
A Conjugate between Lqh-8/6, a Natural Peptide Analogue of Chlorotoxin, and Doxorubicin Efficiently Induces Glioma Cell Death

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
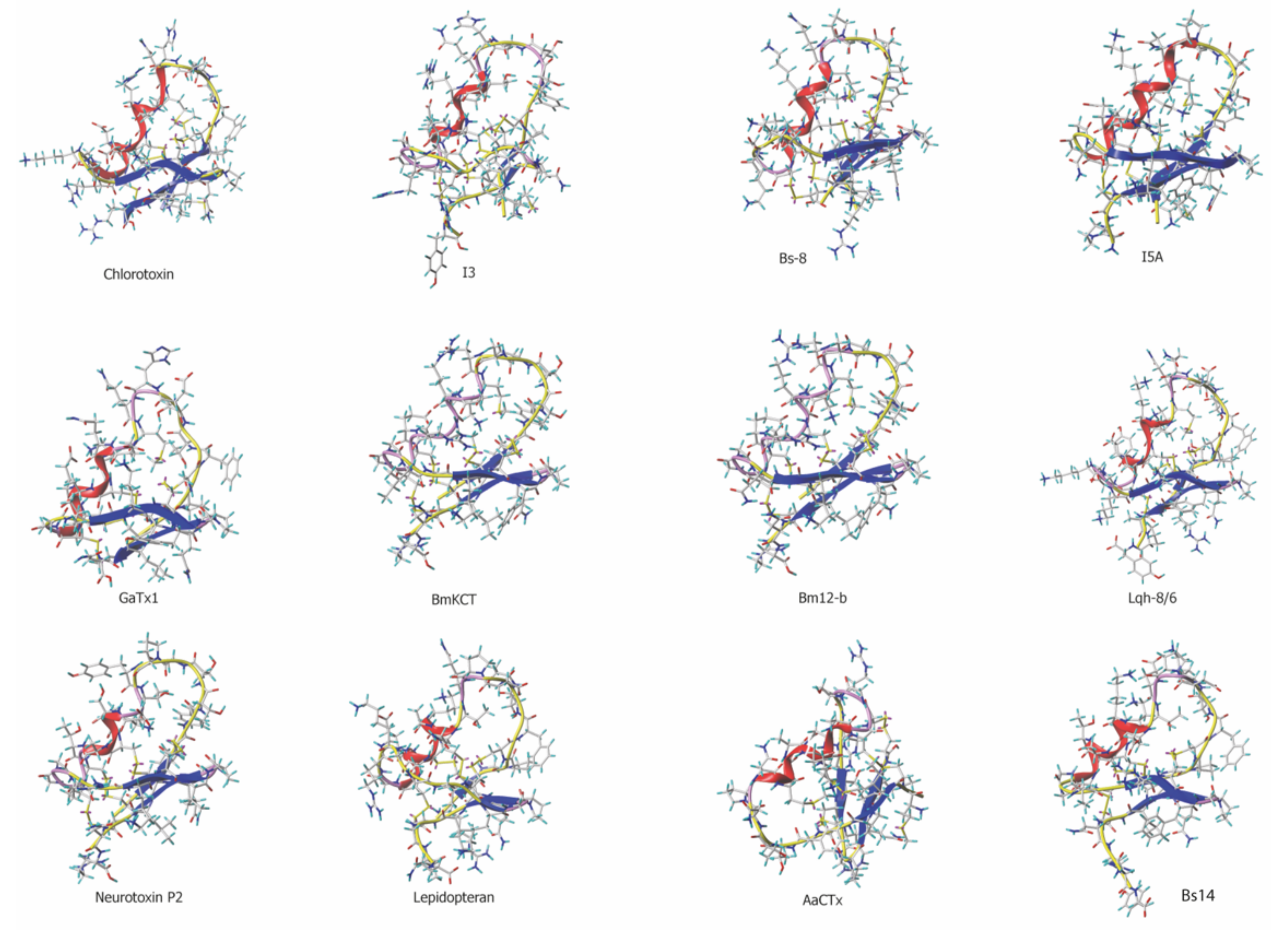
2.2. Molecular Modeling
2.3. Peptide Syntheses
2.4. Cell Cultures
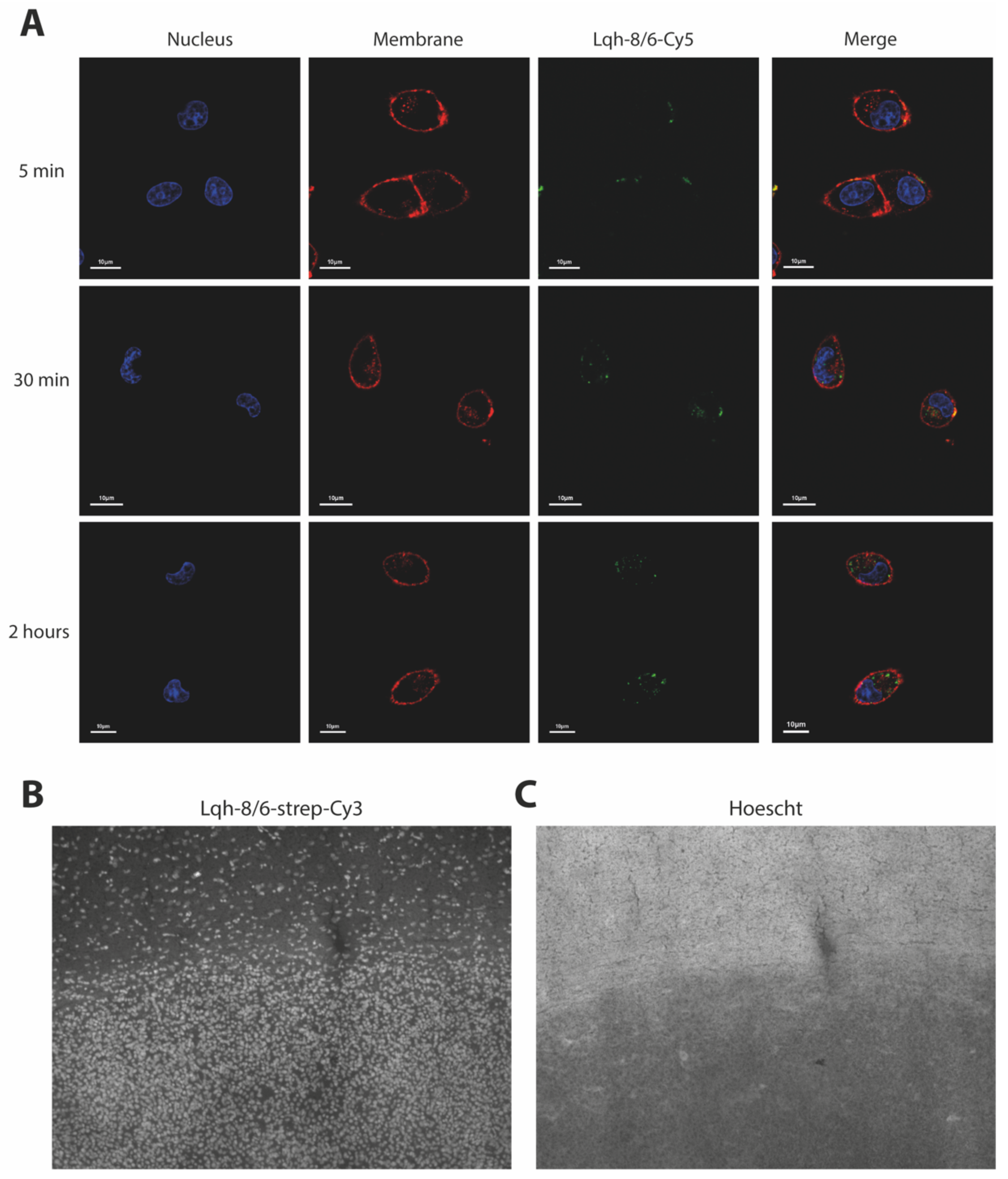
2.5. Confocal Microscope Imaging of GBM Cells
2.6. Cy5-Peptide Labeling of GBM F98 Cells in Culture
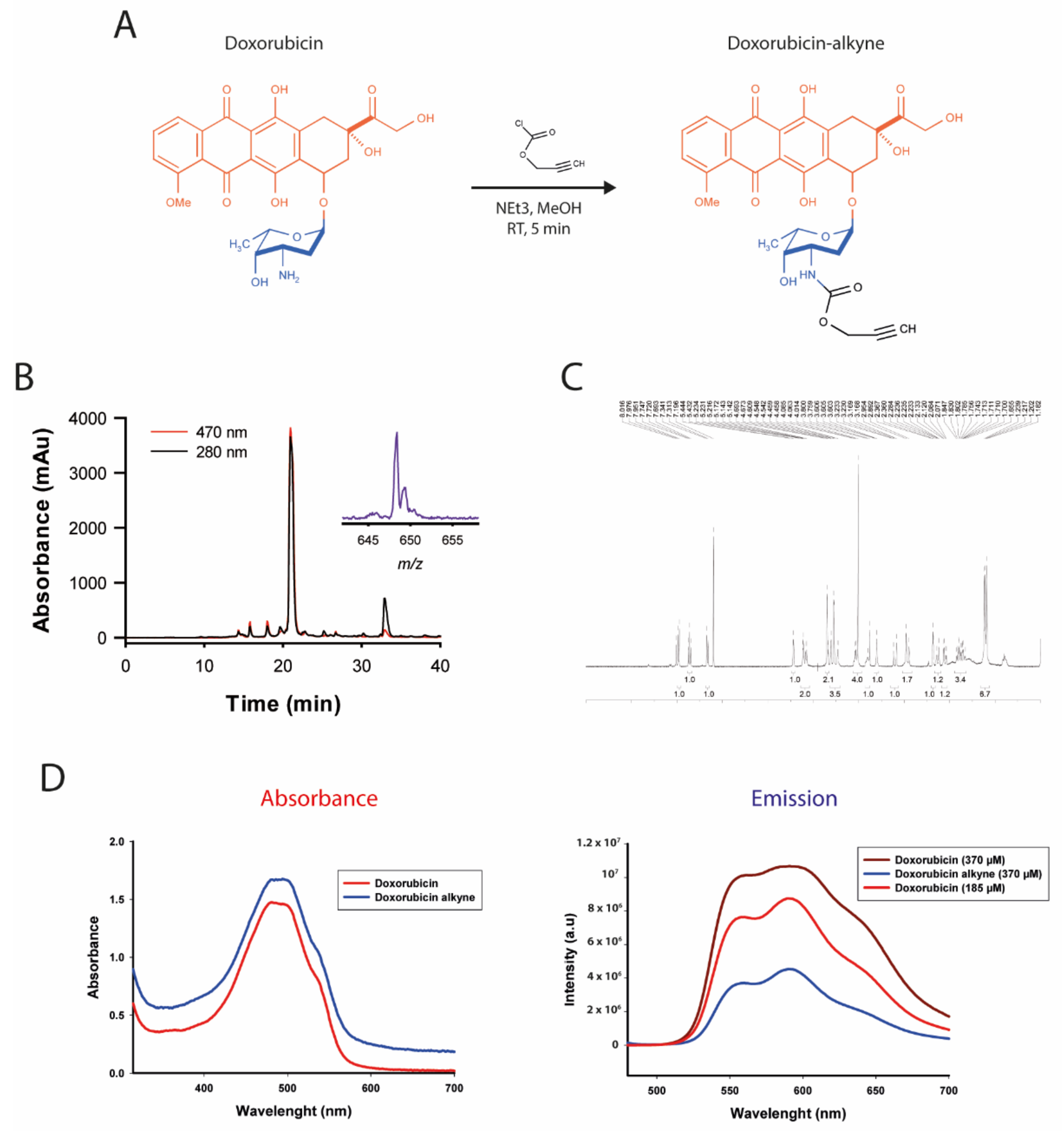
2.7. Synthesis of Doxorubicin-Alkyne
2.8. Conjugation of Doxorubicin-Alkyne to Lqh-8/6-azide
2.9. Excitation and Emission Spectra of Doxorubicin and Doxorubicin-Alkyne
2.10. Stereotaxic Implantation of F98 Cells in Fisher Rat Brain Striatum
2.11. Cy3-Peptide Labeling of F98 GBM Tumors in Brain Slices of Allograft Rats
2.12. Migration and Invasion Assays
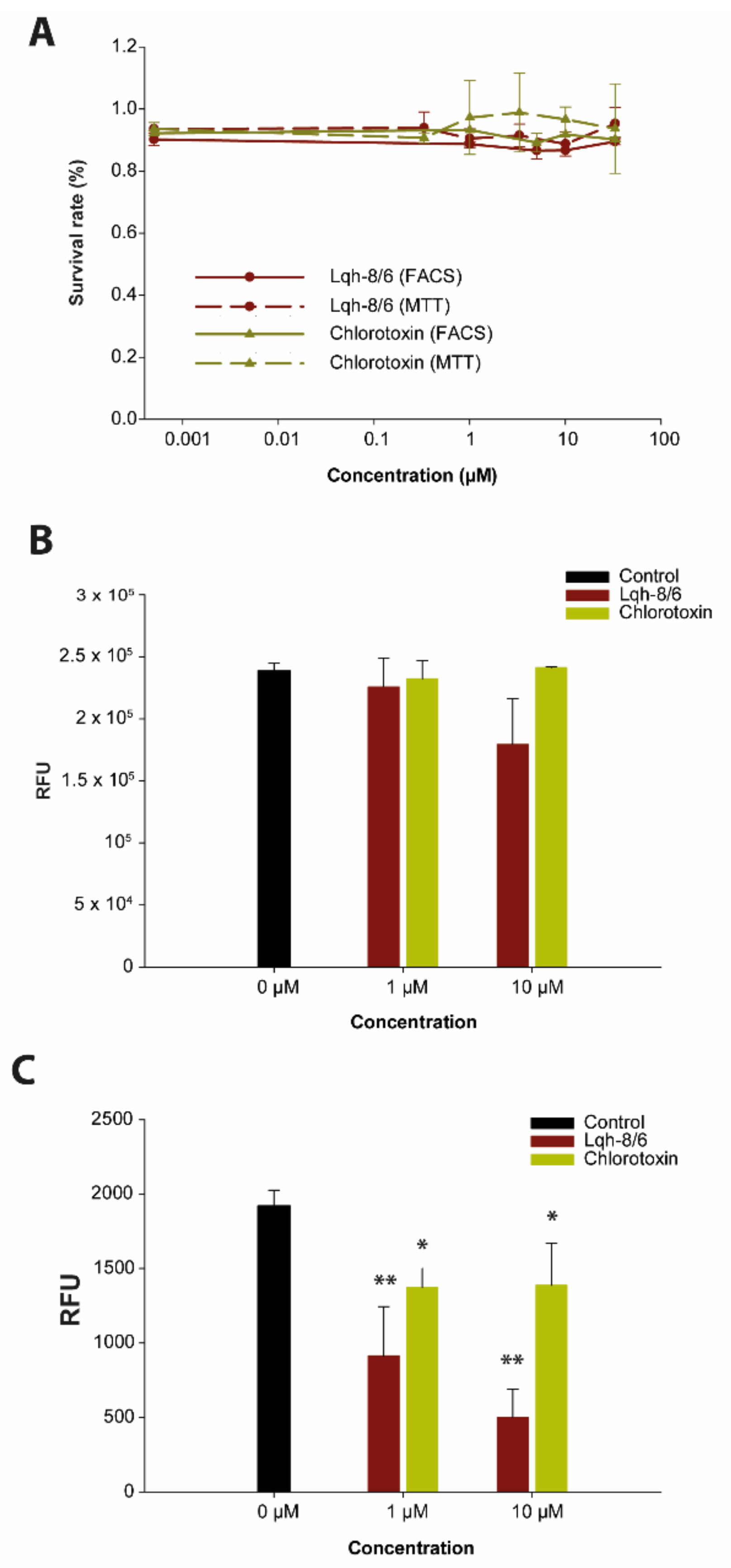
2.13. MTT Assays
2.14. GBM Cell Viability Assays by Flow Cytometry
2.15. Apoptosis Pathway Induced by Lqh-8/6, Doxorubicin-Alkyne and Lqh-8/6-Doxorubicin in U-87 Cells
2.16. Western Immunoblotting Analyses
3. Results
3.1. Lqh-8/6b Efficiently Labels GBM Tumor Cells
3.2. Lqh-8/6 Lacks Intrinsic Cell Toxicity
3.3. Lqh-8/6 Affects Invasion but Spares Migration
3.4. Chemical Synthesis and Characterization of a Click Chemistry-Compatible Doxorubicin Analogue
3.5. Coupling of Doxorubicin-Alkyne to Lqh-8/6-azide Produces an Efficient Anti-GBM Peptide-Drug Conjugate
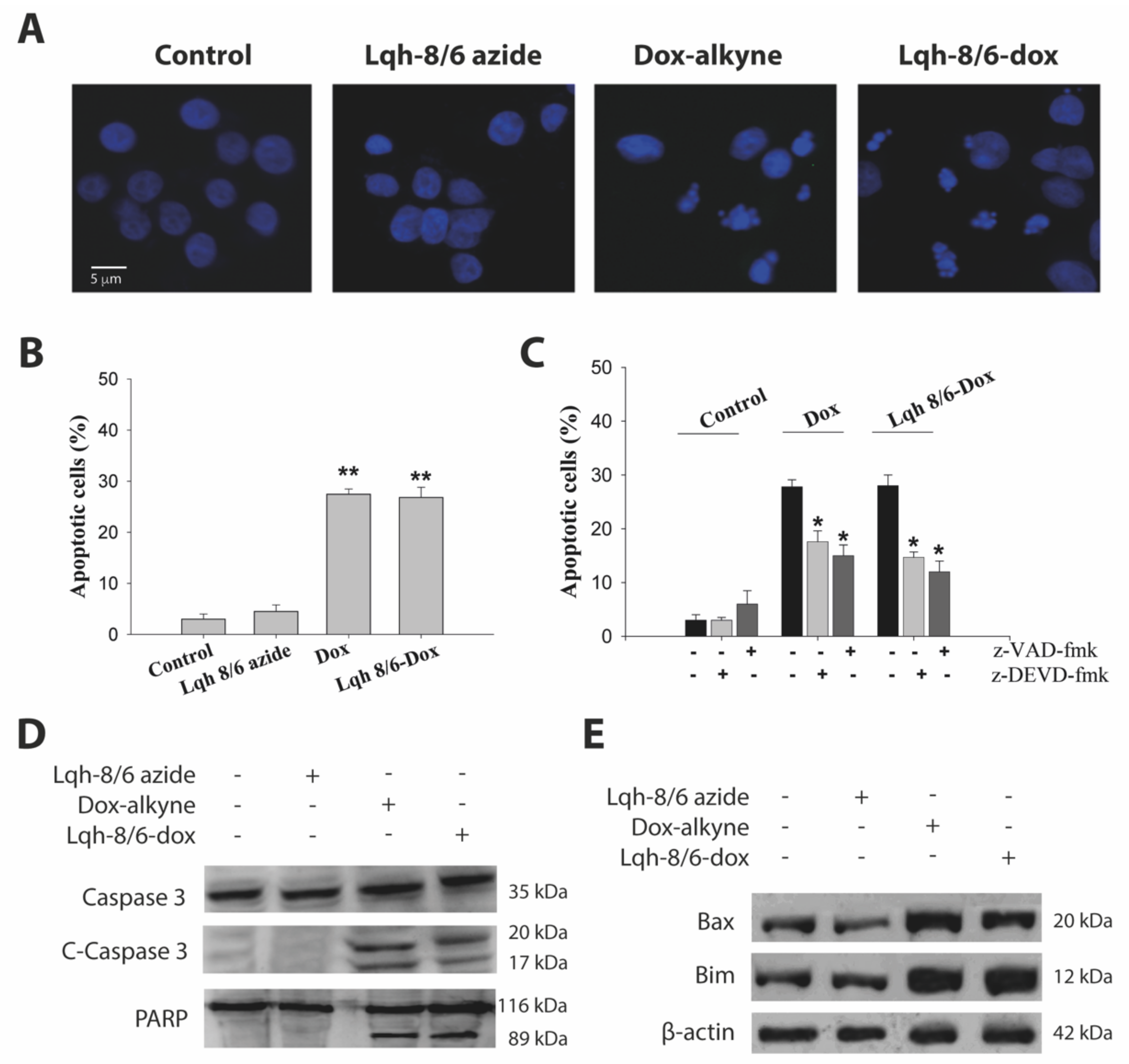
3.6. Lqh-8/6-Doxorubicin Induces GBM Cell Death by Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Glioblastoma. Available online: http://www.abta.org/brain-tumor-information/types-of-tumors/glioblastoma.html (accessed on 1 September 2022).
- De Biase, G.; Garcia, D.P.; Bohnen, A.; Quinones-Hinojosa, A. Perioperative Management of Patients with Glioblastoma. Neurosurg. Clin. N. Am. 2021, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular origin of glioblastoma and its implication in precision therapy. Cell Mol. Immunol. 2018, 15, 737–739. [Google Scholar] [CrossRef] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Behin, A.; Hoang-Xuan, K.; Carpentier, A.F.; Delattre, J.Y. Primary brain tumours in adults. Lancet 2003, 361, 323–331. [Google Scholar] [CrossRef]
- Clarke, J.L.; Chang, S.M. Neuroimaging: Diagnosis and response assessment in glioblastoma. Cancer J. 2012, 18, 26–31. [Google Scholar] [CrossRef]
- Stummer, W.; van den Bent, M.J.; Westphal, M. Cytoreductive surgery of glioblastoma as the key to successful adjuvant therapies: New arguments in an old discussion. Acta Neurochir. 2011, 153, 1211–1218. [Google Scholar] [CrossRef]
- Stupp, R.; Regg, C. New drugs and combinations for malignant glioma. Forum 2003, 13, 61–75. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Homicsko, K.; Negretti, L.; Lhermitte, B.; Stupp, R. Decision making and management of gliomas: Practical considerations. Ann. Oncol. 2012, 23 (Suppl. S10), x33–x40. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Ampie, L.; Woolf, E.C.; Dardis, C. Immunotherapeutic advancements for glioblastoma. Front. Oncol. 2015, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Okonogi, N.; Shirai, K.; Oike, T.; Murata, K.; Noda, S.E.; Suzuki, Y.; Nakano, T. Topics in chemotherapy, molecular-targeted therapy, and immunotherapy for newly-diagnosed glioblastoma multiforme. Anticancer Res. 2015, 35, 1229–1235. [Google Scholar]
- Feron, O. Tumor-penetrating peptides: A shift from magic bullets to magic guns. Sci. Transl. Med. 2010, 2, 34ps26. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [Green Version]
- Alberici, L.; Roth, L.; Sugahara, K.N.; Agemy, L.; Kotamraju, V.R.; Teesalu, T.; Bordignon, C.; Traversari, C.; Rizzardi, G.P.; Ruoslahti, E. De novo design of a tumor-penetrating peptide. Cancer Res. 2013, 73, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Dardevet, L.; Rani, D.; Aziz, T.A.; Bazin, I.; Sabatier, J.M.; Fadl, M.; Brambilla, E.; De Waard, M. Chlorotoxin: A helpful natural scorpion peptide to diagnose glioma and fight tumor invasion. Toxins 2015, 7, 1079–1101. [Google Scholar] [CrossRef]
- Mouhat, S.; Jouirou, B.; Mosbah, A.; De Waard, M.; Sabatier, J.M. Diversity of folds in animal toxins acting on ion channels. Biochem. J. 2004, 378, 717–726. [Google Scholar] [CrossRef]
- DeBin, J.A.; Maggio, J.E.; Strichartz, G.R. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am. J. Physiol. 1993, 264, C361–C369. [Google Scholar] [CrossRef]
- DeBin, J.A.; Strichartz, G.R. Chloride channel inhibition by the venom of the scorpion Leiurus quinquestriatus. Toxicon 1991, 29, 1403–1408. [Google Scholar] [CrossRef]
- Veiseh, O.; Gunn, J.W.; Kievit, F.M.; Sun, C.; Fang, C.; Lee, J.S.; Zhang, M. Inhibition of tumor-cell invasion with chlorotoxin-bound superparamagnetic nanoparticles. Small 2009, 5, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Modulation of glioma cell migration and invasion using Cl− and K+ ion channel blockers. J. Neurosci. 1999, 19, 5942–5954. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, N.; Bordey, A.; Gillespie, G.Y.; Sontheimer, H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience 1998, 83, 1161–1173. [Google Scholar] [CrossRef]
- Olsen, M.L.; Schade, S.; Lyons, S.A.; Amaral, M.D.; Sontheimer, H. Expression of voltage-gated chloride channels in human glioma cells. J. Neurosci. 2003, 23, 5572–5582. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef] [Green Version]
- Kesavan, K.; Ratliff, J.; Johnson, E.W.; Dahlberg, W.; Asara, J.M.; Misra, P.; Frangioni, J.V.; Jacoby, D.B. Annexin A2 is a molecular target for TM601, a peptide with tumor-targeting and anti-angiogenic effects. J. Biol. Chem. 2010, 285, 4366–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiseh, M.; Gabikian, P.; Bahrami, S.B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kievit, F.M.; Veiseh, O.; Fang, C.; Bhattarai, N.; Lee, D.; Ellenbogen, R.G.; Zhang, M. Chlorotoxin labeled magnetic nanovectors for targeted gene delivery to glioma. ACS Nano 2010, 4, 4587–4594. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, O.; Kievit, F.M.; Gunn, J.W.; Ratner, B.D.; Zhang, M. A ligand-mediated nanovector for targeted gene delivery and transfection in cancer cells. Biomaterials 2009, 30, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Ke, W.; Han, L.; Li, J.; Liu, S.; Jiang, C. Targeted delivery of chlorotoxin-modified DNA-loaded nanoparticles to glioma via intravenous administration. Biomaterials 2011, 32, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, O.; Sun, C.; Gunn, J.; Kohler, N.; Gabikian, P.; Lee, D.; Bhattarai, N.; Ellenbogen, R.; Sze, R.; Hallahan, A.; et al. Optical and MRI multifunctional nanoprobe for targeting gliomas. Nano Lett. 2005, 5, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Akcan, M.; Stroud, M.R.; Hansen, S.J.; Clark, R.J.; Daly, N.L.; Craik, D.J.; Olson, J.M. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J. Med. Chem. 2011, 54, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Graf, N.; Mokhtari, T.E.; Papayannopoulos, I.A.; Lippard, S.J. Platinum(IV)-chlorotoxin (CTX) conjugates for targeting cancer cells. J. Inorg. Biochem. 2012, 110, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Smertenko, A. Toxin evolution in scorpion venom: Evidence for toxin divergence under strong negative selection in Leiurus quinquestriatus subspecies. J. Toxicol. Toxin Rev. 2001, 20, 229–244. [Google Scholar] [CrossRef]
- Xu, T.; Fan, Z.; Li, W.; Dietel, B.; Wu, Y.; Beckmann, M.W.; Wrosch, J.K.; Buchfelder, M.; Eyupoglu, I.Y.; Cao, Z.; et al. Identification of two novel Chlorotoxin derivatives CA4 and CTX-23 with chemotherapeutic and anti-angiogenic potential. Sci. Rep. 2016, 6, 19799. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Sun, Z.; Jiang, D.; Dai, C.; Ma, Y.; Zhao, Z.; Liu, H.; Wu, Y.; Cao, Z.; Li, W. BmKCT toxin inhibits glioma proliferation and tumor metastasis. Cancer Lett. 2010, 291, 158–166. [Google Scholar] [CrossRef]
- Pons, J.L.; Labesse, G. @TOME-2: A new pipeline for comparative modeling of protein-ligand complexes. Nucleic Acids Res. 2009, 37, W485–W491. [Google Scholar] [CrossRef]
- Adjadj, E.; Naudat, V.; Quiniou, E.; Wouters, D.; Sautiere, P.; Craescu, C.T. Solution structure of Lqh-8/6, a toxin-like peptide from a scorpion venom—Structural heterogeneity induced by proline cis/trans isomerization. Eur. J. Biochem. 1997, 246, 218–227. [Google Scholar] [CrossRef]
- Tisseyre, C.; Ahmadi, M.; Bacot, S.; Dardevet, L.; Perret, P.; Ronjat, M.; Fagret, D.; Usson, Y.; Ghezzi, C.; De Waard, M. Quantitative evaluation of the cell penetrating properties of an iodinated Tyr-L-maurocalcine analog. Biochim. Biophys. Acta 2014, 1843, 2356–2364. [Google Scholar] [CrossRef]
- Liang, J.F.; Yang, V.C. Synthesis of doxorubicin-peptide conjugate with multidrug resistant tumor cell killing activity. Bioorg. Med. Chem. Lett. 2005, 15, 5071–5075. [Google Scholar] [CrossRef]
- Che, C.; Yang, G.; Thiot, C.; Lacoste, M.C.; Currie, J.C.; Demeule, M.; Regina, A.; Beliveau, R.; Castaigne, J.P. New Angiopep-modified doxorubicin (ANG1007) and etoposide (ANG1009) chemotherapeutics with increased brain penetration. J. Med. Chem. 2010, 53, 2814–2824. [Google Scholar] [CrossRef]
- Meyer-Losic, F.; Quinonero, J.; Dubois, V.; Alluis, B.; Dechambre, M.; Michel, M.; Cailler, F.; Fernandez, A.M.; Trouet, A.; Kearsey, J. Improved therapeutic efficacy of doxorubicin through conjugation with a novel peptide drug delivery technology (Vectocell). J. Med. Chem. 2006, 49, 6908–6916. [Google Scholar] [CrossRef]
- Ai, S.; Duan, J.; Liu, X.; Bock, S.; Tian, Y.; Huang, Z. Biological evaluation of a novel doxorubicin-peptide conjugate for targeted delivery to EGF receptor-overexpressing tumor cells. Mol. Pharm. 2011, 8, 375–386. [Google Scholar] [CrossRef]
- Rousselle, C.; Clair, P.; Lefauconnier, J.M.; Kaczorek, M.; Scherrmann, J.M.; Temsamani, J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol. Pharmacol. 2000, 57, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Rjeibi, I.; Mabrouk, K.; Mosrati, H.; Berenguer, C.; Mejdoub, H.; Villard, C.; Laffitte, D.; Bertin, D.; Ouafik, L.; Luis, J.; et al. Purification, synthesis and characterization of AaCtx, the first chlorotoxin-like peptide from Androctonus australis scorpion venom. Peptides 2011, 32, 656–663. [Google Scholar] [CrossRef]
- Mu, Q.; Lin, G.; Patton, V.K.; Wang, K.; Press, O.W.; Zhang, M. Gemcitabine and Chlorotoxin Conjugated Iron Oxide Nanoparticles for Glioblastoma Therapy. J. Mater. Chem. B 2016, 4, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, M.; Locatelli, E.; Rasile, M.; Monaco, I.; Rodighiero, S.; Corradini, I.; Franchini, M.C.; Passoni, L.; Matteoli, M. A Combined Approach Employing Chlorotoxin-Nanovectors and Low Dose Radiation To Reach Infiltrating Tumor Niches in Glioblastoma. ACS Nano 2016, 10, 2509–2520. [Google Scholar] [CrossRef]
- Zhao, L.; Shi, X.; Zhao, J. Chlorotoxin-conjugated nanoparticles for targeted imaging and therapy of glioma. Curr. Top. Med. Chem. 2015, 15, 1196–1208. [Google Scholar] [CrossRef]
- Fu, Y.; An, N.; Li, K.; Zheng, Y.; Liang, A. Chlorotoxin-conjugated nanoparticles as potential glioma-targeted drugs. J. Neurooncol. 2012, 107, 457–462. [Google Scholar] [CrossRef]
- Yeung, T.K.; Hopewell, J.W.; Simmonds, R.H.; Seymour, L.W.; Duncan, R.; Bellini, O.; Grandi, M.; Spreafico, F.; Strohalm, J.; Ulbrich, K. Reduced cardiotoxicity of doxorubicin given in the form of N-(2-hydroxypropyl)methacrylamide conjugates: And experimental study in the rat. Cancer Chemother. Pharmacol. 1991, 29, 105–111. [Google Scholar] [CrossRef]
- Injac, R.; Strukelj, B. Recent advances in protection against doxorubicin-induced toxicity. Technol. Cancer Res. Treat. 2008, 7, 497–516. [Google Scholar] [CrossRef]
- Aroui, S.; Dardevet, L.; Ben Ajmia, W.; de Boisvilliers, M.; Perrin, F.; Laajimi, A.; Boumendjel, A.; Kenani, A.; Muller, J.M.; De Waard, M. A Novel Platinum-Maurocalcine Conjugate Induces Apoptosis of Human Glioblastoma Cells by Acting through the ROS-ERK/AKT-p53 Pathway. Mol. Pharm. 2015, 12, 4336–4348. [Google Scholar] [CrossRef]
- Aroui, S.; Brahim, S.; De Waard, M.; Breard, J.; Kenani, A. Efficient induction of apoptosis by doxorubicin coupled to cell-penetrating peptides compared to unconjugated doxorubicin in the human breast cancer cell line MDA-MB 231. Cancer Lett. 2009, 285, 28–38. [Google Scholar] [CrossRef]
- Aroui, S.; Brahim, S.; Hamelin, J.; De Waard, M.; Breard, J.; Kenani, A. Conjugation of doxorubicin to cell penetrating peptides sensitizes human breast MDA-MB 231 cancer cells to endogenous TRAIL-induced apoptosis. Apoptosis 2009, 14, 1352–1365. [Google Scholar] [CrossRef]
- Aroui, S.; Brahim, S.; Waard, M.D.; Kenani, A. Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: A comparative study. Biochem. Biophys. Res. Commun. 2010, 391, 419–425. [Google Scholar] [CrossRef]
- Aroui, S.; Dardevet, L.; Najlaoui, F.; Kammoun, M.; Laajimi, A.; Fetoui, H.; De Waard, M.; Kenani, A. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to Human cell glioblastoma apoptosis by platinum-maurocalcin conjugate. Int. J. Biochem. Cell Biol. 2016, 77, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Mili, D.; Brahim, S.; De Waard, M.; Kenani, A. Doxorubicin coupled to penetratin promotes apoptosis in CHO cells by a mechanism involving c-Jun NH2-terminal kinase. Biochem. Biophys. Res. Commun. 2010, 396, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Ram, N.; Appaix, F.; Ronjat, M.; Kenani, A.; Pirollet, F.; De Waard, M. Maurocalcine as a non toxic drug carrier overcomes doxorubicin resistance in the cancer cell line MDA-MB 231. Pharm. Res. 2009, 26, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Perret, P.; Ahmadi, M.; Riou, L.; Bacot, S.; Pecher, J.; Poillot, C.; Broisat, A.; Ghezzi, C.; De Waard, M. Biodistribution, Stability, and Blood Distribution of the Cell Penetrating Peptide Maurocalcine in Mice. Int. J. Mol. Sci. 2015, 16, 27730–27740. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | Primary Sequence | Length | Identity | Disulfide Bridge Pattern | Species |
|---|---|---|---|---|---|
| Chlorotoxin | MC1MPC2 FTTDH QMARK C3DDC4C5 GGK-G RGKC6Y GPQC7L C8-R-- | 36 AA | 100% | C1-C4,C2-C6,C3-C7,C5-C8 | Leiurus quinquestriatus quinquestriatus |
| I3 | MC1MPC2 FTTDH QTARR C3RDC4C5 GGR-G R-KC6F G-QC7L C8GYD | 36 AA | 82% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus eupeus |
| I4 | MC1MPC2 FTTDH NMAKK C3RDC4C5 GGN-- -GKC6F GPQC7L C8NR | 35 AA | 82% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus eupeus |
| Bs-8 | RC1KPC2 FTTDP QMSKK C3ADC4C5 GGK-G KGKC6Y GPQC7L C8 | 35 AA | 80% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus sindicus |
| I5 | MC1MPC2 FTTDP NMANK C3RDC4C5 GGG-K K--C6F GPQC7L C8NR | 35 AA | 79% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus eupeus |
| I5A | MC1MPC2 FTTDP NMAKK C3RDC4C5 GGN-G K--C6F GPQC7L C8NR | 35 AA | 79% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus eupeus |
| GaTx1 | -C1GPC2 FTTDH QMEQK C3AEC4C5 GGI-G K--C6Y GPQC7L C8NR | 34 AA | 79% | C1-C4,C2-C6,C3-C7,C5-C8 | Leiurus quinquestriatus hebraeus |
| BmKCT | -C1GPC2 FTTDA NMARK C3REC4C5 GGI-G K--C6F GPQC7L C8NRI | 35 AA | 76% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus martensii |
| Bm12-b | -C1GPC2 FTTDA NMARK C3REC4C5 GGN-G K--C6F GPQC7L C8NRE | 35 AA | 76% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus martensii |
| Lqh-8/6 | RC1SPC2 FTTDQ QMTKK C3YDC4C5 GGK-G KGKC6Y GPQC7I C8APY | 38 AA | 72% | C1-C4,C2-C6,C3-C7,C5-C8 | Leiurus quinquestriatus hebraeus |
| I1 | MC1MPC2 FTTRP DMAQQ C3RAC4C5 KGR-G K--C6F GPQC7L C8GYD | 36 AA | 71% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus eupeus |
| Neurotoxin P2 | -C1GPC2 FTTDP YTESK C3ATC4C5 GGR-G K--C6V GPQC7L C8NRI | 35 AA | 70% | C1-C4,C2-C6,C3-C7,C5-C8 | Androctonus mauretanicus mauretanicus |
| Lepidopteran | RC1GPC2 FTTDP QTQAK C3SEC4C5 GRK-G G-VC6K GPQC7I C8GIQ | 37 AA | 63% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus tamulus |
| AaCtx | MC1IPC2 FTTNP NMAAK C3NAC4C5 GSRRG S--C6R GPQC7I C8 | 34 AA | 61% | C1-C4,C2-C6,C3-C7,C5-C8 | Androctonus australis |
| Bs-14 | -C1GPC2 FTKDP ETEKK C3ATC4C5 GGI-G R--C6F GPQC7L C8NRGY | 36 AA | 61% | C1-C4,C2-C6,C3-C7,C5-C8 | Buthus sindicus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dardevet, L.; Najlaoui, F.; Aroui, S.; Collot, M.; Tisseyre, C.; Pennington, M.W.; Mallet, J.-M.; De Waard, M. A Conjugate between Lqh-8/6, a Natural Peptide Analogue of Chlorotoxin, and Doxorubicin Efficiently Induces Glioma Cell Death. Biomedicines 2022, 10, 2605. https://doi.org/10.3390/biomedicines10102605
Dardevet L, Najlaoui F, Aroui S, Collot M, Tisseyre C, Pennington MW, Mallet J-M, De Waard M. A Conjugate between Lqh-8/6, a Natural Peptide Analogue of Chlorotoxin, and Doxorubicin Efficiently Induces Glioma Cell Death. Biomedicines. 2022; 10(10):2605. https://doi.org/10.3390/biomedicines10102605
Chicago/Turabian StyleDardevet, Lucie, Feten Najlaoui, Sonia Aroui, Mayeul Collot, Céline Tisseyre, Michael W. Pennington, Jean-Maurice Mallet, and Michel De Waard. 2022. "A Conjugate between Lqh-8/6, a Natural Peptide Analogue of Chlorotoxin, and Doxorubicin Efficiently Induces Glioma Cell Death" Biomedicines 10, no. 10: 2605. https://doi.org/10.3390/biomedicines10102605
APA StyleDardevet, L., Najlaoui, F., Aroui, S., Collot, M., Tisseyre, C., Pennington, M. W., Mallet, J.-M., & De Waard, M. (2022). A Conjugate between Lqh-8/6, a Natural Peptide Analogue of Chlorotoxin, and Doxorubicin Efficiently Induces Glioma Cell Death. Biomedicines, 10(10), 2605. https://doi.org/10.3390/biomedicines10102605