
- Kook, M.G.; Lee, S.-E.; Shin, N.; Kong, D.; Kim, D.-H.; Kim, M.-S.; Kang, H.K.; Choi, S.W.; Kang, K.-S. Generation of Cortical Brain Organoid with Vascularization by Assembling with Vascular Spheroid. Int. J. Stem Cells 2022, 15, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-Y.; Ju, X.-C.; Li, Y.; Zeng, P.-M.; Wu, J.; Zhou, Y.-Y.; Shen, L.-B.; Dong, J.; Chen, Y.-J.; Luo, Z.-G. Generation of Vascularized Brain Organoids to Study Neurovascular Interactions. eLife 2022, 11, e76707. [Google Scholar] [CrossRef]
- Kwak, T.H.; Kang, J.H.; Hali, S.; Kim, J.; Kim, K.-P.; Park, C.; Lee, J.-H.; Ryu, H.K.; Na, J.E.; Jo, J.; et al. Generation of Homogeneous Midbrain Organoids with in Vivo-like Cellular Composition Facilitates Neurotoxin-Based Parkinson’s Disease Modeling. Stem Cells 2020, 38, 727–740. [Google Scholar] [CrossRef]
- Dickson, D.W.; Heckman, M.G.; Murray, M.E.; Soto, A.I.; Walton, R.L.; Diehl, N.N.; van Gerpen, J.A.; Uitti, R.J.; Wszolek, Z.K.; Ertekin-Taner, N.; et al. APOE Ε4 Is Associated with Severity of Lewy Body Pathology Independent of Alzheimer Pathology. Neurology 2018, 91, e1182–e1195. [Google Scholar] [CrossRef]
- Su, C.-J.; Feng, Y.; Liu, T.-T.; Liu, X.; Bao, J.-J.; Shi, A.-M.; Hu, D.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-Interacting Protein Induced α-Synuclein Accumulation via Inhibition of Autophagic Flux: Implications for Parkinson’s Disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef]
- Nogueira, G.O.; Garcez, P.P.; Bardy, C.; Cunningham, M.O.; Sebollela, A. Modeling the Human Brain With Ex Vivo Slices and in Vitro Organoids for Translational Neuroscience. Front. Neurosci. 2022, 16, 838594. [Google Scholar] [CrossRef]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.L.; Ranasinghe, R.T.; et al. Small Soluble α-Synuclein Aggregates Are the Toxic Species in Parkinson’s Disease. Nat. Commun. 2022, 13, 5512. [Google Scholar] [CrossRef]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 558, 464–469. [Google Scholar] [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and Molecular Diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef]
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E.; et al. Time to Redefine PD? Introductory Statement of the MDS Task Force on the Definition of Parkinson’s Disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef]
- Parkkinen, L.; O’Sullivan, S.S.; Collins, C.; Petrie, A.; Holton, J.L.; Revesz, T.; Lees, A.J. Disentangling the Relationship between Lewy Bodies and Nigral Neuronal Loss in Parkinson’s Disease. J. Park. Dis. 2011, 1, 277–286. [Google Scholar] [CrossRef]



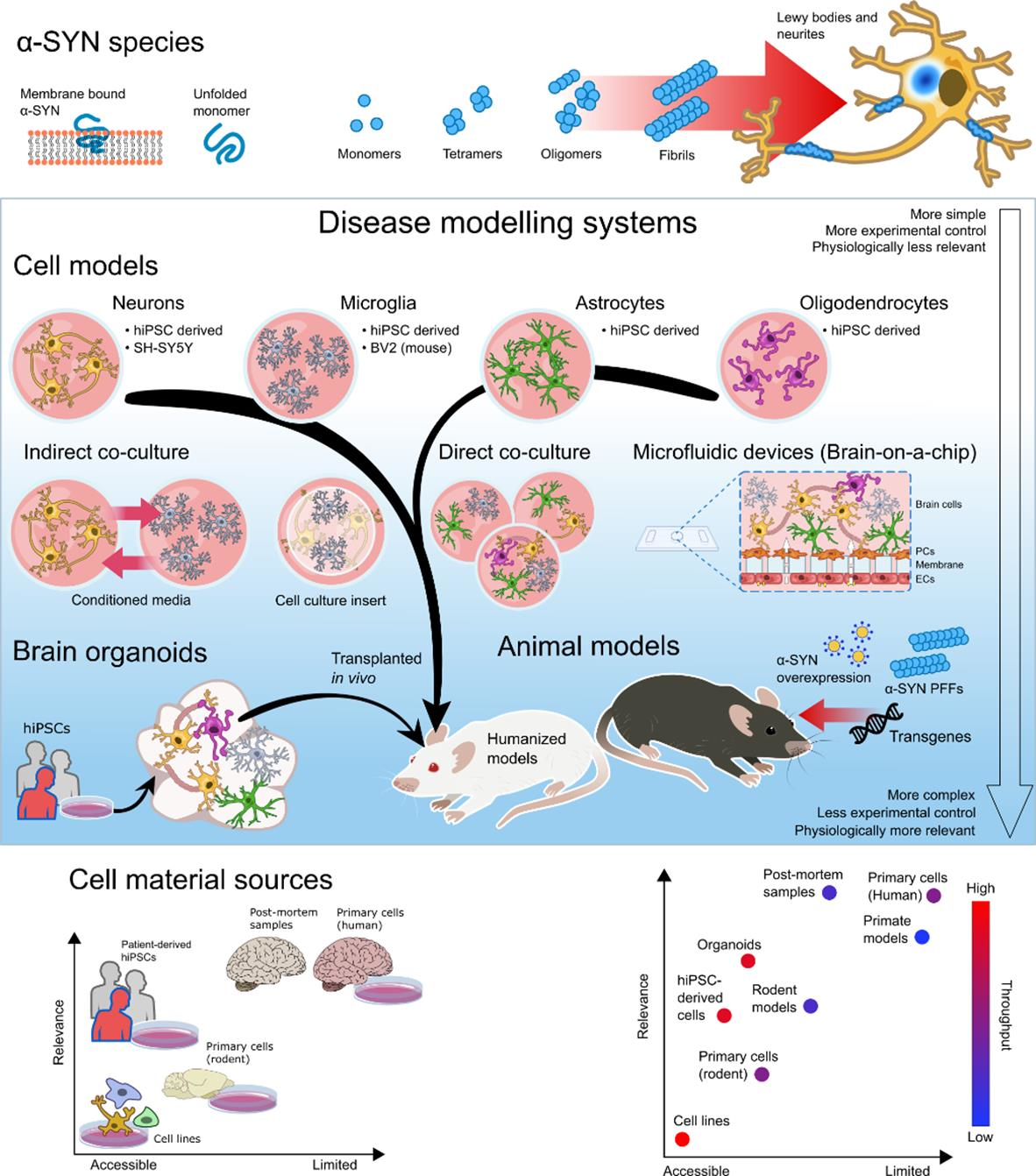{kind=link}
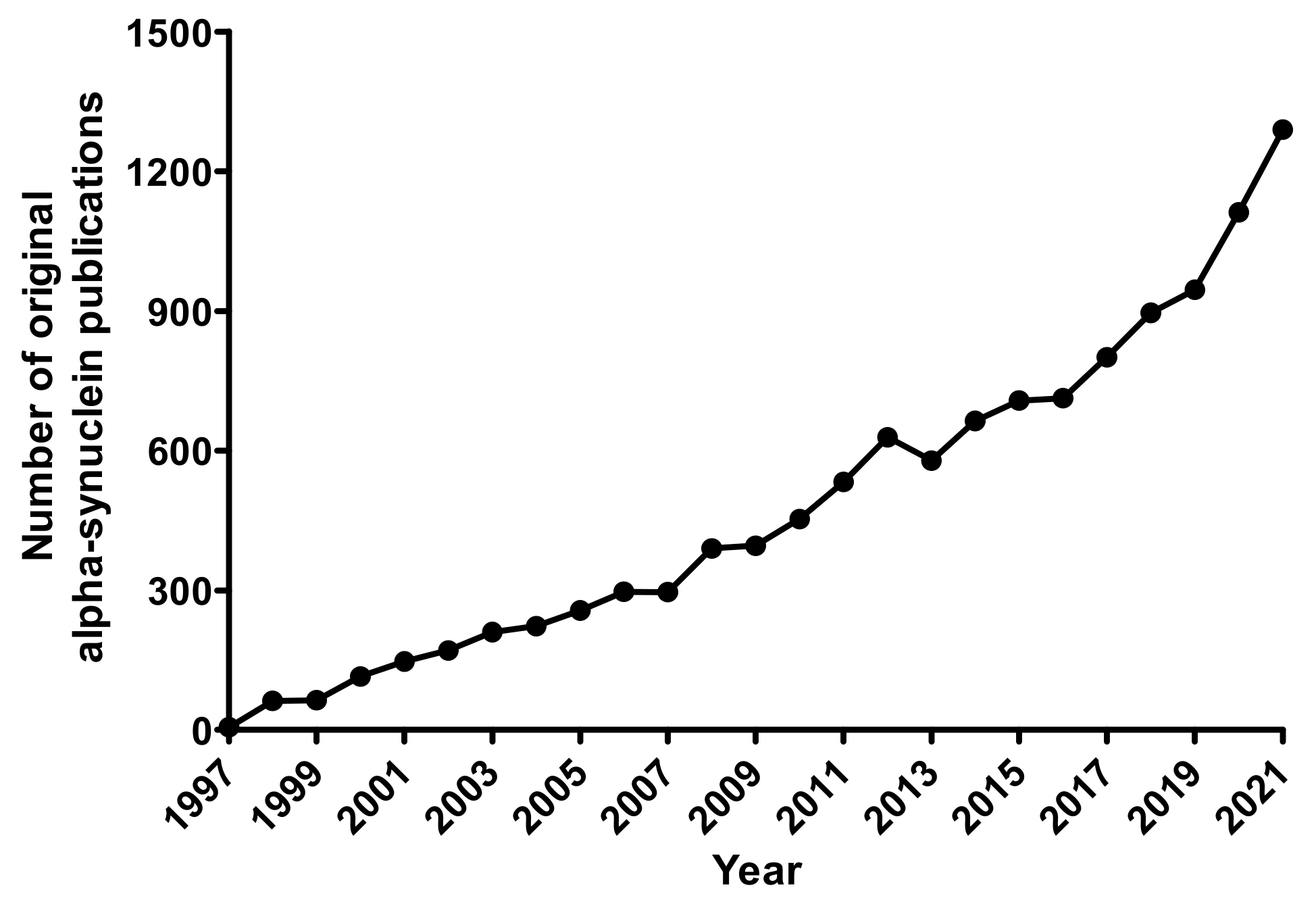{kind=link}
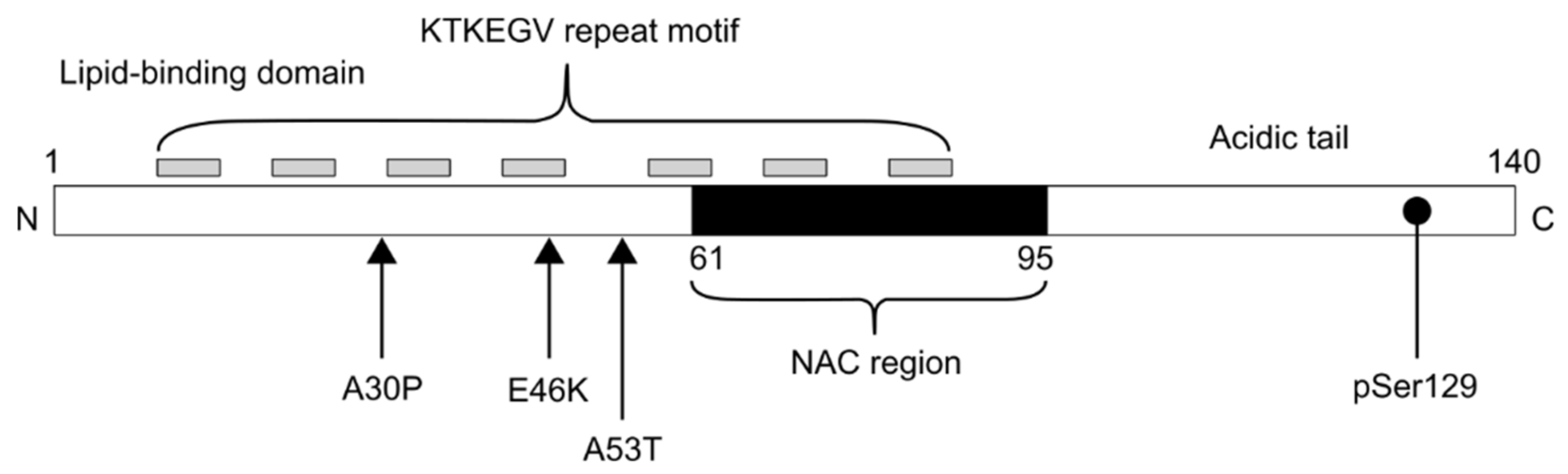{kind=link}
| Model | α-syn Modification | Outcomes of the Study | References |
|---|---|---|---|
| Cell lines | |||
| SH-SY5Y | Overexpression | Recipient microglia suppressed autophagy caused by enhanced expression of miR-19a-3p in exosomes, via the phosphatase and tensin homolog/AKT/mTOR signaling pathway. | [26] |
| Overexpression | miR-335 levels are reduced in PD models and patients. Its overexpression reduced inflammation induced by LPS stimulation or LRRK2 overexpression. | [27] | |
| A53T mutation | Reduced mitochondrial oxygen flow at maximum capacity. | [28] | |
| A53T mutation | Silencing of CK2α results in reduced phosphorylated α-syn at serine129 expression in cells with A53T mutation as well as functionality of dopaminergic neurons and ROS generation. | [29] | |
| PFFs | Micropinocytosis was suggested to be the main pathway of α-syn internalization into SH-SY5Y cells and differentiated neurons. | [30] | |
| PFFs | Higher secretion levels of nanoscopic α-syn aggregates were driven by disrupted protein homeostasis caused by PFF, but it does not lead to aggregation in the cells. | [31] | |
| Monomers, dimers, tetramers | α-syn dimer and tetramer internalization into the cell happened primarily through endocytosis. Aggregated α-syn from PFFs or oligomers displayed more prominent accumulation than monomers. Tetramer structures of α-syn showed more resistance to these processes, which suggested higher infectiousness of higher oligomeric states. | [32] | |
| Monomers, polymers,α-syn-119 | 1:4 ratio of α-syn -119:PQQ (pyrroloquinoline quinone) resulted in neuroprotective effect, showing antioxidant effect of PQQ. PQQ can change the secondary structure of α-syn, inhibiting oligomer formation induced by Cu(II). | [33] | |
| PC12 | Overexpression | Glutamine can enhance Hsp70 expression which is able to promote degradation of α-syn even in the presence of a proteasomal inhibitor. | [34] |
| Overexpression | Increase in oligomerization and aggregation of α-syn might be the result of iron accumulation in neurons. The abnormal iron levels can be caused by higher levels of α-syn concentration in the cells. | [35] | |
| Oligomers and overexpression | Serotonin aldehyde oligomerizes α-syn in vivo and in vitro. | [36] | |
| A53T mutation | Ubiquitin proteasome system dysfunctions caused by α-syn in dopaminergic neurons | [37] | |
| PFFs | PC12 cell line is less resistant to α-syn cytotoxicity than primary hippocampal neurons. | [38] | |
| PFFs | α-syn fibril formation can be inhibited by hydroxytyrosol, and fibrils can be destabilized by hydrotyrosol. | [39] | |
| (MPP+)-treated cells | Low-intensity ultrasounds stimulation results in ROS generation inhibition in MPP+ treated cells, lowering levels of α-syn aggregation. | [40] | |
| LUHMES | Overexpression | Transcriptome and proteasome analyses identified differential regulation of genes associated with PD. Vesicular transport and the lysosome were leading mechanisms. | [41] |
| SNCA knockout | 401 genes associated to the cell cycle had reduced expression after SNCA knockout in dopaminergic neurons. | [42] | |
| Overexpression | During drug screening, PDE1A inhibition showed the most effective results against α-syn toxicity. | [43] | |
| A30P mutation | Overexpression of WT and A30P m α-syn had a significant effect in DNA methylation of genes related to glutamate signaling and locomotor pathways | [44] | |
| MN9D | Normal expression | α-syn accumulation was inhibited by suppression of prolonged adenosine A1 receptor activation. | [45] |
| Normal expression | Damage induced by 6-OHDA can be reduced by icaritin (ICT). ICT increases SOD activity, TH expression, but decreased ROS production and α-syn expression. | [46] | |
| BV2 | A53T mutation | Polygala saponins fractions inhibited NLRP3 inflammasome by AMPK/mTOR and PINK1/parkin pathways, which contribute to the regulation of neuroinflammation decrease and neuronal death via mitophagy | [47] |
| α-syn and MPP+ co-treatment | α-syn and MPP+ co-treatment induced activation of NLRP3 inflammasome. | [48] | |
| Monomers, oligomers | Monomeric α-syn promotes microglial inflammatory phenotype by ERK, NF-κB, and PPARγ pathways. | [49] | |
| α-syn -enriched conditioned media | Neuroinflammation caused by impairment in microglial autophagy is disrupted by α-syn on the Tlr4-dependent p38 and Akt-mTOR pathway. | [50] | |
| PFFs | α-syn fibrils caused a strong inflammatory response. Level of fibrilization is a main trait for its intake. | [51] | |
| A53T mutation | Norepinephrine release caused dopaminergic neuron viability disruption in the noradrenergic system. | [52] | |
| Primary neurons | A53T α-syn T22N Rab7A mutations | Wild type Ras-related in brain 7 (Rab7) reduced α-syn decreased α-syn toxicity, e.g., oxidative stress, mitochondrial perturbations, and DNA damage. | [53] |
| PFFs and α-syn E35K E46K E61K mutants | Lysophosphatidylcholine acyltransferase 1 regulates α-syn pathology. Utilization of α-syn E35K E46K E61K model. | [54] | |
| LRRK2 inhibition | α-syn localization at the presynaptic terminal is connected to the kinase activity of LRRK2. | [55] | |
| PFFs | α-syn aggregation induced by PFFs is not influenced by insulin-related signaling in primary dopamine neurons. | [56] | |
| PFFs | Tannic acid showed the best results in two-step screening for α-syn aggregation inhibitors. | [57] | |
| PFFs and AS69 protein | There is no change in PFF uptake in the presence to AS69 protein, that binds to α-syn, but AS69 decreases α-syn pathology. | [58] | |
| Monomer | GLP-1R-associated neuroprotective and neurotrophic cell signaling can be activated by GLP-1 (9–36). | [59] | |
| Human neural stem cell line (ReNcell) | PFF or overexpression | α-syn and its aggregate degradation can happen with miR-7 use. miR-7 can also decrease α-syn expression. | [60] |
| iPSCs Model | α-syn Modification | Outcomes of the Study | References |
|---|---|---|---|
| iPSC- derived neurons | Overexpression | α-syn binds directly to the bTubIII and it is linked to the neuritic integrity in PD. | [111] |
| A53T mutation | Higher levels (mRNA) of α-syn, early changes in expression of genes related to metabolism, differentiation/development, ion transport, cytoskeleton, extracellular matrix organization, and synaptogenesis. | [112] | |
| A53T mutation | Abnormal accumulation of α-syn disrupts mRNA stability in PD iPSC neurons, disturbs the decapping module in PD brain. | [113] | |
| A53T mutation and isogenic line | Increase in SNCA/α-syn can be enhanced by recombinant pro-cathepsin D. | [114] | |
| A53T mutation | Neuron degradation, protein aggregates, increase in protein synthesis. | [28] | |
| SNCA A53T and GBA1 mutation | ER fragmentation can be caused by the α-syn accumulation in midbrain neurons. | [115] | |
| LRRK2 mutation | Carbosilane dendrimers use can counteract abnormal α-syn accumulation. | [116] | |
| A53T mutation and PFFs | Generation of reliable humanized seeding model for pharmacological research. | [117] | |
| PFFs and ribbons | Spreading of α-syn fibrils and ribbons, aggregation of endogenous α-syn. | [118] | |
| PFFs | α-syn aggregation in the form of phosphorylated α-syn. | ||
| iPSC-derived astrocytes | LRRK2 mutation | LRRK2 and GBA mutations in astrocytes contribute to PD development, manifesting several disease hallmarks. | [119] |
| ATP13A2 mutation | ATP13A2 mutation in astrocytes results in α-syn accumulation in dopaminergic neurons and ATP13A2 deficiency compromises protective astrocytes function from α-syn aggregation. | [120] | |
| LRRK2 mutation | Astrocytes play role in dopaminergic cell death in PD pathogenesis, by dysfunctions in pathway of protein degradation. | [121] | |
| PFFs | Astrocytes and microglia revealed synchronous activity in processing α-syn aggregates. | [122] | |
| PFFs | Exposure of α-syn to PFFs leads to antigen presenting phenotype in astrocytes with upregulation of major histocompatibility complex and antigen molecules, while TNF-α activates pro-inflammatory pathway. | [123] | |
| PFFs | Astrocytic α-syn uptake can be limited by binding to clusterin. A-syn clearance can be improved with lower clusterin levels. | [124] | |
| iPSCs-derived endothelial cells | PFFs | Generation of a substantia nigra brain chip, reproducing α-syn pathology in vivo during PFFs exposure. | [125] |
| iPSCs-derived oligodendrocytes and midbrain spheroids | Overexpression and A53T mutation | PD and MSA can affect oligodendrocytes in early cellular pathways and alterations. Epigenetic, genetic changes, and immune reactivity in MSA can be connected to each other by immune component triggered by α-syn. | [126] |
| Organoids (iPSCs) | Overexpression | SNCA triplication in midbrain organoids revealed pathological hallmarks of synucleinopathies in glial and neuronal cells. | [127] |
| PINK1 mutation | 2-Hydroxypropyl-β-Cyclodextrin treatment resulted in mitophagy improvement and better dopaminergic differentiation by protein levels modifications. | [128] | |
| PARKIN mutation | Mutation in PARKIN results in reduced IF activity. | [129] | |
| PINK1 mutation | Decreased amount of dopamine in vesicles, higher expression of α-syn. | [130] | |
| LRRK2 mutation | Midbrain organoids with LRRK2 mutation showed 3D pathological hallmarks of sporadic PD in patients. Thiol oxidoreductase functions are important in the LRRK2-associated PD development. | [131] | |
| APOE knockout | Lower levels of apoE induce aggregation of insoluble α-syn and phosphorylated α-syn, increased synapse loss, excess lipid droplet formation (hence GBA reduction and endo-lysosomal dysregulation). | [132] | |
| PFFs | Enteroendocrine cells are a key component of gut-brain hypothesis for the outcome and α-syn pathology development, and they show uptake and propagation of PFFs to neurons. | [133] | |
| Organoids (ESC-derived) | GBA1 knockout and overexpression | β-sheet–rich α-syn aggregates can be the result of loss of glucocerebrosidase, linked with α-syn overexpression. | [134] |
| DNAJC6 mutation | Loss of function in DNAJ6 gene results in α-syn aggregation caused by impairment in autophagy. | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albert, K.; Kälvälä, S.; Hakosalo, V.; Syvänen, V.; Krupa, P.; Niskanen, J.; Peltonen, S.; Sonninen, T.-M.; Lehtonen, Š. Cellular Models of Alpha-Synuclein Aggregation: What Have We Learned and Implications for Future Study. Biomedicines 2022, 10, 2649. https://doi.org/10.3390/biomedicines10102649
Albert K, Kälvälä S, Hakosalo V, Syvänen V, Krupa P, Niskanen J, Peltonen S, Sonninen T-M, Lehtonen Š. Cellular Models of Alpha-Synuclein Aggregation: What Have We Learned and Implications for Future Study. Biomedicines. 2022; 10(10):2649. https://doi.org/10.3390/biomedicines10102649
Chicago/Turabian StyleAlbert, Katrina, Sara Kälvälä, Vili Hakosalo, Valtteri Syvänen, Patryk Krupa, Jonna Niskanen, Sanni Peltonen, Tuuli-Maria Sonninen, and Šárka Lehtonen. 2022. "Cellular Models of Alpha-Synuclein Aggregation: What Have We Learned and Implications for Future Study" Biomedicines 10, no. 10: 2649. https://doi.org/10.3390/biomedicines10102649
APA StyleAlbert, K., Kälvälä, S., Hakosalo, V., Syvänen, V., Krupa, P., Niskanen, J., Peltonen, S., Sonninen, T.-M., & Lehtonen, Š. (2022). Cellular Models of Alpha-Synuclein Aggregation: What Have We Learned and Implications for Future Study. Biomedicines, 10(10), 2649. https://doi.org/10.3390/biomedicines10102649